

Abstract
Chemical cross-linking combined with mass spectrometry has been used to elucidate protein structures and protein-protein interactions. However, heterogeneity of the samples and the relatively low abundance of cross-linked peptides make this approach challenging. As an effort to overcome this hurdle, we have synthesized lysine reactive homobifunctional cross-linkers with the biotin in the middle of the linker and used them for enriching cross-linked peptides. The reaction of biotin tagged cross-linkers with purified HIV-1 CA resulted in the formation of hanging and intra-molecular cross-links. The peptides modified with biotinylated cross-linkers were effectively enriched and recovered using a streptavidin coated plate and mass spectrometry friendly buffers. The enrichment of modified peptides and removal of the dominantly unmodified peptides simplify mass spectra and their analyses. The combination of high mass accuracy of FT-ICR MS and MS/MS capability of linear ion trap allows us to unambiguously identify the cross-linking sites and additional modification, such as oxidation.
Introduction
Characterization of the protein-protein interactions and conformational changes in large supramolecular complexes presents a substantial challenge to structural biology due to limitations on obtaining atomic resolution structures with traditional methods such as X-ray crystallography and nuclear magnetic resonance (NMR). While the recent development of cryo-electron microscopy (Cryo-EM) image reconstruction techniques can provide high resolution information on the protein fold and subunit disposition in the macromolecular complexes,1–6 they typically do not achieve high enough resolution to identify key side chain interactions.
Mass spectrometry (MS) has become a central part of biological research with the invention of electrospray ionization (ESI) and matrix-assisted laser desorption ionization (MALDI).7, 8 Both ESI MS and MALDI MS can provide molecular mass information for large proteins in complex biological samples with high speed, accuracy and sensitivity. Thus, MS has been used mainly to identify unknown components of biological samples as well as to detect post-translational modifications.
Beyond protein identification, various MS based techniques have been introduced to study structural biology problems. Limited proteolysis and chemical modification combined with MS has been used to detect the local dynamics of proteins and to map protein-protein interfaces.9 MS based hydrogen/deuterium exchange is one of the most powerful and popular techniques to monitor protein dynamics and local stability in solution. It has been used to investigate structural features accompanying protein folding and unfolding, local dynamics of a protein or complexes, functionally different forms of a single protein, and protein-protein interactions.10–15 Native ESI MS also has been used to detect intact macromolecular complexes and their intermediates and monitor assembly/disassembly processes of macromolecular complexes.16–20 Recently chemical cross-linking combined with MS has been used to elucidate protein structures and protein-protein interactions.21–26 The use of cross-linkers of different lengths provides distance constraints between reactive groups within and between protein subunits.24, 27 This information can be used to improve the resolution of molecular models of the protein complexes of interest.21, 23 Cross-linking reagents have been used to assist in determination of protein-protein interactions, ligand-receptors, and 3-D structures of proteins.21–23, 25, 26, 28
Chemical cross-linking reactions generally produce both inter- and intra-molecular cross-linkings in the macromolecular complexes. In most cases, size-exclusion chromatography or SDS-PAGE is applied to separate monomers and different types of oligomers.14, 24, 28, 29 To identify cross-linking sites and partners, separated fractions are subsequently subjected to proteolytic cleavages and the cross-linked peptides are analyzed by MS. The actual detection of cross-linked peptides presents a technical challenge due to sample heterogeneity and the relatively low abundance of cross-linked peptides.
In an effort to overcome these obstacles, cross-linking reagents carrying affinity tags and stable isotope labels have been developed.24, 25, 30 Affinity tagged cross-linked peptides can be isolated from complex peptide pools and the enriched cross-linked peptides can be used for further analyses.25 Stable isotope labeling on the cross-linkers provides a recognizable mass signature in the complex mass spectra.30 Mixed samples of isotope labeled and unlabeled cross-linked peptides produce regular isotopic distribution pairs in the mass spectra simplifying identification of cross-linked peptides.30
In this study, we have synthesized biotin derivatives containing homo-bifunctional lysine-reactive affinity labeled chemical cross-linkers of different lengths, and evaluated their application for mass spectrometric analysis using HIV-1 capsid protein (CA) and hybrid ion trap-FT-ICR MS. HIV-1 CA which has a molecular weight of 25,602 Da and contains 11 lysines was chosen for study because the structures of the individual N- and C- terminal domains are known crystallographically but the disposition and dynamics between the domains both in solution and within the virus remains an unsolved problem in virus structure.
Experimental
Synthetic Chemistry General
Melting points (m.p.) were determined using an Electrothermal 9100 apparatus and were uncorrected. All 1H and 13C NMR spectra were recorded on a Brucker 300 MHz spectrometer using TMS as internal standard. Elemental analyses were performed by Atlantic Microlab (Norcross, GA) and the results indicated by symbols for the elements were within ± 0.4% of theoretical values. Reactions were monitored by TLC (Whatmann, silica gel, UV254, 25 μm plates), and flash column chromatography utilized ‘BAKER’ silica gel (40 μm) in the solvent systems indicated. Anhydrous solvents used for reactions were purchased in Sure-Seal™ bottles from Aldrich Chemical Company. Other reagents were purchased from Aldrich, Lancaster or Acros chemical companies and used as received.
