

Abstract
Amelogenins make up a class of proteins associated with the formation of mineralized enamel in vertebrates, possess highly conserved N- and C-terminal sequence regions, and represent an interesting model protein system for understanding biomineralization and protein assembly. Using bioinformatics, we report here the identification of molecular traits that classify 12 amelogenin proteins as members of the intrinsically disordered or unstructured protein family (IDPs), a group of proteins that normally exist as unfolded species but are capable of transformation to a folded state as part of their overall function. Using biophysical techniques (CD and NMR), we follow up on our bioinformatics studies and confirm that one of the amelogenins, recombinant porcine rP172, exists in an extended, unfolded state in the monomeric form. This protein exhibits evidence of conformational exchange between two states, and this exchange may be mediated by Pro residues in the sequence. Although the protein is globally unfolded, we detect the presence of local residual secondary structure [α-helix, extended β-strand, turn/loop, and polyproline type II (PPII)] that may serve several functional roles within the enamel matrix. The extended, labile conformation of rP172 amelogenin is compatible with the known functions of amelogenin in enamel biomineralization, i.e., self-assembly, associations with other enamel matrix proteins and with calcium phosphate biominerals, and interaction with cell receptors. It is likely that the labile structure of this protein facilitates interactions of amelogenin with other macromolecules or with minerals for achievement of internal protein stabilization.
The formation of inorganic compounds by organisms (biomineralization) is a substantial scientific puzzle. The ability of cells to employ proteins to control nucleation, crystal morphology, polymorphism, and the material properties of living tissues requires precise molecular control and efficient mechanisms (1). One such protein, amelogenin, is found in mammalian tooth enamel, one of the most highly mineralized materials of vertebrates (1–3). Amelogenin is essential for normal enamel development and is capable of protein self-association, forming supramolecular assemblies under defined conditions in the laboratory (4–6). These supramolecular assemblies (nanospheres) are believed to exert control over the morphology, organization, and directionality of hydroxyapatite crystal growth (7, 8). Primary sequence analysis of 26 mammalian lineages indicates that the N-terminus (Tyr-rich) and C-terminus (charged) of amelogenin are highly conserved, whereas variations occur in the central regions (9). Amelogenin sequence mutations lead to defective enamel crystal formation and organization (10,11), and deletion of the conserved terminal domains leads to the formation of ill-defined enamel crystals, highlighting the importance of these conserved domains in protein–protein or protein–mineral interactions (12). It has been suggested that amelogenin can interact with other important enamel matrix proteins that participate in enamel formation (13). Hence, the amelogenin protein family represents a highly relevant model for understanding protein-mediated biomineralization processes in nature.
One of the keys to understanding amelogenin function is to establish the structure of this protein. However, the determination of amelogenin structure has been limited by both the intrinsic lability of this protein and the self-association phenomenon. These traits are also common to other unfolded proteins, and recent studies have established that unfolded domains are functionally important for these proteins (14–19). Proteins or domains that lack organization and exist as a dynamic ensemble of interconverting structures have been classified as intrinsically disordered or natively unfolded proteins (IDPs)1 (18, 19). The amino acid compositions of IDPs are less complex and are dominated by disorder-promoting residues (E, K, R, G, Q, S, P, and A) (19). The functional attributes of IDPs include self-assembly into organized structures or binding to multiple targets with concomitant structural alterations (18, 19). These compositional and functional attributes are also common to the amelogenins as well (3–9, 20–23).
Hence, we believe that the functions of amelogenin will be better understood once we understand the molecular behavior of the unfolded, preassociative monomeric form of this protein. Such information will allow us to clarify conformational transitions and nonbonding interactions that occur when amelogenin interacts with itself or with other enamel matrix targets (i.e., non-amelogenins, cell surfaces, and mineral phase). This can be achieved by employing methods such as bioinformatics (24, 25) and biophysical techniques (14–17, 21) to directly link amelogenin to the IDP protein family and establish folded and unfolded regions of this protein. For biophysical experiments to be successful, care must be taken to stabilize the monomeric form of amelogenin and avoid problems that are encountered by aggregation (20–23). Recently, it has been shown that aggregation problems in proteins can be successfully minimized by utilizing unbuffered deionized distilled water (UDDW), where buffer-induced salting out effects that drive hydrophobic interactions are negligible (26). Under these conditions, a high degree of protein solubility, minimal hydrophobic intermolecular interactions, excellent NMR signal dispersion, and the appearance of important intrinsic conformational states have been reported (26). Hence, with this approach, a joint investigation using bioinformatics and IDP-specific structural techniques such as NMR spectroscopy (14–18) is now more feasible.
This study establishes a link between amelogenin and the IDP protein class. We first employ bioinformatic approaches (24, 25) to fingerprinting amelogenins as IDPs. We then utilize NMR spectroscopy, an IDP “friendly” technique (14–18, 26), to determine the global and local conformational states of a specific amelogenin (recombinant porcine amelogenin, rP172) in solution. Our bioinformatics data confirm that the majority of amelogenins from different species fall within the IDP category. This conclusion is also supported by our NMR data, where we observe that monomeric porcine rP172 amelogenin exists in an extended molecular configuration that consists of random coil conformation and partially structured regions (PPII, helix, extended β-strand, turn, or loop). These molecular features are consistent with both the in vitro behavior and the known in vivo enamel matrix functions of amelogenins.
MATERIALS AND METHODS
Calculation of Mean Hydrophobicity and Net Charge
The normalized values of the Kyte and Doolittle hydrophobicity scale for individual residues were obtained from http://www.expasy.org/tools/protscale.html. The amino acid sequences of the intrinsic disorder and folded proteins were obtained from ref 24. The mean hydrophobicity is the sum of the hydrophobicity of all amino acid residues divided by the total number of residues, and the mean net charge is the absolute net charge at pH 7.0 divided by the total number of residues.
Preparation of rP172 Recombinant Amelogenin
Recombinant porcine amelogenin [U-13C,15N]rP172 was expressed in Bio-Express Cell Growth Media (U-13C, 98%, U-15N, 98%) using Escherichia coli strain BL21-codon plus (DE3- RP, Stratagene, La Jolla, CA) as previously described (3, 4). The protein in the cell lysate was precipitated by 20% ammonium sulfate and purified using a reversed-phase column, in a Varian Prostar HPLC system. The precipitate was dissolved in 0.1% TFA and loaded onto a C4 semi-preparative column (214TP510, Vydac, Hesperia, CA) and fractionated using a linear gradient of 60% (v/v) acetonitrile at a flow rate of 2 mL/min. The homogeneity of the protein was confirmed by analytical chromatography using a C4 analytical column (214TP54, Vydac). The purity of the protein was more than 98%. rP172 has 172 amino acids and is an analogue of native P173 porcine amelogenin without the first methionine and the phosphate on Ser16 (27).
Dynamic Light Scattering Experiments
DLS measurements were performed on a DynaPro-99EMS/X instrument equipped with a solid-state laser operating at 655 nm with a temperature-controlled MicroSampler at 10 °C (Wyatt Technologies, Santa Barbara, CA). Amelogenin rP172 was dissolved in unbuffered double-distilled water (UDDW) to a final concentration of 1.5 mg/mL (75 µM, pH 6.0). The normalized intensity correlation function was analyzed by a regularization method included in the data analysis software package (Dynamics version 6.3.01), to give the information about the distribution of the exponential decay function with decay rate, Γ. The acquisition time for each run was set at 10 s. The experiment continued for 50–60 runs which corresponded to 8–10 min. The data analysis software analyzed the correlation function and provided the hydrodynamic radii and mass distribution of particles.
Circular Dichroism Experiments
CD measurements for amelogenin in UDDW were conducted on a JASCO J-810 spectropolarimeter calibrated using a 0.06% (+)-10-camphorsulfonic acid solution. The instrument optics and sample chamber were continually flushed with dry N2 gas at a rate of 20 L/min. The CD spectra were recorded at 10 °C in a 1 mm path length quartz cell (300 µL) by using a scanning speed of 50 nm/min, a response time of 1 s, a bandwidth of 1 nm, and an average of eight scans.
The variable-temperature (VT) experiments were performed on this same spectropolarimeter using a Peltier setup. For these VT experiments, the instrument optics and sample chamber were continually flushed with dry N2 gas at a rate of 20 L/min. The CD spectra were recorded at 5 and 10 °C in a 1 mm path length quartz cell (300 µL) by using a scanning speed of 50 nm/min, a response time of 1 s, a bandwidth of 1 nm, and an average of eight scans. A total of 16 scans were recorded to reduce the signal-to-noise ratio, and the resultant spectra were baseline-subtracted, and smoothed by the Savitzky–Golay method. The thermal dependency of the ellipticity at 224 nm was monitored at a heating or cooling rate of 60 °C/h.
