

Abstract
Multiple sclerosis (MS) is a T cell-mediated autoimmune disease of the CNS, possessing both immune and neurodegenerative events that lead to disability. Adoptive transfer (AT) of myelin basic protein (MBP)-specific T cells into naïve female SJL/J mice results in a relapsing-remitting (RR) form of experimental autoimmune encephalomyelitis (EAE). Blocking the mechanisms by which MBP-specific T cells are activated before AT may help characterize the immune arm of MS and offer novel targets for therapy. One such target is calpain, which is involved in activation of T cells, migration of immune cells into the CNS, degradation of axonal and myelin proteins, and neuronal apoptosis. Thus, the hypothesis that inhibiting calpain in MBP-specific T cells would diminish their encephalitogenicity in RR-EAE mice was tested. Incubating MBP-specific T cells with the calpain inhibitor SJA6017 before AT markedly suppressed the ability of these T cells to induce clinical symptoms of RR-EAE. These reductions correlated with decreases in demyelination, inflammation, axonal damage, and loss of oligodendrocytes and neurons. Also, calpain:calpastatin ratio, production of tBid, and Bax:Bcl-2 ratio, and activities of calpain and caspases, and internucleosomal DNA fragmentation were attenuated. Thus, these data suggest calpain as a promising target for treating EAE and MS.
Keywords: apoptosis, EAE, encephalitogenicity, inflammation, MBP-specific T cells
Introduction
Multiple sclerosis (MS) is a debilitating autoimmune disease of the central nervous system (CNS) and MS appears as a relapsing-remitting (RR) disease in 85% of patients, characterized by episodes of varying clinical disability interspersed with periods of complete or partial recovery (Noonan et al. 2002). MS is thought to develop as a result of an infiltration of myelin-reactive T cells and other immune cells into the CNS, resulting in inflammation, myelin degradation, axonal damage, and loss of neurons and oligodendrocytes (Peterson et al. 2001; Keegan and Noseworthy 2002). Whether or not immune components of MS pathology occur before or in conjunction with neurodegenerative components is still under debate, and studies have demonstrated that axonal damage occurs early in disease progression and correlates with disease severity (Trapp et al. 1999). Since the complete etiology is not clearly understood, long-term effective therapies for MS have not yet been developed; however, only treatments are anti-inflammatory drugs that provide temporary relief by reducing the inflammatory responses. Nevertheless, the degradation of myelin proteins in the CNS of MS patients has previously implicated the involvement of various proteases in the pathogenesis of this disease (Einstein et al. 1972; Cuzner et al. 1975; Banik et al. 1979) and thus proteases are therapeutic targets utilizing protease inhibitors as intervening agents (Govindarajan et al. 1974; Marks et al. 1974). The calcium (Ca2+)-dependent protease calpain was postulated to be involved in MS more than two decades ago (Banik et al. 1985; de Rosbo and Bernard 1989).
Calpain exists as ubiquitous and tissue-specific isoforms that are dependent on Ca2+ for activation and the ubiquitous isoforms of calpain, μcalpain and mcalpain, are activated by μM and mM Ca2+ concentrations, respectively (Hassen et al. 2006). Over the years, support for the involvement of ubiquitous isoforms of calpain in demyelinating diseases has accumulated (Shields et al. 1999; Schaecher et al. 2001a). Calpain expression and activity are increased in spinal cord and optic nerve of animals with experimental autoimmune encephalomyelitis (EAE), an animal model of MS (Shields et al. 1998a; Shields et al. 1998b), as well as in postmortem tissues from the patients with MS (Shields and Banik 1999; Diaz-Sanchez et al. 2006). Increased calpain activity correlated with disease onset, T cell and macrophage migration into the CNS, axonal damage, and neuronal loss in an acute EAE rat model (Schaecher et al. 2002; Guyton et al. 2005). Calpain is also involved in the activation of T cells (Deshpande et al. 1995b; Schaecher et al. 2001b) and when released from activated T cells, it degrades myelin basic protein (MBP) and other myelin components (Deshpande et al. 1995a) strongly suggesting a role for calpain in perpetuating immune-mediated demyelination by calpain-cleaved antigenic peptides. Nuclear factor kappa-B (NF-κB) is a nuclear transcription factor that plays a key role in increasing the expression of many pro-inflammatory mediators, including inducible nitric oxide synthase (iNOS) and cyclooxygenase-2 (COX-2) (Surh et al. 2001). Calpain causes indirectly the activation and nuclear translocation of NF-κB via degradation of the inhibitor of κB-alpha (IκBα) (Schaecher et al. 2004). Calpain has also been linked to neurodegenerative events such axonal damage and loss of neurons and oligodendrocytes (Guyton et al. 2005; Cerghet et al. 2006), at least partially through modulation of proteins involved in classical receptor and mitochondrial apoptotic pathways (Das et al. 2008).