5-Amino-1,9-nonanedioic acid (2b)
To a solution of 5-oxoazelaic acid (1b) (0.60 g, 3.0 mmol) in methanol (60 ml) was added ammonium chloride (0.80 g, 15 mmol) followed by NaCNBH3 (0.40 g, 6.0 mmol). The pH of above mixture was adjusted to 5~6 by periodical addition of ammonium hydroxide. The reaction mixture was stirred at room temperature for 4 days. 1N HCl was added to adjust the pH of the reaction mixture to 1~2 and this was stirred at room temperature for 40 min. Solvent was removed and the residue was purified by ion-exchange chromatography. A column containing resin (Dowex-50 × 8–100, 25 × 3 cm) was washed with water (2 L) before a solution of the crude product in water (10 ml) was placed on top of the resin. The resin was eluted with water (1.5 L), and then with 1N NH4OH to exchange the product off column. The collected fractions were concentrated to provide 2b (0.50 g, 86%) as a white solid: m.p. 161.8–162.5°C. 1H NMR (CD3OD) δ:1.59–1.76 (m, 8H), 2.26–2.28 (brm, 4H), 3.23–3.32 (m, 1H); 13C NMR (CD3OD) δ: 23.05, 33.89, 38.13, 53.01, 182.48; MS: m/z 204 (M+H)+.
Biotin N-Hydroxysuccinimide (biotin-NHS)
To a solution of biotin (0.98 g, 4.0 mmol) in DMF (120 ml) was added triethylamine (2.80 ml, 20.0 mmol) followed by N, N′-disuccinimidyl carbonate (1.30 g, 4.80 mmol). The reaction mixture was stirred at room temperature for 6 hrs. Solvent was removed by rotary evaporation (rotovap) and the residue was triturated with ether and ethyl acetate. Filtration and washing with ether provided rather pure biotin-NHS (1.34 g, 98%) as a solid: m.p. 198–200°C. This was used in the next step without further purification. 1H NMR (CDCl3) δ:1.55–1.86 (m, 6H), 2.59–2.72 (m, 3H), 2.82–2.94 (m, 4H), 2.95–2.97 (m, 1H) 3.13–3.20 (m, 1H), 4.34–4.38 (m, 1H), 4.52–4.57 (m, 1H), 5.30 (brs, 1H), 5.75 (brs, 1H); 13C NMR (CDCl3) δ:24.52, 25.42, 25.63, 27.88, 27.93, 30.68, 40.49, 55.28, 60.27, 61.85, 163.81, 168.62, 169.42,172.48; MS: m/z 342 (M+H)+.
3-Amino-N-biotinoyl-1,5-pentanedioic acid (3a)
To a solution of β-glutamic acid (2a) (0.53 g, 3.6 mmol) in a mixture of triethylamine (8.8 ml), water (2 ml) and acetonitrile (2 ml) was added biotin-NHS (1.4 g, 4.0 mmol) in DMF (25 ml). The reaction mixture was stirred at room temperature for 3.5 hrs. Additional β-glutamic acid (22 mg, 0.15 mmol) in a mixture of triethylamine (0.3 ml) and water (0.1 ml) was added and the reaction mixture stirred for another 1 hr. The solvent was removed on a rotovap and the residue was triturated with dichloromethane. After filtration and washing with dichloromethane, the crude solid was recrystillized from methanol/water (2/1) to provide 3a (1.0 g, 91%) as a white solid: m.p. 255.7–257.2°C. 1H NMR (DMSO) δ: 1.28–1.51 (m, 6H), 2.01 (t, 2H, J = 7.5 Hz), 2.43 (d, 4H, J = 6.6 Hz), 2.57 (d, 1H, J = 13.2 Hz), 2.85 (dd, 1H, J1 = 13.2 Hz, J2 = 5.0 Hz), 3.06–3.10(m, 1H), 4.14–4.16 (m, 1H), 4.29–4.35 (m, 2H), 6.39 (brs, 1H), 6.43 (brs, 1H), 7.81(d, 1H, J = 7.5 Hz); 13C NMR (DMSO) δ:18.90, 25.60, 28.32, 36.15, 38.66, 43.42, 55.72, 56.34, 59.57, 61.34, 163.12, 171.83, 172.55, 172.61; MS: m/z 374 (M+H)+; Anal. (C15H23N3O6S) C, H, N.
5-Amino-N-biotinoyl-1,9-nonanedioic acid (3b)
The procedure was the same as for 3a but utilized 5-amino-1,9-nonanedioic acid (2b). This provided 3b (0.70 g, 54%) as a solid after recrystallization from methanol/water (10/1): m.p. 182.8–185.3°C. 1H NMR (DMSO) δ: 1.29–1.51 (m, 14H), 2.06 (t, 2H, J = 7.5 Hz), 2.16–2.20 (m, 4H), 2.57 (d, 1H, J = 12.0 Hz), 2.84 (dd, 1H, J1 = 12.0 Hz, J2 = 5.0 Hz), 3.06–3.12 (m, 1H), 3.68–3.70 (m, 1H), 4.14–4.22 (m, 1H), 4.28–4.30 (m, 1H), 6.37 (brs, 1H), 6.42 (brs, 1H), 7.52 (d, 1H, J = 9.0 Hz); 13C NMR (DMSO) δ:21.51, 25.85, 28.42, 28.51, 33.82, 34.34, 35.75, 47.40, 55.79, 59.53, 61.37, 163.06, 171.97, 174.78; MS: m/z 430 (M+H)+; Anal. (C19H31N3O6S) C, H, N.