NMR Spectroscopy
All experiments were performed at 283 K on a Bruker AVANCE 700 MHz four-channel NMR system equipped with an x,y,z axis PFG 5 mm HXY cryoprobe. The low temperature was chosen to stabilize the sample against aggregation and to slow conformational exchange. A rP172 sample was created using lyophilized [U-13C,15N]rP172 dissolved in Milli-Q pure deionized distilled water and 10% (v/v) D2O (99.99 atom % D, Cambridge Isotope Laboratories) in the absence of any buffer (i.e., the protein acts as its own buffer). This creates a 75 µM, pH 3.8 sample as measured with a NMR pH microelectrode using a standardized Radiometer Copenhagen pH meter. Monitoring of sample pH was performed periodically between experiments, and it was found that the sample pH remained stable near 4. Samples were placed in 5 mm symmetrical D2O-matched Shigemi NMR microtubes (Shigemi, Inc., Alison Park, PA). No visible aggregation was evident, and periodic 15N HSQC experiments were conducted to verify that line width broadening or chemical shift changes associated with aggregation did not occur. Sequential assignments were obtained using a suite of multidimensional NMR experiments (backbone-specific, HNCO, HNCACO, CBCACONH, and HNCACB; side chain-specific, CCCONH and HCCCONH) (15, 16). In addition to the backbone and side chain experiments, 15N-1H HSQC NOESY experiments (16) were conducted at optimal mixing times of 25, 50, 75, and 100 ms. NOE assignments were made using Sparky visualization software (SPARKY 3, University of California, San Francisco, CA). HNHA experiments (28) were conducted to determine 3JCH-NH coupling constants, and corresponding dihedral ϕ angles were calculated using the equation in the accompanying reference. All data were processed using NMR Pipe (National Institutes of Health, Bethesda, MD). The spectra were referenced with respect to the temperature-corrected water resonance (29), and 13C and 15N chemical shifts were referenced on the basis of the 1H reference following IUPAC guidelines using the unified chemical shift scale (29, 30). Acquisition and processing parameters are provided in the appropriate figure legends and Supporting Information. Sequential assignments were made using NMRViewJ, version 8.0.a11 (One Moon Scientific, Inc., Newark, NJ).
rP172 Amelogenin Model Building
The initial rP172 amelogenin Protein Data Bank files were generated using MacPyMol (DeLano Scientific, Palo Alto, CA). Torsion angles of the initial structures were not deliberately specified. Two structures were generated: one in which a neutral pH scenario was envisioned, where aspartate and glutamate side chains were negatively charged, and another assuming an acidic environment, in which the aspartate and glutamate side chains were protonated. In both cases, all cationic (Arg, His, and Lys) side chain groups were positively charged and the terminal regions were represented as NH3+ and COO−. All calculations were carried out using XPLOR-NIH. Protein parameters were defined using the default XPLOR protein parameter file, protein.par, which is based on the CHARMM19 force field. NMR interproton distance restraints based on experimental NOESY data were generated using the noe2xplor.py Python script (http://pbil.ibcp.fr/~nsapay/tools/script.html). Peak intensities were categorized as strong (1.8–2.8 Å), medium (1.8–3.6 Å), weak (1.8–5.0 Å), and very weak (1.8–6.0 Å). Pseudoatom corrections were used where necessary, and the corresponding corrections were included as part of the restraint input file. The NOE restraining function was defined as a soft-square well potential with a force constant of 50 kcal mol−1 Å−2. A 4.5 Å cutoff for nonbonding interactions was used. A total of 239 NOE restraints were used for the calculations. In addition to NOE distance restraints, residue-specific backbone dihedral angle restraints (ϕ and ψ) were used as part of the simulations. These restraints were generated from 1Hα, 13Cα, 13Cβ, 15N, and 13CO using TALOS. A total of 149 dihedral angle restraints were generated. The molecules were solvated implicitly through the use of a distance-dependent dielectric constant of 78.5.
The simulated annealing experiments were performed using a modified version of the XPLOR sa.inp file wherein each structure’s coordinates are randomized by a chaotic variation of each ϕ and ψ angle. This was followed by 30 ps of equilibrium dynamics at 300 K that were added after the initial minimization and before the structure was heated to 2000 K. The system was cooled from 2000 K at 100 K per step to 300 K. The resultant structures were then energy minimized with the Powell method. Calculations were conducted with a Verlet algorithm integrator in 1 fs time steps. Temperature coupling was achieved with a Langevin-type dynamics with zero random forces and a scaled friction coefficient. The accept.inp script was used to search the 1000 calculated structures for the 100 lowest-energy structures with zero NOE restraint violations and a minimum number of dihedral angle violations. From these 100 structures, the 10 lowest-energy structures were chosen for a conformer library; the final lowest-energy structure was then identified for additional comparisons. Visualization of structures, Ramachandran maps, and backbone alignment and rmsd calculations were performed using the VMD 1.8.6 software package.
RESULTS
Amino Acid Sequence Analysis of Vertebrate Amelogenins
Mammalian amelogenin sequences are highly conserved, and for the purposes of this study, we chose to analyze the sequences from 12 representative species. Interestingly, all 12 vertebrate amelogenins contain 50–60% disorder-promoting residues (Table 1) (19). In the absence of experimental assignments, the mean net charge (〉R〈) versus mean hydrophobicity (〉H〈) correlation (CH plot) has been developed for binary classification of proteins and provides perhaps the most intuitive description of overall protein conformation (18, 23). As shown in the CH plot (Figure 1), the IDPs are separated from folded proteins (indicated by the line) by the relation 〉H〈boundary = (〉R〈 + 1.151)/2.785. In the CH plot, porcine amelogenin is located in the region of intrinsically disordered proteins.
Table 1.
Analysis of Disorder- and Order-Promoting Residues in Amelogenin Sequences
| amelogenin (species) |
% disorder- promoting residues |
% order-promoting residues |
|---|---|---|
| bovine | 56.9 | 25.4 |
| chimpanzee | 56 | 24.6 |
| guinea pig | 55.2 | 27.1 |
| goat | 53.8 | 26.7 |
| golden hamster | 58.3 | 24.4 |
| horse | 52.3 | 31.3 |
| human | 56 | 24.6 |
| macaque | 55.4 | 24.6 |
| marmoset | 55.7 | 24.4 |
| orangutan | 55.4 | 24.6 |
| pig | 54.9 | 25.4 |
| rat | 58.3 | 24.4 |
FIGURE 1.
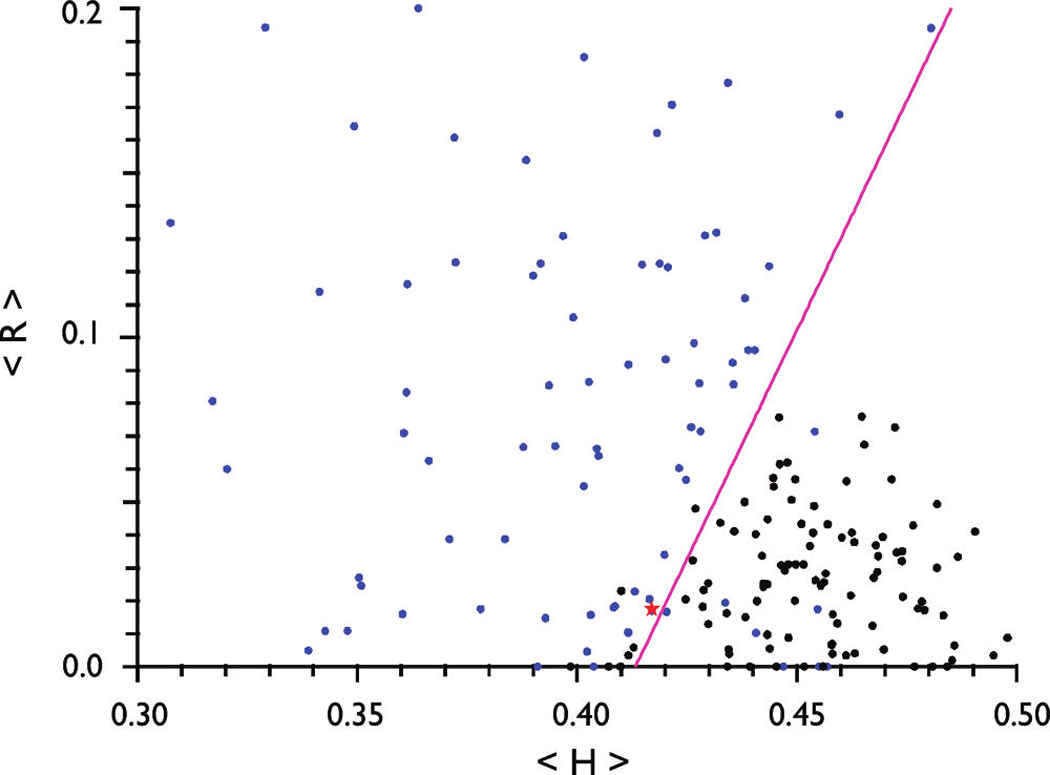
Computational analysis (CH plot) of porcine amelogenins. The mean net charge (〉R〈) vs mean hydrophobicity (〉H〈) plot for IDPs (blue circles), folded proteins (black circles), and the boundary [magenta line; 〉H〈boundary = (〉R〈 + 1.151)/2.785] that demarcates the two classes of proteins. The red star indicates the position of porcine amelogenin in the CH plot.