Targeting multiple pathogenic events of a disease has been postulated to offer better therapy in heterogeneous diseases such as MS. Since calpain has been implicated in both immune and neurodegenerative arms of MS and EAE, blocking this protease may inhibit multiple pathways linked to disability. Although calpain is regulated in vivo by its endogenous inhibitor calpastatin, in reality calpastatin is too large to be used as a therapeutic agent (Higuchi et al. 2005). Therefore, synthetic cell-permeable calpain inhibitors have been developed for using in the treatment of neurodegenerative diseases including animal models of Parkinson’s disease (PD), Alzheimer’s disease (AD), spinal cord injury (SCI), and traumatic brain injury (TBI) (Ray and Banik 2003). Calpain inhibitors have also proven effective in reducing clinical symptoms of EAE in acute animal models by reducing inflammation, axonal damage, and neuronal loss (Guyton et al. 2006; Hassen et al. 2006).
Our current study expands previous investigations by examining the effects of incubating MBP-specific T cells with the water-soluble calpain inhibitor SJA6017 before adoptive transfer (AT) in the SJL/J mouse model of RR-EAE. These studies are important as they incorporate an RR model of EAE, which is clinically more relevant, and allow dissecting the effects of specifically targeting the T cell component in the immune arm of EAE development on inflammatory and neurodegenerative events in EAE. The EAE animals that received MBP-specific T cells incubated with SJA6017 before AT demonstrated a dose-dependent reduction in clinical symptoms of disability during acute and chronic relapses, as well as a reduction in number of relapses. These findings correlated well with decreases in calpain:calpastatin ratio, demyelination, inflammation, axonal damage, and neuronal and oligodendrocyte death in spinal cord tissues from the RR-EAE mice. Results also indicated that reductions in neuronal and oligodendrocyte death were achieved by inhibiting the activity of various pro-apoptotic proteins. These findings strongly implicate calpain to be a therapeutic target for the treatment of MS.
Materials and methods
Isolation of MBP-specific T cells and incubation with SJA6017
Specific pathogen-free female SJL/J mice (3 to 4 weeks old) were purchased from the commercial vendor (Harlan Laboratories, Indianapolis, IN). Animal maintenance and experimental protocols were approved by the Institutional Animal Care and Use Committee (IACUC) of the Rush University Medical Center (Chicago, IL). Isolation of MBP-specific T cells for AT was performed as previously described (Dasgupta et al. 2003). Briefly, donor mice were immunized subcutaneously (s.c.) with bovine MBP (400 μg) and Mycobacterium tuberculosis (60 μg) in IFA, sacrificed on day 10–12 post-immunization, and the draining lymph nodes were harvested. Single-cell suspensions were treated with red blood cell (RBC) lysis buffer, washed, and cultured at a concentration of 4–5×106 cells/mL in RPMI 1640 (supplemented with 10% fetal bovine serum, 50 μmol/L 2-mercaptoethanol, 2 mmol/L L-glutamine, 100 U/mL penicillin, and 100 μg/mL streptomycin) and incubated with 50 μg/mL MBP plus the vehicle dimethyl sulfoxide (DMSO) or 10–100 μmol/L SJA6017 (Senju Laboratory, Japan). On day 4, cells were harvested and resuspended in Hank’s balanced salt solution (HBSS).
Induction of RR-EAE by passive transfer of MBP-specific T cells
A total of 2 × 107 viable cells in a volume of 200 μL were injected into the tail vein of naïve SJL/J mice and Pertussis toxin (150 ng/mouse) was injected once via intraperitoneal (i.p.) injection on 0 day post-transfer (dpt). Cells isolated from donor mice immunized with complete Freund’s adjuvant (CFA) or incomplete FA (IFA) alone were not viable after 4 days in culture with MBP and therefore were not transferred. The viability of MBP-specific T cells incubated with SJA6017 was similar to the viability of cells incubated with vehicle alone (>95% viability). Animals were observed daily for clinical symptoms and scored (by a blinded investigator) as follows: 0, no clinical disease; 0.5, piloerection; 1, tail weakness; 1.5, tail paralysis; 2, hind limb weakness; 3, hind limb paralysis; 3.5, forelimb weakness; 4, forelimb paralysis; and 5, moribund or death. Some animals in each group were monitored until day 94 dpt to determine the effect of SJA6017 treatment of MBP-specific T cells on relapse rate and demyelination during the chronic phase of the disease. In other studies, animals were sacrificed on 21 dpt, which corresponds to peak of the initial acute episode in this RR-EAE model and spinal cord tissues were collected for biochemical and immunohistochemical analyses of pathophysiological changes in RR-EAE animals.