Bis-(N-Hydroxysuccinimidyl) 3-Amino-N-biotinoyl-1,5-pentanedioate (BCCL1)
To a solution of disuccinimidyl carbonate (0.48 g, 1.8 mmol) in DMF (5 ml) was added dropwise triethylamine (0.62 ml, 4.4 mmol) followed by a solution of diacid 3a (0.16 g, 0.44 mmol) in DMF (5 ml). The reaction mixture was stirred at room temperature overnight. Solvent was removed on a rotovap and the residue was triturated with ether. The residue was purified on a silica flash column (5 × 1 cm, 15% iso-PrOH in CHCl3) to provide BCCL1 (0.16 g, 64%) as a hygroscopic solid: m.p. 118.5–122.1°C (decomposed). 1HNMR (CDCl3/DMSO) δ: 1.30–1.51 (m, 6H), 2.13 (t, 2H, J = 7.5 Hz), 2.50–2.60 (m, 1H), 2.76–2.82 (brm, 9H), 2.96–3.13 (m, 5H), 4.17–4.20 (m, 1H), 4.35–4.39 (m, 1H), 4.59–4.62 (m, 1H), 5.84 (s, 1H), 5.96 (brs, 1H), 7.76 (d, 1H, J = 8.1 Hz); 13C NMR (DMSO) δ: 25.34, 25.58, 25.81, 28.34, 28.42, 34.99, 35.54, 42.82, 55.68, 59.55, 61.30, 163.07, 166.64, 170.43, 172.25; MS: m/z 568 (M+H)+; Anal. (C23H29N5O10S) C, H, N.
Bis-(N-Hydroxysuccinimidyl) 5-Amino-N-biotinoyl-1,9-nonanedioate, (BCCL2)
The same procedure as BCCL1 was used but started with 3b to give BCCL2 (0.16 g, 66%) as a hygroscopic solid: m.p. 80.4–83.6°C (decomposed). 1H NMR (DMSO) δ: 1.29–1.71(m, 14H), 2.15(t, 2H, J = 6.6 Hz), 2.55–2.68(m, 5H), 2.76–2.85(brm, 9H), 3.02–3.11(m, 1H), 4.12–4.16(m, 1H), 4.28–4.36(m, 2H),), 6.06(s, 1H), 6.09(s, 1H), 7.59(d, 1H); 13C NMR (DMSO) δ:21.31, 25.79, 25.84, 28.35, 28.52, 30.25, 33.81, 35.53, 47.50, 55.75, 59.54, 61.34, 62.37, 163.05, 169.27, 170.61, 172.24; MS: m/z 624 (M+H)+; Anal. (C27H37N5O10S) C, H, N.
Cross-linking reactions and sample preparations for mass spectrometry
HIV CA were purified as previously described.14 Purified CA in 50 mM sodium phosphate, 100 mM NaCl, pH 8.0 was mixed with freshly prepared 0.1 M BCCL cross-linkers (1 mM final) in DMSO and the reaction mixtures were incubated at room temperature for 30 sec and quenched with 60 mM Tris. Purified CA was mixed with neat DMSO (1% final conc. by volume) as a control and cross-linker treated samples were run in parallel. Free BCCLs were removed by a Bio-Gel P-6 spin column (Micro Bio-spin P-6, Bio-Rad). One half of the desalted reactions were stored for whole protein mass analysis and the other half was digested with sequencing grade trypsin (Roche) at 37 °C overnight. Tryptic digests (200 μl) of each reaction were applied to the 96 well streptavidin coated plate (Sigma), incubated at room temperature for 2 hrs, and the unbound fractions were collected for further analysis. The plates were washed three times with 0.05% Triton-X containing water to remove non-specifically bound peptides and subsequently washed three more times with plain water to remove residual contaminants. Bound samples were extracted with elution solution (47.5% water: 47.5% acetonitrile: 5% formic acid) at 37 °C for 2 hrs. Recovered samples were concentrated by vacuum evaporation prior to mass spectrometric analysis.
Mass spectrometry
The whole protein masses of the cross-linking reactions were analyzed by LC-ESI on a time-of-flight (TOF) mass spectrometer (LCT, Waters) as described previously.28, 31 10 μl samples were manually injected using a gas-tight syringe into a 10 μl sample loop attached to a Rheodyne injection valve. Applied samples were loaded onto a C4 trap (Michrom BioResources, Inc.). The proteins were rapidly desalted and eluted with a 5–95% acetonitrile gradient with 36 μl/min flow. Spectra were acquired in the range of m/z 200–1650.28, 31 Mass spectra were processed using the MaxEnt 1 algorithm from MassLynx version 4.0 (Waters) to obtain average masses from multiple charge state distributions.
Tryptic digests were analyzed on a LC coupled ESI ion trap-FT ICR hybrid mass spectrometer (LTQ FT, Thermo Finnigan) equipped with a nanospray source and 7 T magnet. 1 μl samples were manually injected using a gas-tight syringe into a 5 μl sample loop attached to a Rheodyne injection valve. Tryptic digests were separated on a prepacked nano MAGIC C18 reverse phase column (0.1 × 50 mm, 200 Å) (Michrom BioResources) using a linear gradient from 5% to 95% acetonitrile containing 0.1% formic acid for 40 min and electrosprayed from a 20 μm i.d. PicoTip emitter (New Objective). An Agillent HPLC pump system was used to deliver the solvent and the flow was split after column to achieve the output flow rate of approximately 0.5 μl/min.