Biophysical Characterization of Amelogenin rP172 in Pure Water
Confirming the observations obtained for other aggregation-prone proteins (26), we found recombinant porcine amelogenin rP172 to be highly soluble in unbuffered, deionized distilled water (UDDW) at 10 °C; the protein solution appeared transparent and exhibited no observable turbidity. When the sample was dissolved in UDDW, the final pH of the unbuffered amelogenin sample was weakly acidic (i.e., the pH ranges from 3.8 to 6.0 but is stable over time). We believe that the variations in pH from sample to sample can be attributed to a number of factors, such as the source of deionized distilled water and variations in residual buffer salt retained by each protein preparation after purification (26).
Dynamic light scattering analysis of an amelogenin solution (concentration of 75 µM in UDDW, pH 6.0, 10 °C) revealed the monomeric state of the protein, although trace amounts of aggregates were also detected (Table 2). The CD spectra of rP172 were recorded at 5 and 10 °C (Figure 2A), and both spectra exhibit an intense negative minimum around 202 nm with an isoelliptic point around 209 nm. For the sake of comparison, we note that IDPs exhibit characteristic CD spectra with an intense negative minimum around 195–203 nm (31). Strong absorption bands in the n-π* region are typically associated with the presence of an ordered secondary structure (21, 31, 32), and the absence of these bands for rP172 in UDDW suggests that amelogenin exists in a disordered conformation (21, 25, 26, 31, 32).
Table 2.
Dynamic Light Scattering Data for rP172 Amelogenin in UDDWa
| item | R (nm) | polydispersity | MW-R (kDa) |
scattering intensity |
percentage mass |
|---|---|---|---|---|---|
| peak 1 | 1.4 | 0 | 7 | 1.5 | 100 |
| peak 2 | 103.2 | 15 | 173423 | 76.1 | 0 |
| peak 3 | 587.5 | 13.7 | 10140000 | 13.2 | 0 |
| peak 4 | 7970.4 | 0 | 4.53 × 109 | 9.2 | 0 |
Regularization data representing particle size distribution of unlabeled amelogenin rP172 in water at a concentration of 1.5 mg/mL (75 µM) and 10 °C. Note that unlabeled amelogenin rP172 was used. R is the particle radius; MW-R is the molecular weight. While the percentage of scattering intensity is mostly contributed by the larger aggregates, the majority of the mass is contributed by the monomers of amelogenin (percentage mass = 100). Amelogenin aggregates are present in trace amounts, but their scattering is very large.
FIGURE 2.
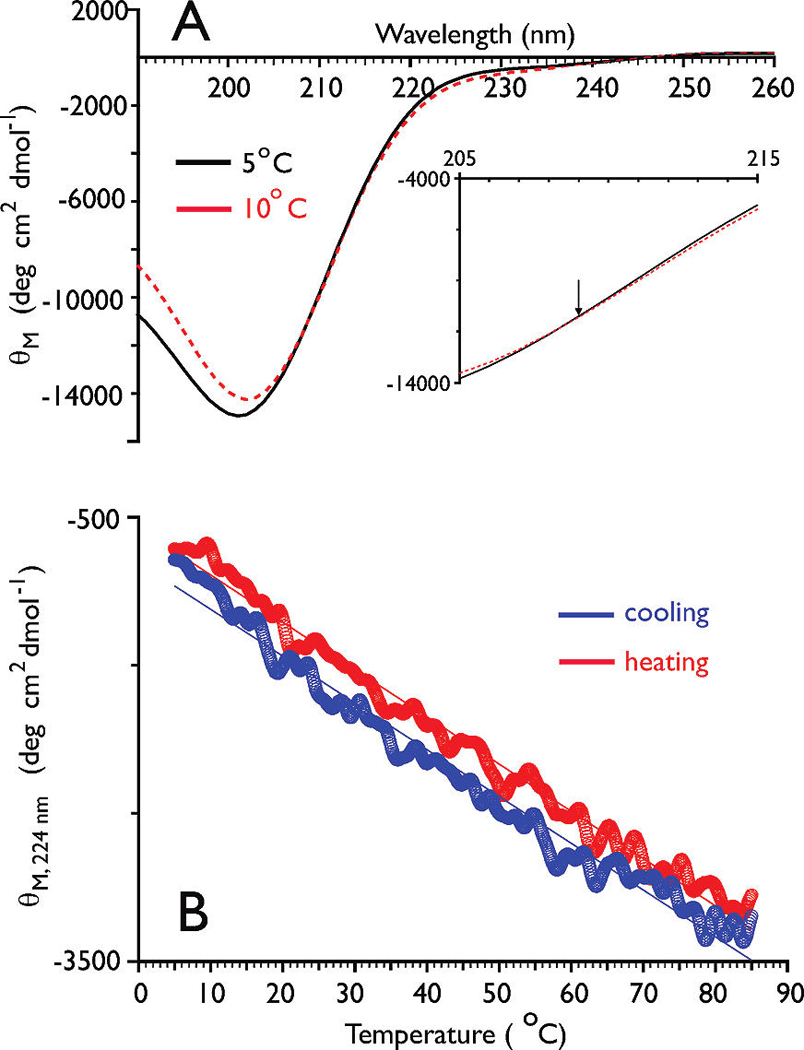
(A) CD spectra of rP172 (0.4 mg/mL) in UDDW. The inset spectrum is an enlarged region close to the isoelliptic point (denoted by the arrow). (B) Thermal dependency of ellipticity values at 224 nm for rP172 (0.4 mg/mL) in UDDW. The red circles indicate heating cycles, while the blue circles indicate cooling cycles.
The presence of an isoelliptic point in the CD spectra for rP172 indicates that an equilibrium exists between unordered and PPII conformations (33). A characteristic feature of proteins or polypeptides that exhibit an unordered PPII equilibrium is the presence of a linear dependency of the ellipticity values at 222 nm on temperature, in contrast to the sigmoidal dependence exhibited by proteins with α-helix/β-strand structures (33). To probe this, we measured the temperature-dependent melting and annealing behavior of rP172 and monitored the ellipticity intensity at 224 nm (Figure 2B). The presence of aggregation was ruled out by the absence of sample turbidity during heating as well as the reproducibility of ellipticity curves between a 10 °C sample and a parallel sample that prior to CD measurement was first heated to 85 °C and then cooled to 10 °C (Figure S1 of the Supporting Information). In UDDW, rP172 exhibits a linear transition over 5–85 °C with a slope value of −31.6 deg cm2 dmol−1 C−1 for heating and −31.9 deg cm2 dmol−1 C−1 for annealing (Figure 2B). The absence of any cooperative transition within this temperature range indicates that PPII structure is present at low temperatures and transforms to unordered/β-strand structure at elevated temperatures. An estimation of the PPII content was obtained from the limiting ellipticity values based on the assumption that the linear transition is an approximation to a broad sigmoidal curve (32, 33). From these values, the PPII content for rP172, in pure water at 10 °C, is estimated to be 22.7 ± 1.2%.
Global Conformational Features of Monomeric rP172
Intrinsically disordered or unfolded proteins present a challenge for structure determination by NMR due to the absence of stable, unique structures and the presence of conformational averaging, resulting in compromises in chemical shift dispersion, resonance broadening, and NOE cancellation (14–17, 26). Following the protocol utilized in the study of other unfolded proteins (14–17), we utilized a suite of triple-resonance NMR experiments (see Materials and Methods) that take advantage of the comparatively wide chemical shift dispersions of 15N and 13C spins and analyzed the backbone structure of rP172. We utilized low temperatures (10 °C) to slow conformational exchange (26, 34, 35) and prevent aggregation. Using this approach, the majority of the backbone resonances in [U-13C,15N]rP172 were resolved and sequentially assigned except for E40, Q126, and H132, which could not be identified (Table S1 and Figures S2 and S3 of the Supporting Information).
Having obtained backbone structure-sensitive data [i.e., 13Cα, 13Cβ, 13CO, and 1Hα backbone chemical shifts (36, 37); 3JCH-NH coupling constants (38, 39)], we next determined the extent of deviation of these rP172-specific NMR parameters from protein database random coil values (Figure 3), such that a global picture of rP172 folding could be obtained. Here, we note that the majority of secondary shift amplitudes are not particularly intense (Figure 3), indicating that there are no well-defined or stable helical or folded β-sheet secondary structures within rP172 in UDDW. We also analyzed the Pro 13Cβ and 13Cγ chemical shifts (Table S1 of the Supporting Information) and found that all of these are within the canonical chemical shift ranges of both the trans and cis configuration (40–42). However, if we apply the more stringent chemical shift difference (δ[13Cβ] – δ[13Cγ]) as a reference-independent descriptor of the Xaa–Pro peptide bond conformation (40), then we find that the majority of Pro residues adopt the trans configuration, with the exception of P41, P52, P96, P130, and P154, which resonate within the 13C chemical shift range for the cis configuration (40). Thus, we conclude the following. (a) The global structure of rP172 in the monomeric form is largely unfolded, and (b) the majority of Pro residues in rP172 appear to adopt the trans configuration, similar to what is observed within the Pro-rich titin PEVK entropic chain segment (37), a member of the IDP protein class (19).
FIGURE 3.