Automated MS/MS experiments used a survey scan, from m/z 400–2000, to select the three most abundant ions as MS/MS precursors with three min exclusion duration. Three consecutive MS/MS were performed in the linear ion trap with the collision energy of 35V. The acquired MS and MS/MS spectra were analyzed and processed by the embedded software, Qual Browser and Bioworks 3.2 (Thermo Finnigan).
Results and discussion
Syntheses of BCCL1 and BCCL2
The syntheses of targets BCCL1 and BCCL2 are summarized in Scheme 1. In this procedure 2a was obtained commercially and 2b was synthesized. The reductive amination of keto-diacid 1b was carried out using sodium cyanoborohydride and the resulting amino diacid 2b was purified by ion-exchange chromatography (93% yield). Due to the poor solubility of amino-diacids 2a and 2b in organic solvents, several amidation conditions were tried but failed. It was found that water had to be used as co-solvent with acetonitrile and DMF. Thus the amidations of 2a and 2b with biotin-NHS in the presence of triethylamine were completed within 4 hrs. The yields of 3a and 3b were 91% and 54%, respectively. The analytically pure samples were obtained by recrystallization from methanol/water.
Scheme 1.

Synthesis of BCCL1 and BCCL2
With the diacids 3a and 3b in hand, target compounds BCCLs were prepared using activation of the diacids with N,N′-disuccinimidyl carbonate in DMF in the presence of triethylamine. Since the desired target compounds contain the N-hydroxysuccinimide ester (NHS) group, which is sensitive to moisture, the purification had to avoid aqueous work-up. The final products of BCCLs were purified by chromatography. The yields of BCCL1 and BCCL2 were 64% and 66%, respectively, with > 95% purity as determined by 1H-NMR spectroscopy using an internal standard.
Cross-linking HIV-1 capsid protein (CA) with different lengths of BCCLs
NHS cross-linkers are well known to result in the formation of an amide bond between the ε-amino of lysine and the cross-linker. The extent of modification was determined by relative abundances of the deconvoluted masses of each species (Figure 1). There are two possible modifications of the monomer, the formation of an intra-molecular cross-link and the formation of a hanging cross-link in which one arm is cross-linked to the protein and the other is hydrolyzed by reaction with the solvent. Intra-molecular cross-links of BCCL1 and BCCL2 are calculated to result in mass increases of 337.10962 and 393.17222 Da, respectively, whereas the formation of a hanging cross-link should result in mass increases of 355.12019 Da for BCCL1 and 411.18179 Da for BCCL2. Consistent with this expectation, LC-ESI-TOF-MS analyses showed serial increases of 337 and 355 Da in the BCCL1 treated CA proteins (Figure 1b) and 393 and 411 Da in the BCCL2 treated CA proteins (Figure 1c). Approximately 50% of the monomers were intra-molecularly cross-linked with two BCCL1 molecules. Approximately half of this subpopulation (20% of the total) also carried a hanging cross-linker. Thirty-five percent of the molecules were cross-linked with one BCCL1. The bulk of this subpopulation (22% of the total) also carried a hanging cross-link. Fifteen percent of the monomers were cross-linked with three BCCL1s (Figure 1b) and carried no hanging cross-link. BCCL2 also modified the monomers to similar extents as BCCL1 although with a slightly different distribution (Figure 1c).
Figure 1.

Deconvoluted mass spectra of untreated (a), BCCL1 treated (b), and BCCL2 treated (c) HIV CA. The numbers of intra-molecular and hanging cross-linkings were assigned based on the observed masses and indicated as numbers and numbers with asterisks, respectively.
Enrichment of the cross-linked peptides with streptavidin coated plates
To enrich for BCCL modified peptides and remove the predominating unmodified peptides, we distributed the tryptic digests into the wells of a streptavidin coated 96 well plate and incubated them at room temperature for 2 hrs. To avoid competition between the residual free BCCL cross-linker and cross-linked peptides for the streptavidin binding sites on the plate, the residual cross-linkers were removed using a Bio-Gel P-6 spin desalting column prior to trypsin digestion. Following incubation, the unbound fraction was removed and reserved and the plate was washed extensively to remove any non-specifically bound peptides (see experimental). The bound samples were recovered by elution with a solution composed of 47.5% water: 47.5% acetonitrile: 5% formic acid at 37 °C for 2 hrs. The eluted samples were concentrated using a speed-vac for subsequent LC-MS analysis. For comparison, DMSO only treated CA protein was treated in parallel.
The un-fractionated digest, the unbound fraction, and the eluted fraction were analyzed by nanoLC-ESI ion-trap FT-ICR hybrid MS (LTQ FT, Thermo Finnigan). Figure 2 represents the total ion count (TIC) chromatographs of tryptic digests of untreated (DMSO only) (Figure 2a) and BCCL1 cross-linker treated CA (Figure 2b). The top, middle, and bottom panels are the TIC chromatographs of the un-fractionated digests, the unbound fraction, and the eluted fraction, respectively (Figure 2). The elution profiles of the un-fractionated untreated and BCCL1 treated CA samples were slightly different (Figure 2a, b, top panel) presumably because cross-linking alters the digestion preference and efficiency of the trypsin. While coverage of the untreated CA approached 100% of the primary sequence, coverage of the BCCL1 treated CA was limited to approximately 70% of primary sequence (data not shown). However, the elution profiles of both the un-fractionated and unbound samples were very similar (compare figure 2a, b, top and middle panels) suggesting that modified (intra-molecular cross-linked and hanging) peptides are not abundant and underscoring the challenge of detecting cross-linked species.
Figure 2.

Total ion count (TIC) chromatographs of tryptic digests of untreated (a) and BCCL1 treated (b) HIV CA. Un-fractionated cross-linking reaction (top), unbound fraction (middle), and eluted fraction (bottom) from a 96 well streptavidin coated plate were analyzed by nanoLC-ESI ion-trap FT-ICR hybrid mass spectrometry (LTQ FT, Thermo Finnigan).
In contrast, multiple peaks were observed in the eluted samples of BCCL1 (Figure 2b, bottom panel) and BCCL2 (data not shown) treated CA, whereas no significant peaks were detected in the eluted samples of untreated sample (Figure 2a, bottom panel). The absolute intensities of the eluted peaks were one order of magnitude lower than the typical intensities of the un-fractionated sample supporting their low abundance in the un-fractionated tryptic digests. The multiple distinct peaks observed in the eluted samples suggested the capture and elution of a variety of modified species.
Identification of the enriched cross-linked peptides
Modification of a lysine side-chain by cross-linking prevents tryptic cleavage at that site. To identify any peptides carrying hanging cross-links, the masses of all possible tryptic peptides with a single missed cleavage site plus a hydrolyzed cross-linker (hanging) were calculated. To identify cross-linked peptides, a matrix containing the masses of all possible combinations of two peptides (each with a single missed cleavage site) plus the cross-linker was constructed. The experimentally observed masses were compared to the values in these tables using an acceptance criterion of 2 ppm. Surprisingly, the observed masses did not match any of predicted modified tryptic fragments. Furthermore, none of the ions found in the eluted sample could be detected in the un-fractionated sample. This suggested that covalent modifications were occurring during sample preparation.
Both methionine and biotin have reported tendencies to oxidize to the sulfoxide and sulphone forms.32, 33 We therefore considered the possibility that either the methionines or the biotin moiety in the cross-linker became oxidized upon elution from the plate resulting in mass increases of 16 Da for each sulfur oxidized. Indeed this proved to be the case. For example, a doubly charged ion eluting at 21.9 min in the eluted fraction had an m/z of 599.79989 (Figure 3b), whereas a doubly charged ion found in the un-fractionated sample had an m/z of 591.80255 (Figure 3a). The observed monoisotopic mass difference of 15.99468 Da is in an excellent agreement with the mass of molecular oxygen (15.99491 Da). Based on mass matching, the best fit to the observed mass was obtained by assuming a hanging cross-link at Lys 170 of the peptide spanning residues 168–173 with or without oxidation. As there is no methionine in this peptide, it is likely that the biotin is oxidized.
Figure 3.

FT-ICR MS (a,b) and ion trap MS/MS (c,d) spectra of a peptide from digested BCCL1 cross-linker treated CA from the un-fractionated cross-linking reaction (a,c) and the eluted fraction (b,d). The monoisotopic masses of precursor ions of the peptide from the un-fractionated and eluted samples were detected at m/z = 591.80255 (theoretical m/z = 591.80243) and m/z = 599.79989 (theoretical m/z = 599.79974), respectively (a,b). The monoisotopic mass increment (15.99468 Da) corresponded precisely to the mass of molecular oxygen (15.99491 Da). Mass measurement errors are less than 1 ppm on the FT-ICR MS. Ion trap MS/MS spectra of [M+2H]2+ ion of the peptide from the un-fractionated reaction (c) and eluted fraction (d). Corresponding b and y ions are indicated.
To determine the site of modification, we performed MS/MS on the precursor ions (monoisotopic m/z = 591.80255 and m/z = 599.79989). The resulting series of b and y ions were assigned as in Figure 3c and 3d, respectively. A large mass gap (483 Da) whose mass matched the sum of the masses of Lys (128 Da) and a hanging BCCL1 cross-linker (355 Da) was observed between y3 and y4 (or b2 and b3) ions confirming that the modification occurs at Lys 170 (Figure 3c). The site of oxidation was identified by comparing the two MS/MS spectra (Figure 3c, d). While the daughter ion masses from y1-y3 and b2 were the same in both spectra, those from y4-y5 and b3-b5 in the eluted sample showed a 16 Da mass increase strongly suggesting that the biotin is oxidized to biotin sulfoxide (Figure 3d).