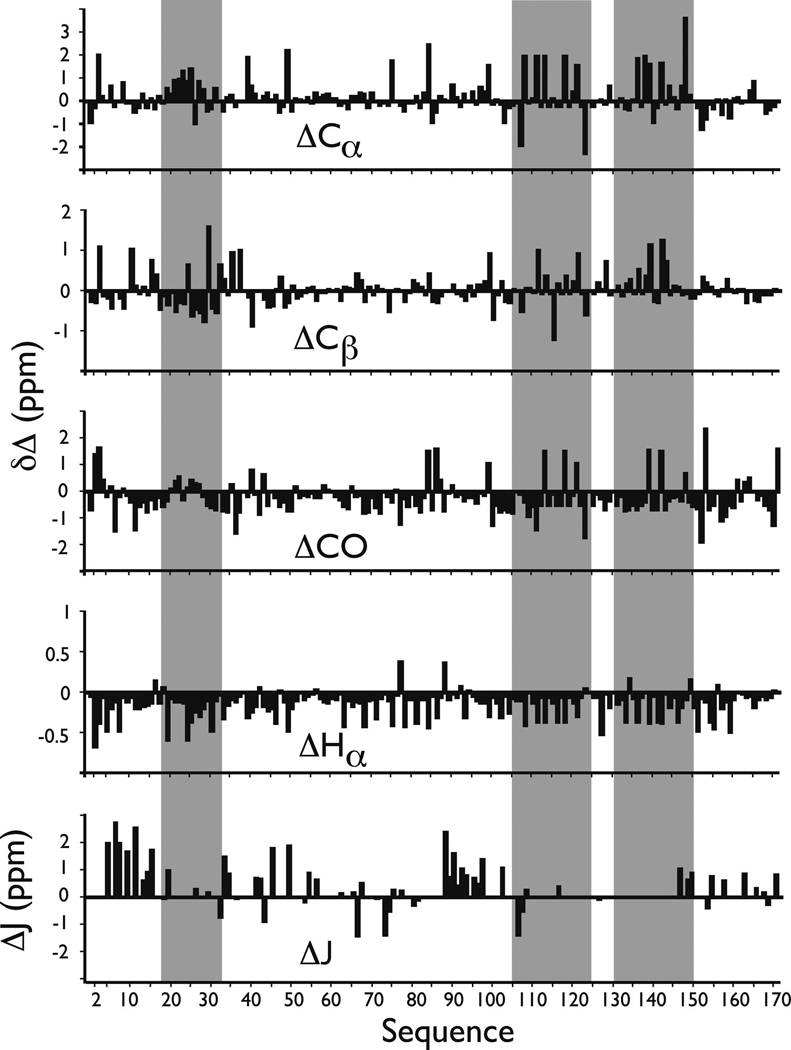
13C and 1H backbone conformational shifts and ΔJ values for rP172 amelogenin in UDDW. Values were determined from the NMR data presented in Table S1 of the Supporting Information. Gray stripes indicate sequence regions that exhibit non-random coil characteristics. In general, (+) secondary shifts for 13Cα and 13CO indicate helical structures whereas (−) shifts indicate a propensity for extended conformations such as β-strand (36, 37). For 1Hα and 13Cβ, (−) values indicate helical conformations and (+) values indicating extended conformations (36, 37). Deviations of ≥1 Hz from database 3JCH-NH random coil values are characteristic of structured domains (37, 38).
Evidence of Conformational Exchange within rP172 Amelogenin
Given that rP172 is largely unfolded, one would suspect that this protein would exhibit labile conformational qualities. This is confirmed by an examination of the HSQC spectra for rP172, where we note the presence of duplicated HSQC cross-peaks for residues V54, A63, W161, and T164 (Figures S2 and S3 of the Supporting Information). Duplication suggests the presence of two rP172 conformations, presumably in slow exchange with one another (43–45). Of these residues, only T164 exhibited well-defined cross-peaks whose volume integrations corresponding to the two conformations of T164 are approximately 2:1. Coincidentally, V54, A63, and W161 all lie within the proximity of an isolated proline residue (P59, P66, and P162, respectively). As reported elsewhere, duplication of HSQC resonances can arise from conformational exchange directed by cis–trans proline isomerization within proteins (43–45). Alternatively, it is known that Pro residues can function as a molecular “hinge” within protein sequence segments and influence the dynamics, conformation, and/or motion of sequence segments (43–45), which in turn could give rise to duplicated NMR cross-peaks (46). Thus, the lability of the rP172 protein is confirmed by the presence of conformational exchange, and it is possible that three of the conformational exchange sites may be modulated by Pro cis–trans interconversion and/or Pro-mediated sequence segment dynamics.
Assignment of Residual Secondary Structure within the rP172 Sequence
It is known that unfolded proteins can possess local regions where residual secondary structures reside (14–17). An examination of our qualitative NMR data set reveals that, although there is no well-defined, continuous regions of ordered structure, there are three short sequence segments that possess non-random coil J coupling and conformational shift deviations (Figure 3). The first is the V19-P33 segment within the tyrosine-rich amelogenin polypeptide domain (TRAP, P2-W45), whose negative 13Cα, 13Cβ, and 1Hα conformational shifts show consistent deviations from random coil values that correlate with helical propensities. We also note significant deviations from ΔJ values for residues which flank this segment (Figure 3) (38, 39). The second region is the P105–L125 segment which contains the redundant Pro- and Gln-rich sequence, and the third is the M131–S151 segment which contains the redundant Pro, Met sequence. Both of these regions possess consistent (+) and (−) 13Cα, 13Cβ, and 13CO conformational shifts and primarily (−) 1Hα shifts, indicating mixed propensities for either helix or sheet structure in both sequence blocks (36, 37).
The coexistence of global unfolded structure with residual local structure was also confirmed by quantitative NMR data [NOEs and J couplings (Figure 4)]. The absence of global folded structure is confirmed by (1) the lack of consistent sequential backbone, medium-range, and long-range backbone and sequential, medium-range, and long-range backbone–side chain NOEs (14–17, 26, 47, 48) and (2) 3J coupling constants which predominantly fall within the random coil range (38). However, this data set also confirms that rP172 does possess short, dispersed sequence regions which exhibit partially ordered secondary structure traits (Figure 4). These are summarized as follows. (a) For helix, using the medium-range dα-N(i,i+4) NOEs as a guide to α-helix occurrence (47, 48), we tentatively assign a short helical region at L23–Q27, which correlates with conformational shift deviations in that region (Figure 3). (b) For extended β-strand, using the extended β-strand criteria of 3J > 8 Hz, NOE intensity ratios, and the presence of short-range dα-N(i,i+1) and dNN(i,i+1) and the absence of medium-range backbone and backbone–side chain NOEs (47, 48), we tentatively assign extended β-strand structure to sequence regions of P2–sP22, L46–H62, and G90–Q101. This correlates with the low Pro content in these three sequence segments and the large deviation in ΔJ values (Figure 3). (c) For turn or loop regions, using the criteria of sequential and medium-range dα-N(i,i+2) and dNN(i,i+2) NOEs (48) and 3J < 6 Hz (38), we assign β-turn or loop structure to the Y37–W45 segment. Not surprisingly, this sequence segment, -YGYEPMGGW-, contains turn- or loop-forming amino acids Y, G, and M (34, 35).
FIGURE 4.