By considering the mass increase resulting from the oxidation of the biotin and methionines, we could identify hanging and intra-molecular cross-linked peptides from BCCL1 treated CA as listed in table 1. While many peptides with hanging cross-linkers were recovered and identified, only one intra-molecular cross-linked peptide was identified. Since we detected as many as three intra-molecular cross-links in the whole protein analyses we expected to see more intra-molecular cross-linked peptides. There are several possible explanations for not detecting all of the intra-molecular cross-linked peptides. The simplest one is incomplete digestion of the intra-molecular cross-linked peptides due to limited access of trypsin to the cleavage sites close to the cross-linked peptides. A second possible explanation is poor binding of the intra-molecular cross-linked peptides to the plates due to steric hindrance of the biotin moiety by the cross-linked peptide arms.
Table 1.
| Modification | Peptide(s) | Charge | Retention time (min) | Observed m/z | Calculated m/z | Error (ppm) | Oxidations |
|---|---|---|---|---|---|---|---|
| Hanging | 19–30 (K25) | 2+ | 26.7 | 893.95501 | 893.95584 | < 1 | biotin |
| Hanging | 31–97 (K70) | 6+ | 28.7 | 1282.43711 | 1282.43857 | 1.1 | biotin and Mets |
| Hanging | 133–143 (K140) | 2+ | 28.7 | 848.48503 | 848.48437 | < 1 | biotin |
| Hanging | 133–167 (K140 & K158) | 5+ | 25.6 | 987.0509 | 987.0511 | < 1 | 2 biotins |
| Hanging | 155–162 (K158) | 2+ | 20.9 | 665.31668 | 665.31644 | < 1 | biotin |
| Hanging | 155–167 (K158) | 3+ | 23.5 | 659.97553 | 659.97563 | < 1 | biotin |
| Hanging | 168–173 (K170) | 2+ | 21.9 | 599.79989 | 599.79974 | < 1 | biotin |
| Intra-molecular | 31–97(K70):155–162(K158) | 7+ | 30 | 1220.88184 | 1220.88026 | 1.3 | biotin and Mets |
Stable isotopic labeling of cross-linkers has been used to obtain a mass tag signature of cross-linking.30 It is also possible to obtain a signature of cross-linking using mass tagging if two different lengths of cross-linkers are used and they cross-link the same sites. In this study, regular mass differences (56 Da) between BCCL1 and BCCL2 treated CA samples (eluted from streptavidin plate) were observed arising from the longer carbon chain (C4H8 addition) in BCCL2. For example, a 56.06257 Da mass difference was observed between ions in BCCL1 and BCCL2 treated CA samples eluting at 21.9 min (Figure 4). In MS/MS experiments, the fragment ion masses corresponding to y1-y3 and b2 were the same for BCCL1 and BCCL2 treated samples whereas those of y4 and b3 were 56 Da higher in BCCL2 cross-linked samples than in BCCL1 treated samples confirming that the mass increase arose from the longer cross-linker (data not shown).
Figure 4.
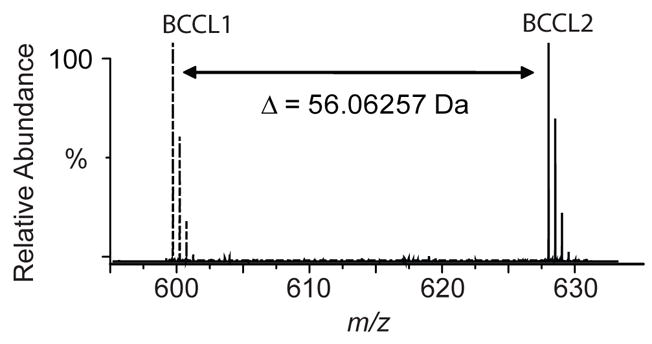
Superposition of the FT-ICR MS spectra of a peptide from a digest of BCCL1 (dashed lines) and BCCL2 (solid lines) cross-linker treated CA. The monoisotopic mass increment (56.06257 Da) corresponds precisely to the mass difference between the two cross-linkers (C4H8, 56.06260 Da). Mass measurement errors are less than 1 ppm on the FT-ICR MS.
As mentioned earlier, methionines in the peptide are prone to be oxidized and all the methionine residues in the eluted peptides turned out to be oxidized. For example, the peptide spanning residues 31–97 and carrying a hanging cross-linker was un-oxidized in the un-fractionated cross-linking sample but the same peptide was heavily oxidized in the eluted sample (Figure 5). Accurate mass measurement of the un-oxidized (m/z = 1266.44365, 6+) and oxidized (m/z = 1282.43711, 6+) peptides were in an excellent agreement with the calculated monoisotopic masses (m/z = 1266.44640, 6+ and m/z = 1282.43771, 6+, respectively).
Figure 5.

FT-ICR MS spectra of a peptide from a digest of BCCL1 cross-linker treated CA. The un-fractionated reaction (top) and eluted fraction (bottom). Accurate mass measurement of the un-oxidized (m/z = 1266.44365, 6+) and completely oxidized (m/z = 1282.43711, 6+) hanging peptides were in an excellent agreement with the calculated monoisotopic masses (m/z = 1266.44640, 6+ and m/z = 1282.43771, 6+, respectively).