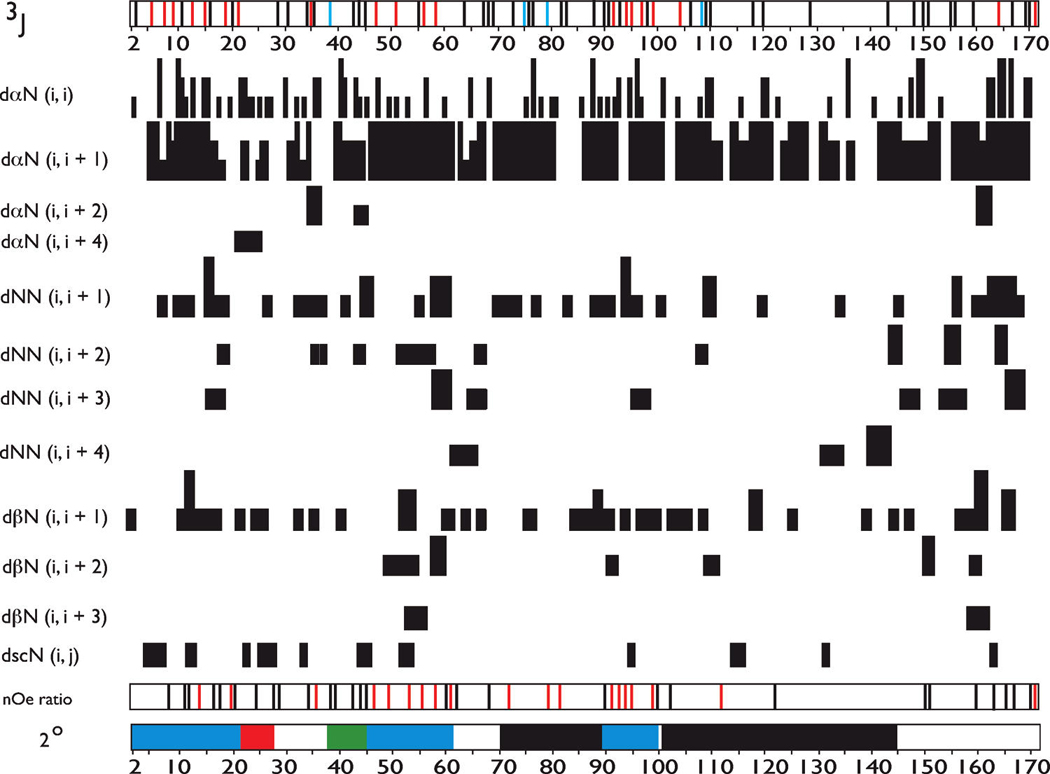
Summary of sequence-specific NMR parameters for rP172 amelogenin at pH 3.8 and 283 K in UDDW. The summary includes intraresidue NOEs; interresidue sequential, medium-range, and long-range backbone NOEs; and 3JHNHα coupling constants (3J). 3J coupling constants were determined using HNHA experiments and are denoted as color-coded bars along the sequence number line [blue for R-helical (<6 Hz), red for extended chain (>8 Hz), and black for random coil (6.0–8.0 Hz)]. The absence of a bar denotes that the 3J could not be obtained for this residue. For the sequential proton–proton NOE connectivities, the three different intensities of connecting bars indicate strong, medium, and weak NOE intensities. “nOe ratio” refers to backbone NOE intensity ratios [αN(i,i+1)/αN(i,i)] and/or [αN(i,i+1)/NN(i,i+1)] that were calculated for observable, resolvable NOE cross-peaks in the 15N–1H HSQC-NOESY spectra of rP172 and are denoted as color-coded bars along the sequence number line {black for random coil [αN(i,i+1)/NN(i,i+1) ≤ 1.4, and αN(i,i+1)/αN(i,i) ≤ 2.3] and red for values larger than these limits} (41, 47). The absence of a bar indicates that an NOE ratio could not be obtained for this residue. 2° indicates the secondary structure assignment based upon NOE and 3J data in this figure (black for PPII, red for α-helix, blue for extended β-strand, green for β-turn or loop, and white for random coil or unstructured). The terminology and presentation of this figure follow the standardization set forth in ref 30 with some modifications.
Our CD studies suggested that rP172 possesses PPII structure. Logically, we would consider that PPII structure would most likely occur within the sequence regions of I70–P89 and P102–P145, which possess a high content of the PPII-forming amino acids, Pro and Gln (17, 49, 50). Relative to other regions within rP172, these Pro-rich regions exhibit few if any medium-range NOEs and a low percentage of sequential nOes (Figure 4). These features have been associated with the presence of PPII secondary structure (17,41). Further evidence of the presence of non-random coil structure in these two segments is found in the NOE ratios (Figure 4), where we observe that the ratios obtained for the I70–P89 region exceed random coil values. This correlates with the consistent non-random coil conformational shifts obtained for the P102–P145 region (Figure 3). On the basis of this evidence, we tentatively assign PPII structure to these two Pro regions. Thus, from NMR data, we would estimate that at 10 °C the PPII content in rP172 amelogenin is 37%, and thus, we believe that the true PPII content in this protein lies somewhere between our CD (22.7%) and NMR (37%) estimates. The remaining regions of rP172, i.e., N28–S36, A63–H69, and M146–D173, are presumed to be random coil in structure (Figure 4). Overall, our structural findings are consistent with previous biophysical studies which identified helical, sheet, and PPII structures within amelogenin in solution (21).
Model Building
To gain insights into the possible molecular configurations that rP172 amelogenin adopts in the monomeric form in solution, we utilized the NOE data set along with ϕ and ψ backbone torsion angles generated by TALOS from our NMR chemical shift data set (Table S1 of the Supporting Information) as restraints for molecular dynamics simulated annealing using XPLOR-NIH. Due to the limited number of backbone restraints obtained for labile rP172 amelogenin (Figure 4), we advise that the conformations generated by our simulations should be considered qualitative in nature.
The experimental pH of the amelogenin NMR sample is 3.8, which is near the pKa values of Asp and Glu carboxylate groups. Thus, it is likely that some percentage of the Asp and Glu residues within the protein may exist in either the deprotonated or fully protonated state. Since we cannot pinpoint which Asp and Glu residues are protonated or deprotonated within rP172, we chose to model rP172 in both the fully protonated and deprotonated states, such that the effect of Asp and Glu side chain charge representation on the global structure of the protein could be estimated. Thus, we performed parallel runs in which Asp and Glu carboxylate were represented as either fully protonated or fully deprotonated species (see Materials and Methods). A comparison of the fully protonated and deprotonated backbone aligned structural ensemble (n = 10) that fit the target NMR data set shows overall agreement in terms of global, extended conformation (Figure 5), and we note that an extended structure for amelogenin was also reported in earlier X-ray scattering studies (51). Thus, the representation of Asp and Glu side chain charge does not have a significant impact on the global backbone structure of rP172.
FIGURE 5.
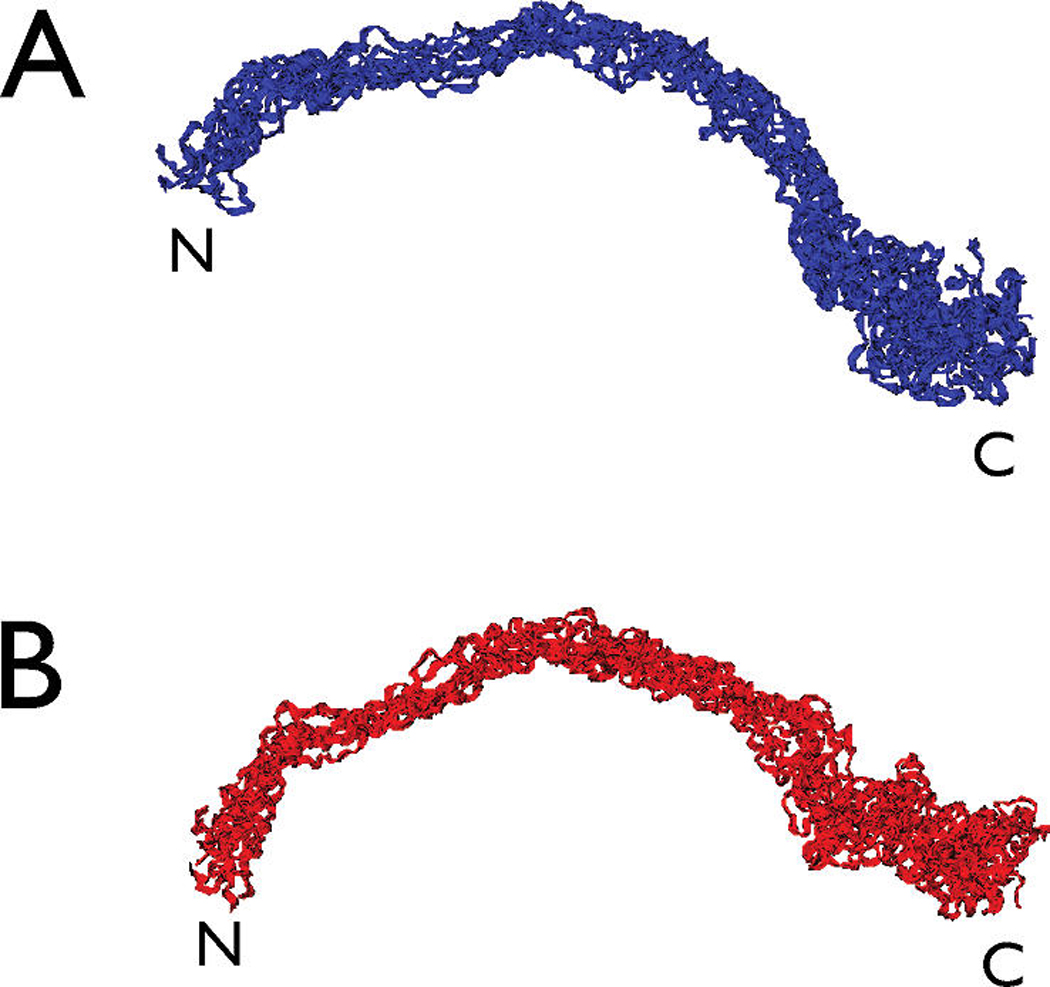
(A) Backbone-aligned ensemble (n = 10; overall backbone rmsd = 5.6 Å) of the low-energy structures obtained for rP172 amelogenin in the fully deprotonated form. (B) Same as panel A, but for the fully protonated form (n = 10; overall backbone rmsd = 5.0 Å).
Analysis of the best or lowest-energy structures for each protonation state reveals overall good agreement in terms of backbone configuration (Figure 6). As one would expect, there are minor backbone variations which reflect differences in local chain structure in the vicinity of Asp or Glu [i.e., E18 and E40 within the tyrosine-rich amelogenin peptide (TRAP) (52) or A-domain (53)] and in the charged C-terminal region (Figure 6). This is also reflected in the Ramachandran plot for these two structures, where we note a similar distribution of backbone torsion angles within right- and left-handed helix, β-turn, and β-strand regions (Figure 6), but with slightly different values. Thus, the representation of Asp and Glu side chain charge does impact the local structure of rP172.
FIGURE 6.
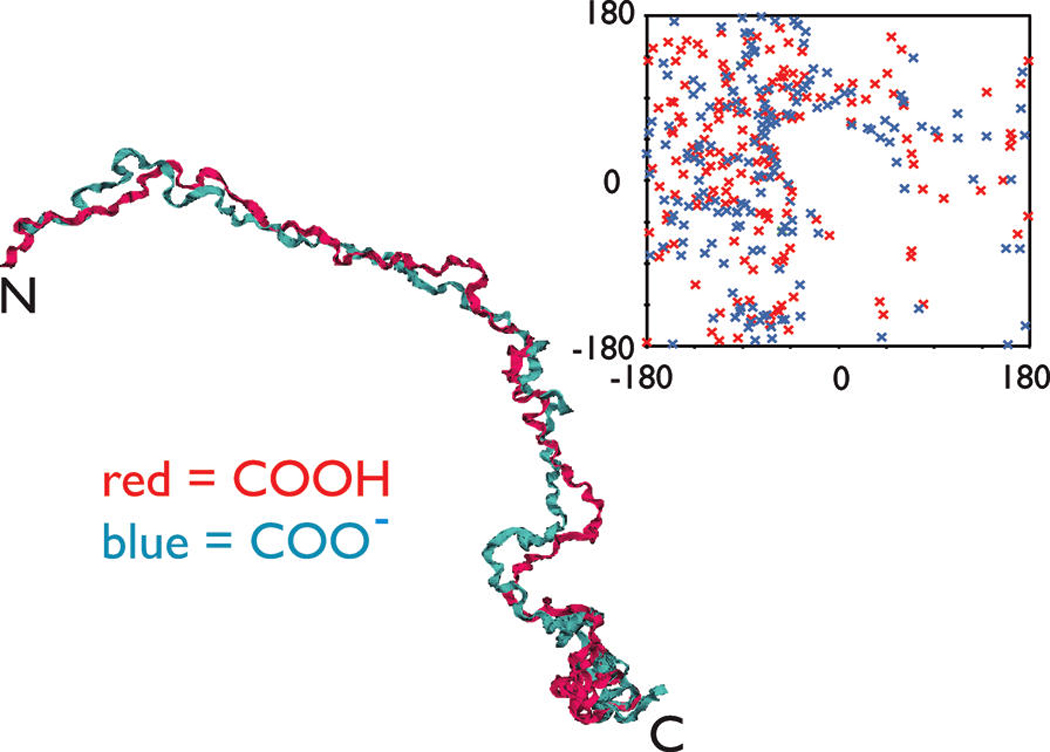
Backbone-aligned best structures obtained for the fully protonated and deprotonated forms (backbone rmsd = 4.7 Å). Ribbon representations, torsion angle plot, and rmsd backbone fitting were performed using VMD 1.8.6 for all figures. Backbonealigned best structures obtained for the fully protonated and deprotonated forms (backbone rmsd = 4.7 Å). Ribbon representations, torsion angle plot, and rmsd backbone fitting were performed using VMD 1.8.6 for all figures. The inset is a Ramachandran plot of the XPLOR-NIH lowest-energy structures for amelogenin rP172 in the fully protonated (red) and deprotonated (blue) states.
A comparison of the best protonated/deprotonated models of rP172 (Figure 6) with the general secondary structure features found in our NMR data set (Figure 4) shows good agreement with regard to important secondary structure features. rP172 amelogenin adopts an extended β-conformation within the P2–P22, L46–H62, and G90–Q101 regions. There is evidence of a turn or loop region near the C-terminal end of the TRAP region (Y37–W45) and extended helical or PPII-like structure in the A63–H69 and M146–D173 regions. The C-terminal domain appears to be coil-like and more condensed in configuration, which is consistent with other NMR studies of the amelogenin C-terminal region (54).
DISCUSSION
Our studies confirm that rP172 amelogenin is an IDP (Figure 1 and Figure 2) that is globally extended (Figure 3 and Figure 4) and possesses some degree of residual secondary structure, including PPII (Figure 2–Figure 6). We note that earlier studies proposed an extended configuration for amelogenin and postulated that this configuration is important for self-assembly (51). Our current work supports this hypothesis, and we believe that the extended, unfolded state of rP172 amelogenin offers conformational freedom (19) to allow interactions and multiple contacts to occur between amelogenins, with other enamel matrix proteins, or with the mineral phase (8, 20–23, 53). This conformational freedom is mirrored not only in the general unfolded structure of amelogenin (Figure 3–Figure 6) but also in the presence of conformational exchange (Figures S2 and S3 of the Supporting Information) where more than one protein structural state coexists in solution. Unfolded or disordered conformations also offer advantages in terms of entropy: the disorder-to-order transition that occurs upon binding of unfolded IDP to targets would decrease conformational entropy and make highly specific interactions possible (18, 19). If this is true, then the interaction of rP172 amelogenin with other molecular entities would be partially driven by the need to achieve internal structural stabilization within the individual amelogenin molecules. Preliminary studies indicate that disorder-to-order transitions do occur within amelogenin during the self-assembly process in vitro (E. Beniash, personal communication), and we intend to explore this phenomenon in more detail in a subsequent report.
Although there are a diverse set of functional subcategories within the IDP class (19), one common trait noted for many of the IDP proteins is binding promiscuity; i.e., the protein adopts different structures upon interaction with different molecular entities (18, 19). We argue that the unfolded structure of rP172 amelogenin would enable a similar promiscuity in terms of structural adaptability in response to different enamel matrix targets. In turn, this promiscuity would enable rP172 amelogenin to behave in a multifunctional capacity within the enamel matrix in the following way. For example, the labile structure of amelogenin may permit interactions with cell receptor proteins that trigger amelogenin-initiated cell signaling and regulation of gene expression activities (55, 56). At the same time, the amelogenin protein, via its extended, unfolded N-terminal TRAP domain (amino acids 1–45), offers sufficient molecular contacts for assembly within the protein–protein interaction process (21, 53). Likewise, the random coil-like structure of the polar, charged C-terminal domain (amino acids 157–173) would provide enhanced multiple charged contacts with the mineral surface (23). Hence, the unfolded nature of monomeric amelogenin appears to be a molecular design asset, given the number of diverse activities in which this protein reportedly participates. Since assembly is the primary process associated with rP172 amelogenin, we would tentatively classify this protein as a member of the IDP “assembler” class (19), members of which are noted for their ability to form macromolecular complexes (18, 19). However, we acknowledge that additional information regarding amelogenin function may come to light at a later date, which may lead to reclassification of this protein within the IDP functional series.
Intriguing features of rP172 amelogenin are the presence and location of residual secondary structure within the protein sequence and the potential role that residual structure may play in terms of functionality. We believe that there are at least three important functions that residual structure contributes to rP172 amelogenin. First, as noted for other unfolded proteins (14–17), residual structure can serve as initiation or nucleation sites for conformational transformation during protein–target binding processes (14–17, 19). In other words, target-induced folding could proceed from these nascent structural regions. This may hold true for rP172 amelogenin with regard to forming supramolecular assemblies within the enamel matrix, where nascent helical or extended β-strand regions (Figure 4) may propagate in response to external stabilization. Second, the location of residual structure and the boundaries between residual structural elements within rP172 amelogenin may have an important impact with regard to molecular recognition, and in particular by amelogenin-specific proteases. It is known that amelogenin is cleaved by specific proteases at specific sites within the primary sequence (57, 58). Coincidentally, the common conserved cleavage sites within porcine amelogenin, W45/H46, S148/M149, W161/A162, and H62/H63 (57, 58), are positioned at the boundaries of secondary structure regions (Figure 4). Given the unfolded nature of the rP172 amelogenin protein, these cleavage sites are located at the starting points of residual secondary structure and thus would not be obscured by any residual folding whatsoever but would be accessible for protease recognition. Finally, one of the most interesting types of secondary structure, PPII, is well-represented within monomeric rP172 amelogenin [I70–P89 and P102–P145 (Figure 4)], and we note that PPII structure has been linked to protein–protein interaction processes (49) and to the stabilization of sequences against conformational transformation (50). Thus, the presence of PPII structure within the rP172 amelogenin protein may serve dual roles in terms of promoting molecular recognition and stabilizing unfolded regions against transformation until appropriate conditions are realized. Obviously, additional experimentation will be necessary to firmly establish how these residual structure regions assist in amelogenin functionality.
Supplementary Material
rP172 1H, 13C, and 15N temperature-corrected (283 K) backbone chemical shifts and HNHA-determined 3JNHCHα coupling constants (Table S1), CD spectra of rP172 in UDDWat pH 6.0 and 10 °C and heated to 85 °C and then cooled to 10 °C (Figure S1), and 15N, 1H HSQC spectra of rP172 in UDDW at pH 3.8 (Figures S2 and S3). This information is available free of charge via the Internet at http://pubs.acs.org.
ACKNOWLEDGMENT
Portions of this work represent contribution 49 from the Laboratory for Chemical Physics (J.S.E.) and were funded by a grant from the Department of Energy (DE-FG02-03ER46099 to J.S.E.). Prof. John Evans is a member of the New York Structural Biology Center, which is a STAR center supported by the New York State Office of Science, Technology, and Academic Research. NMR resources were supported by NIH Grant P41 GM66354. We thank Prof. Uversky for helpful discussions and suggestions regarding amelogenin sequence analysis, Prof. Ralph Lengan for allowing access to the CD instrument, and Kaushik Dutta for assistance in the NMR experimental setup.