Despite the extensive oxidation observed, removal of predominating unmodified peptides allowed us to identify the cross-linked peptide in the eluted sample (Figure 6). A septuply charged ion (m/z = 1233.59177) eluted with a retention time of approximately 30 min, the position where either very hydrophobic or large peptides would be expected to elute. The best fit to the observed mass was obtained by assuming lysine 70 of the peptide spanning residues 31–97 was cross-linked to lysine 158 of the peptide spanning residues 155–162 with complete oxidation of all five methionines and the biotin. The calculated m/z for this combination was 1233.59019 in an excellent agreement (1.3 ppm) with the observed monoisotopic m/z = 1233.59177. To confirm the presence of the cross-linked peptides in the un-fractionated reaction, we searched for both the observed ion and the putative un-oxidized form. As expected, the oxidized form was not found in the un-fractionated reaction, whereas a septuply charged ion of m/z = 1220.88184 which corresponds to 96 fewer Da was observed. The observed m/z was in an excellent agreement (1.3 ppm) with the calculated monoisotopic m/z (1220.88026, 7+) of the un-oxidized form of the cross-linked peptides. The formation of a crosslink between lysine 70 which lies in the N-terminal domain of HIV-1 CA and lysine 158 which lies in the C-terminal domain implies considerable flexibility of the linker region between the domains in solution. Linker flexibility appears to be required for the CA protein to adopt the different conformations required for the formation of closed viral cores.34, 35
Figure 6.

FT-ICR MS spectra of the BCCL1 cross-linked peptides from the eluted fraction (top) and un-fractionated cross-linking reaction (bottom). (Top) The observed monoisotopic m/z = 1233.59177 is in an excellent agreement (1.3 ppm) with the calculated m/z = 1233.59019 for BCCL1 cross-linked peptides 31–97 and 155–162 with complete oxidation of the five methionines and the biotin. (Bottom) The observed m/z was in an excellent agreement (1.3 ppm) with the calculated monoisotopic m/z (1220.88026, 7+) of above two peptides cross-linked through BCCL1 cross-linker without oxidation.
Conclusions
In this study, we have successfully synthesized biotinylated lysine reactive homobifunctional cross-linkers. Cross-linking of purified HIV-1 CA resulted in the formation of both hanging and intra-molecular cross-links. The cross-linked peptides were effectively enriched by streptavidin coated plates and successfully recovered using a MS friendly buffer composition.
However, extra care must be taken to account for covalent modifications during sample preparation such as oxidation, amidation, and carboxylation, etc. Although extensive oxidation of methionines and biotin was observed in the recovered samples, the enrichment of modified peptides and removal of the predominating unmodified peptides simplified the mass spectrometric analyses. The combination of the high mass accuracy of FT-ICR MS and the MS/MS capability of the linear ion trap allowed us to unambiguously identify the cross-linking sites and oxidation points.
In spite of highly specific binding and recovery of the biotinylated cross-linkers, preferred binding of hanging peptides over the intra-molecularly cross-linked peptides was observed. Intra-molecularly cross-linked peptides have two long peptides attached to the cross-linker which may screen the biotin moiety and prevent binding. To avoid selective enrichment of hanging cross-linkers, further optimization should be undertaken. Altering the length of the linker between the biotin moiety and the reactive sites on the cross-linker might be one such approach. Various proteases could also be tested to enhance digestion and result in shorter peptides which may improve binding. In this regard, FT-ICR MS may contribute significantly to reduce numbers of candidate possibilities through accurate mass measurement.
Acknowledgments
This work was supported by NIH grants AI44626 and AI53821 to PEP.
References
- 1.Jiang W, Baker ML, Jakana J, Weigele PR, King J, Chiu W. Nature. 2008;451:1130. doi: 10.1038/nature06665. [DOI] [PubMed] [Google Scholar]
- 2.Jiang W, Li Z, Zhang Z, Baker ML, Prevelige PE, Jr, Chiu W. Nat Struct Biol. 2003;10:131. doi: 10.1038/nsb891. [DOI] [PubMed] [Google Scholar]
- 3.Lander GC, Tang L, Casjens SR, Gilcrease EB, Prevelige P, Poliakov A, Potter CS, Carragher B, Johnson JE. Science. 2006;312:1791. doi: 10.1126/science.1127981. [DOI] [PubMed] [Google Scholar]