Footnotes
This work was supported by funding from the Department of Energy (DE-FG02-03ER46099) and the National Institute of Dental and Craniofacial Research (DE-013414).
Abbreviations: UDDW, unbuffered deionized distilled water; IDP, intrinsically disordered or unfolded protein; CD, circular dichroism; rP172, recombinant porcine amelogenin (172 amino acids); PPII, polyproline type II; rmsd, root-mean-square deviation.
REFERENCES
- 1.Lowenstam HA, Weiner S. On Biomineralization. New York: Oxford University Press; 1989. [Google Scholar]
- 2.Snead ML, Zeichner-David M, Chandra T, Robson KJ, Woo SL, Slavkin HC. Construction and identification of mouse amelogenin cDNA clones. Proc. Natl. Acad Sci. U.S.A. 1983;80:7254–7258. doi: 10.1073/pnas.80.23.7254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fincham AG, Moradian-Oldak J, Simmer JP. The structural biology of the developing dental enamel matrix. J. Struct. Biol. 1999;126:270–299. doi: 10.1006/jsbi.1999.4130. [DOI] [PubMed] [Google Scholar]
- 4.Fincham AG, Moradian-Oldak J, Diekwisch TG, Lyaruu DM, Wright JT, Bringas P, Jr, Slavkin HC. Evidence for amelogenin “nanospheres” as functional components of secretory-stage enamel matrix. J. Struct. Biol. 1995;115:50–59. doi: 10.1006/jsbi.1995.1029. [DOI] [PubMed] [Google Scholar]
- 5.Moradian-Oldak J, Du C, Falini G. On the formation of amelogenin microribbons. Eur. J. Oral Sci. 2006;114 (Suppl. 1):289–296. doi: 10.1111/j.1600-0722.2006.00285.x. discussion 327 – 329, 382. [DOI] [PubMed] [Google Scholar]
- 6.Wiedemann-Bidlack FB, Beniash E, Yamakoshi Y, Simmer JP, Margolis HC. pH triggered self-assembly of native and recombinant amelogenins under physiological pH and temperature in vitro. J. Struct. Biol. 2007;160:57–69. doi: 10.1016/j.jsb.2007.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ijima M, Moradian-Oldak J. Control of octacalcium phosphate and apatite crystal growth by amelogenin matrices. J. Mater. Chem. 2004;14:2189–2199. [Google Scholar]
- 8.Beniash E, Simmer JP, Margolis HC. The effect of recombinant mouse amelogenins on the formation and organization of hydroxyapatite crystals in vitro. J. Struct. Biol. 2005;149:182–190. doi: 10.1016/j.jsb.2004.11.001. [DOI] [PubMed] [Google Scholar]
- 9.Delgado S, Girondot M, Sire JY. Molecular evolution of amelogenin in mammals. J. Mol. Evol. 2005;60:12–30. doi: 10.1007/s00239-003-0070-8. [DOI] [PubMed] [Google Scholar]
- 10.Gibson CW, Yuan ZA, Hall B, Longenecker G, Chen E, Thyagarajan T, Sreenath T, Wright JT, Decker S, Piddington R, Harrison R, Kulkarni AB. Amelogenin-deficient mice display an amelogenesis imperfecta phenotype. J. Biol. Chem. 2001;276:31871–31875. doi: 10.1074/jbc.M104624200. [DOI] [PubMed] [Google Scholar]
- 11.Paine ML, Lei YP, Dickerson K, Snead ML. Altered amelogenin self-assembly based on mutations observed in human X-linked amelogenesis imperfecta (AIH1) J. Biol. Chem. 2002;277:17112–17116. doi: 10.1074/jbc.M110473200. [DOI] [PubMed] [Google Scholar]
- 12.Zhu DH, Paine ML, Luo W, Bringas P, Snead ML. Altering biomineralization by protein design. J. Biol. Chem. 2006;281:21173–21182. doi: 10.1074/jbc.M510757200. [DOI] [PubMed] [Google Scholar]
- 13.Bartlett JD, Ganss B, Goldberg M, Moradian-Oldak J, Paine ML, Snead ML, Wen X, White SN, Zhou YL. Protein-protein interactions of the developing enamel matrix. Curr. Top. Dev. Biol. 2006;74:57–115. doi: 10.1016/S0070-2153(06)74003-0. [DOI] [PubMed] [Google Scholar]
- 14.Eliezer D, Yao J, Dyson HJ, Wright PE. Structural and dynamic characterization of partially folded states of apomyoglobin and implications for protein folding. Nat. Struct. Biol. 1998;5:148–155. doi: 10.1038/nsb0298-148. [DOI] [PubMed] [Google Scholar]
- 15.Dyson HJ, Wright PE. Equilibrium NMR studies of unfolded and partially folded proteins. Nat. Struct. Biol. 1998;7:499–503. doi: 10.1038/739. [DOI] [PubMed] [Google Scholar]
- 16.Dyson HJ, Wright PE. Nuclear magnetic resonance methods for elucidation of structure and dynamics in disordered states. Methods Enzymol. 1998;339:258–270. doi: 10.1016/s0076-6879(01)39317-5. [DOI] [PubMed] [Google Scholar]
- 17.Ishijima J, Nagasaki N, Maeshima M, Miyano M. RVCaB, a calcium binding protein in radish vacuoles, is predominantly an unstructured protein with a polyproline type II helix. J. Biochem. 2007;142:201–211. doi: 10.1093/jb/mvm130. [DOI] [PubMed] [Google Scholar]
- 18.Uversky VN. Natively unfolded proteins: A point where biology waits for physics. Protein Sci. 2002;11:739–756. doi: 10.1110/ps.4210102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tompa P. Intrinsically unstructured proteins. Trends Biochem. Sci. 2002;27:527–533. doi: 10.1016/s0968-0004(02)02169-2. [DOI] [PubMed] [Google Scholar]
- 20.Lakshminarayanan RY, Hegde BG, Du C, Fan D, Moradian-Oldak J. Analysis of secondary structure and self-assembly by variable temperature circular dichroism and isothermal titration calorimetry. Proteins: Struct., Funct., Bioinf. 2009 doi: 10.1002/prot.22369. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lakshminarayanan R, Fan D, Du C, Moradian-Oldak J. The role of secondary structure in the entropically driven amelogenin self-assembly. Biophys. J. 2007;93:3664–3674. doi: 10.1529/biophysj.107.113936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Moradian-Oldak J, Simmer JP, Lau EC, Sarte PE, Slavkin HC, Fincham AG. Detection of monodisperse aggregates of a recombinant amelogenin by dynamic light scattering. Biopolymers. 1994;34:1339–1347. doi: 10.1002/bip.360341006. [DOI] [PubMed] [Google Scholar]
- 23.Moradian-Oldak J, Bouropoulos N, Wang LL, Gharakhanian N. Analysis of self-assembly and apatite binding properties of amelogenin proteins lacking the hydrophilic C-terminal. Matrix Biol. 2002;21:197–205. doi: 10.1016/s0945-053x(01)00190-1. [DOI] [PubMed] [Google Scholar]
- 24.Uversky VN, Gillespie JR, Fink AL. Why are “natively unfolded ” proteins unstructured under physiologic conditions? Proteins. 2000;41:415–427. doi: 10.1002/1097-0134(20001115)41:3<415::aid-prot130>3.0.co;2-7. [DOI] [PubMed] [Google Scholar]
- 25.Lim KH, Collver HH, Le YTH, Nagchowdhuri P, Kenney JM. Characterizations of distinct amyloidogenic conformations of the Aβ(1–40) and (1–42) peptides. Biochem. Biophys. Res. Commun. 2007;353:443–449. doi: 10.1016/j.bbrc.2006.12.043. [DOI] [PubMed] [Google Scholar]
- 26.Li M, Liu J, Ran X, Fang M, Shi J, Qin H, Goh JM, Song J. Resurrecting abandoned proteins with pure water: CD and NMR studies of protein fragments solubilized in salt-free water. Biophys. J. 2006;91:4201–4209. doi: 10.1529/biophysj.106.093187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fincham AG, Moradian-Oldak J. Amelogenin post-translational modification: Carboxy-terminal processing and the phosphorylation of bovine and porcine “TRAP” and “LRAP” amelogenins. Biochem. Biophys. Res. Commun. 1993;197:248–255. doi: 10.1006/bbrc.1993.2468. [DOI] [PubMed] [Google Scholar]
- 28.Vuister GW, Bax A. Quantitative J correlation: A new approach for measuring homonuclear three-bond J(HNHα) coupling constants in 15N-enriched proteins. J. Am. Chem. Soc. 1993;115:7772–7777. [Google Scholar]
- 29.Wang Y, Wishart DS. A simple method to adjust inconsistently referenced 13C and 15N chemical shift assignments of proteins. J. Biomol. NMR. 2005;31:143–148. doi: 10.1007/s10858-004-7441-3. [DOI] [PubMed] [Google Scholar]
- 30.Markley JL, Bax A, Arata Y, Hilbers CW, Kaptein R, Sykes BD, Wright PE, Wüthrich K. Recommendations for the presentation of NMR structures of proteins and nucleic acids. J. Biomol. NMR. 1998;12:1–23. doi: 10.1023/a:1008290618449. [DOI] [PubMed] [Google Scholar]
- 31.Receveur-Brechot V, Bourhis J-M, Uversky VN, Canard B, Longhi S. Assessing protein disorder and induced folding. Proteins. 2006;62:24–25. doi: 10.1002/prot.20750. [DOI] [PubMed] [Google Scholar]
- 32.Danielsson J, Jarvet J, Damberg P, Graslund A. The Alzheimer β-peptide shows temperature-dependent transitions between left-handed 31-helix, β-strand and random coil secondary structures. FEBS J. 2005;272:3938–3949. doi: 10.1111/j.1742-4658.2005.04812.x. [DOI] [PubMed] [Google Scholar]
- 33.Bienkiewicz EA, Woody AYM, Woody RW. Conformation of the RNA polymerase IIC-terminal domain: Circular dichroism of long and short fragments. J. Mol. Biol. 2000;297:119–133. doi: 10.1006/jmbi.2000.3545. [DOI] [PubMed] [Google Scholar]
- 34.Wustman B, Santos R, Zhang B, Evans JS. Identification of a “glycine loop”-like coiled structure in the 34- AA Pro, Gly, Met repeat domain of the biomineral-associated protein, PM27. Biopolymers. 2002;65:1305–1318. doi: 10.1002/bip.10274. [DOI] [PubMed] [Google Scholar]
- 35.Zhang B, Wustman B, Morse DE, Evans JS. Model peptide studies of sequence regions in the elastomeric biomineralization protein, Lustrin A. I. The C-domain consensus-PG-, -NVNCT- motif. Biopolymers. 2002;64:358–369. doi: 10.1002/bip.10069. [DOI] [PubMed] [Google Scholar]
- 36.(a) Wishart DS, Bigham CG, Holm A, Hodges RS, Sykes BD. 1H, 13C and 15N random coil NMR chemical shifts of the common amino acids. I. Investigations of nearest-neighbor effects. J. Biomol. NMR. 1995;5:67–81. doi: 10.1007/BF00227471. [DOI] [PubMed] [Google Scholar]; (b) Wishart DS, Sykes Bd, Richards FM. Relationship between nuclear magnetic resonance chemical shift and protein secondary structure. J. Mol. Biol. 1991;222:311–333. doi: 10.1016/0022-2836(91)90214-q. [DOI] [PubMed] [Google Scholar]
- 37.Wishart DS, Case DA. Use of chemical shifts in macromolecular structure determination. Methods Enzymol. 2001;338:3–34. doi: 10.1016/s0076-6879(02)38214-4. [DOI] [PubMed] [Google Scholar]
- 38.Serrano L. Comparison between the ϕ distribution of the amino acids in the protein database and NMR data indicates that amino acids have various ϕ propensities in the random coil conformation. J. Mol. Biol. 1995;254:322–333. doi: 10.1006/jmbi.1995.0619. [DOI] [PubMed] [Google Scholar]
- 39.Smith LJ, Bolin KA, Schwalbe H, MacArthur MW, Thornton JM, Dobson CM. Analysis of main chain torsion angles in proteins: Prediction of NMR coupling constants for native and random coil conformations. J. Mol. Biol. 1996;255:494–506. doi: 10.1006/jmbi.1996.0041. [DOI] [PubMed] [Google Scholar]
- 40.Schubert M, Labudde D, Oschkinat H, Schmieder P. A software tool for the prediction of Xaa-Pro bond conformations in proteins based on 13C chemical shift statistics. J. Biomol. NMR. 2002;34:149–154. doi: 10.1023/a:1020997118364. [DOI] [PubMed] [Google Scholar]
- 41.Ma K, San LS, Wang K. Polyproline II helix is a key structural motif of the elastic PEVK segment of titin. Biochemistry. 2001;40:3427–3438. doi: 10.1021/bi0022792. [DOI] [PubMed] [Google Scholar]
- 42.Hinck AP, Eberhardt ES, Markley JL. NMR strategy for determining Xaa-Pro peptide bond configurations in proteins: Mutants of staphylococcal nuclease with altered configuration at proline 117. Biochemistry. 1993;32:11810–11818. doi: 10.1021/bi00095a009. [DOI] [PubMed] [Google Scholar]
- 43.Sarkar P, Reichman C, Saleh T, Birge R, Kalodimos CG. Proline cis-trans isomerization controls autoinhibition signalling protein. Mol. Cell. 2007;25:413–426. doi: 10.1016/j.molcel.2007.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Andreotti AH. Native state proline isomerization: An intrinsic molecular switch. Biochemistry. 2003;42:9515–9524. doi: 10.1021/bi0350710. [DOI] [PubMed] [Google Scholar]
- 45.Nicholson LK, Lu KP. Prolyl cis-trans isomerization as a molecular timer in Crk signalling. Mol. Cell. 2007;25:483–486. doi: 10.1016/j.molcel.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 46.Kulp JL, Shiba K, Evans JS. Probing the conformational features of a phage display polypeptide sequence directed against single-walled carbon nanohorn surfaces. Langmuir. 2005;21:11907–11914. doi: 10.1021/la050961x. [DOI] [PubMed] [Google Scholar]
- 47.Fiebig KM, Schwalbe H, Buck M, Smith LJ, Dobson CM. Towards a description of the conformations of denatured states of proteins. Comparison of a random coil model with NMR measurements. J. Phys. Chem. 1996;100:2661–2666. [Google Scholar]
- 48.Wuthrich K. NMR of proteins and nucleic acids. New York: Wiley-Interscience; 1986. [Google Scholar]
- 49.Cubelis MV, Caillez F, Blundell TL, Lovell SC. Properties of polyproline II, a secondary structure element implicated in protein-protein interactions. Proteins: Struct., Funct., Bioinf. 2005;58:880–892. doi: 10.1002/prot.20327. [DOI] [PubMed] [Google Scholar]
- 50.Darnell G, Orgel JPRO, Pahl R, Meredith SC. Flanking polyproline sequences inhibit β-sheet structure in poly-glutamine segments by inducing PPII-like helix structure. J. Mol. Biol. 2007;374:688–704. doi: 10.1016/j.jmb.2007.09.023. [DOI] [PubMed] [Google Scholar]
- 51.Matsushima N, Izumi Y, Aoba T. Small angle X-ray scattering and computer aided molecular modeling studies of 20 kDA fragment of porcine amelogenin. Does amelogenin adopt an elongated bundle structure? J. Biochem. 1998;123:150–156. doi: 10.1093/oxfordjournals.jbchem.a021902. [DOI] [PubMed] [Google Scholar]
- 52.Fincham AG, Belcourt AB, Termine JD, Butler WT, Cothran WC. Dental enamel matrix: Sequences of two amelogenin polypeptides. Biosci. Rep. 1981;1:771–778. doi: 10.1007/BF01114799. [DOI] [PubMed] [Google Scholar]
- 53.Paine ML, Snead ML. Protein interactions during assembly of the enamel extracellular matrix. J. Bone Miner. Res. 1997;12:221–227. doi: 10.1359/jbmr.1997.12.2.221. [DOI] [PubMed] [Google Scholar]
- 54.Shaw WJ, Ferris K, Tarasevich B, Larson JL. The structure and orientation of the C-terminus of LRAP. Biophys. J. 2008;94:3247–3257. doi: 10.1529/biophysj.107.119636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gibson CW. The amelogenin “enamel proteins” and cells in the periodontium. Crit. Rev. Eukaryotic Gene Expression. 2008;18:345–360. doi: 10.1615/critreveukargeneexpr.v18.i4.30. [DOI] [PubMed] [Google Scholar]
- 56.Veis A. Amelogenin gene splice products: Potential signaling molecules. Cell. Mol. Life Sci. 2003;60:38–55. doi: 10.1007/s000180300003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ryu OH, Fincham AG, Hu CC, Zhang C, Qian Q, Bartlett JD, Simmer JP. Characterization of recombinant pig enamelysin activity and cleavage of recombinant pig and mouse amelogenins. J. Dent. Res. 1999;78:743–750. doi: 10.1177/00220345990780030601. [DOI] [PubMed] [Google Scholar]
- 58.Moradian-Oldak J, Jimenez I, Maltby D, Fincham AG. Controlled proteolysis of amelogenins reveals exposure of both carboxy- and amino-terminal regions. Biopolymers. 2001;58:606–616. doi: 10.1002/1097-0282(200106)58:7<606::AID-BIP1034>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
rP172 1H, 13C, and 15N temperature-corrected (283 K) backbone chemical shifts and HNHA-determined 3JNHCHα coupling constants (Table S1), CD spectra of rP172 in UDDWat pH 6.0 and 10 °C and heated to 85 °C and then cooled to 10 °C (Figure S1), and 15N, 1H HSQC spectra of rP172 in UDDW at pH 3.8 (Figures S2 and S3). This information is available free of charge via the Internet at http://pubs.acs.org.