- 4.Marles-Wright J, Grant T, Delumeau O, van Duinen G, Firbank SJ, Lewis PJ, Murray JW, Newman JA, Quin MB, Race PR, Rohou A, Tichelaar W, van Heel M, Lewis RJ. Science. 2008;322:92. doi: 10.1126/science.1159572. [DOI] [PubMed] [Google Scholar]
- 5.Simonetti A, Marzi S, Myasnikov AG, Fabbretti A, Yusupov M, Gualerzi CO, Klaholz BP. Nature. 2008;455:416. doi: 10.1038/nature07192. [DOI] [PubMed] [Google Scholar]
- 6.Yu X, Jin L, Zhou ZH. Nature. 2008;453:415. doi: 10.1038/nature06893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fenn JB, Mann M, Meng CK, Wong SF, Whitehouse CM. Science. 1989;246:64. doi: 10.1126/science.2675315. [DOI] [PubMed] [Google Scholar]
- 8.Tanaka K, Waki H, Ido Y, Akita S, Yoshida Y, Yoshida T, Matsuo T. Rapid Commun Mass Spectrom. 1988;2:151. [Google Scholar]
- 9.Bothner B, Dong XF, Bibbs L, Johnson JE, Siuzdak G. J Biol Chem. 1998;273:673. doi: 10.1074/jbc.273.2.673. [DOI] [PubMed] [Google Scholar]
- 10.Arrington CB, Robertson AD. J Mol Biol. 2000;300:221. doi: 10.1006/jmbi.2000.3859. [DOI] [PubMed] [Google Scholar]
- 11.Deng Y, Smith DL. Biochemistry. 1998;37:6256. doi: 10.1021/bi972711o. [DOI] [PubMed] [Google Scholar]
- 12.Kang S, Poliakov A, Sexton J, Renfrow MB, Prevelige PE., Jr J Mol Biol. 2008;381:772. doi: 10.1016/j.jmb.2008.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kang S, Prevelige PE., Jr J Mol Biol. 2005;347:935. doi: 10.1016/j.jmb.2005.02.021. [DOI] [PubMed] [Google Scholar]
- 14.Lanman J, Lam TT, Barnes S, Sakalian M, Emmett MR, Marshall AG, Prevelige PE., Jr J Mol Biol. 2003;325:759. doi: 10.1016/s0022-2836(02)01245-7. [DOI] [PubMed] [Google Scholar]
- 15.Miranker A, Robinson CV, Radford SE, Aplin RT, Dobson CM. Science. 1993;262:896. doi: 10.1126/science.8235611. [DOI] [PubMed] [Google Scholar]
- 16.Benjamin DR, Robinson CV, Hendrick JP, Hartl FU, Dobson CM. Proc Natl Acad Sci USA. 1998;95:7391. doi: 10.1073/pnas.95.13.7391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Esteban O, Bernal RA, Donohoe M, Videler H, Sharon M, Robinson CV, Stock D. J Biol Chem. 2008;283:2595. doi: 10.1074/jbc.M704941200. [DOI] [PubMed] [Google Scholar]
- 18.Fandrich M, Tito MA, Leroux MR, Rostom AA, Hartl FU, Dobson CM, Robinson CV. Proc Natl Acad Sci USA. 2000;97:14151. doi: 10.1073/pnas.240326597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kang S, Oltrogge LM, Broomell CC, Liepold LO, Prevelige PE, Young M, Douglas T. J Am Chem Soc. 2008;130:16527. doi: 10.1021/ja807655t. [DOI] [PubMed] [Google Scholar]
- 20.Kitagawa N, Mazon H, Heck AJR, Wilkens S. J Biol Chem. 2008;283:3329. doi: 10.1074/jbc.M707924200. [DOI] [PubMed] [Google Scholar]
- 21.Back JW, de Jong L, Muijsers AO, de Koster CG. J Mol Biol. 2003;331:303. doi: 10.1016/s0022-2836(03)00721-6. [DOI] [PubMed] [Google Scholar]
- 22.Cai K, Itoh Y, Khorana HG. Proc Natl Acad Sci USA. 2001;98:4877. doi: 10.1073/pnas.051632898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schulz DM, Ihling C, Clore GM, Sinz A. Biochemistry. 2004;43:4703. doi: 10.1021/bi036149f. [DOI] [PubMed] [Google Scholar]
- 24.Sinz A. J Mass Spectrom. 2003;38:1225. doi: 10.1002/jms.559. [DOI] [PubMed] [Google Scholar]
- 25.Trester-Zedlitz M, Kamada K, Burley SK, Fenyo D, Chait BT, Muir TW. J Am Chem Soc. 2003;125:2416. doi: 10.1021/ja026917a. [DOI] [PubMed] [Google Scholar]
- 26.Young MM, Tang N, Hempel JC, Oshiro CM, Taylor EW, Kuntz ID, Gibson BW, Dollinger G. Proc Natl Acad Sci USA. 2000;97:5802. doi: 10.1073/pnas.090099097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Green NS, Reisler E, Houk KN. Pro Sci. 2001;10:1293. doi: 10.1110/ps.51201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kang S, Hawkridge AM, Johnson KL, Muddiman DC, Prevelige PE., Jr J Proteome Res. 2006;5:370. doi: 10.1021/pr050356f. [DOI] [PubMed] [Google Scholar]
- 29.Rosenfeld J, Capdevielle J, Guillemot JC, Ferrara P. Anal Biochem. 1992;203:173. doi: 10.1016/0003-2697(92)90061-b. [DOI] [PubMed] [Google Scholar]
- 30.Petrotchenko EV, Olkhovik VK, Borchers CH. Mol Cell Proteomics. 2005;4:1167. doi: 10.1074/mcp.T400016-MCP200. [DOI] [PubMed] [Google Scholar]
- 31.Kang S, Lander GC, Johnson JE, Prevelige PE. Chembiochem. 2008;9:514. doi: 10.1002/cbic.200700555. [DOI] [PubMed] [Google Scholar]
- 32.Melville DB. J Biol Chem. 1954;208:495. [PubMed] [Google Scholar]
- 33.Musker WK, Wolford TL, Roush PB. J Am Chem Soc. 1978;100:6416. [Google Scholar]
- 34.Datta SAK, Curtis JE, Ratcliff W, Clark PK, Crist RM, Lebowitz J, Krueger S, Rein A. J Mol Biol. 2007;365:812. doi: 10.1016/j.jmb.2006.10.073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cardone G, Purdy JG, Cheng N, Craven RC, Steven AC. Nature. 2009;457:694. doi: 10.1038/nature07724. [DOI] [PMC free article] [PubMed] [Google Scholar]