Determination of cell viability
In order to determine cell viability via the 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyl tetrazolium bromide (MTT) assay (Sigma Chemical, St. Louis, MO), cells were incubated with MBP ± SJA6017 (10 or 50 μmol/L) for 3 days as described above. Specific volume (10 μL) of MTT (1 mg/mL) was added to each well and the plate was incubated for 1 h. All culture media were then removed and the cells were resuspended in 100 μL of DMSO and incubated for 1 h. Cell viability was assessed via colorometric changes in absorbance at a wavelength of 570 nm using an ELx800 Microplate Reader (Bio-Tek Instruments, Winooski, VT). All treatments were performed in triplicate. Lactade dehydrogenase (LDH), an enzyme released in cell supernatant as cells die, was also used as an indicator of cell viability. Culture supernatants (100 μL) were also collected from each well, and LDH activity was determined with a colorimetric LDH assay kit (Cayman Chemicals, Ann Arbor, MI) reading absorbance at 490 nm.
Determination of demyelination
The effects of AT of MBP-specific T cells incubated with SJA6017 on reducing demyelination during the late chronic phase of RR-EAE were assessed via Luxol Fast Blue (LFB), which stains myelin, as described previously (Tyor et al. 2002; Sribnick et al. 2005). Briefly, animals were sacrificed on 94 dpt and brain and spinal cord tissues were fixed overnight in 95% paraformaldehyde at 4°C. After fixation, samples were embedded in paraffin, sliced into 10 μm sections, and placed on slides. Slides were stained with LFB, and then counter-stained with nuclear fast red stain. Stained sections were used to capture pictures on an Olympus microscope and imaged with the Magna Fire SP CCD camera.
Western blot analyses of protein expression
Protein extraction and Western blot analyses of proteins in spinal cord homogenate from EAE mice have been described previously (Das et al. 2008). All primary IgG antibodies for Western blotting were purchased from Santa Cruz Biotechnology (Santa Cruz, CA) and diluted at a concentration of 1:200, except for MBP that was diluted 1:1000 and SMI-32 (de-phosphorylated neurofilament protein or de-NFP) that was purchased from Sternberger Monoclonals (Lutherville, MD) and diluted at 1:400. Blots were incubated for 24 h with primary IgG antibody against mcalpain, calpastatin, capase-8, tBid, Bax, Bcl-2, caspase-3, MBP, or SMI-32, diluted in TBS with 0.1% Tween-20 (TBST) plus 5% (w/v) fat-free milk (FFM), and then incubated with horseradish peroxidase (HRP)-conjugated anti-rabbit (1:2000) or anti-mouse (1:2000) secondary IgG antibody in 0.1% TBST for 45 min. Protein bands were detected by alkaline HRP-catalyzed oxidation of luminol in the presence of H2O2 using enhanced chemiluminescence (ECL) system (Amersham Life Science, Arlington Heights, IL). Blots were exposed to X-OMAT XAR-2 films and autoradiograms were scanned and imaged using Adobe Photoshop software (Adobe Systems, San Jose, CA). Bands were quantified using NIH ImageJ software, normalized to β-actin, and expressed as a ratio or as % change in protein level, compared with an exposure to SJA6017-0 μmol/L (ES-0) set at 100%.
Cell-specific immunohistochemical labeling
Spinal cord tissues from EAE mice were flash-frozen in tissue freezing media and stored at −70°C until processed. Immunohistochemical staining for pathophysiological changes in spinal cord tissue sections was performed as previously described (Guyton et al. 2005). Microgliosis and astrogliosis were determined using primary IgG antibodies specific for ED-2 (1:100) and glial fibrillary acidic protein (GFAP, 1:400). Briefly, sections were incubated for 1 h in blocking buffer containing 2% horse serum in phosphate-buffered saline (PBS, pH 7.4), followed by 3–4 h incubation with antibody ED-2 or GFAP. For detection of axonal degeneration, slides were first autoclaved for 5 min in 0.1 mol/L citrate buffer (pH 6.0) and then blocked for 1 h, as described above. Next, tissue sections were incubated overnight at 4°C with SMI-311, a pan-monoclonal antibody (1:1000, Sternberger Monoclonals) for detection of de-NFP. Notably, NFPs are de-phosphorylated before axonal degeneration following injury (Pitt et al. 2000). The sections were incubated for 30 min in the dark with (1:100) horse anti-mouse secondary IgG antibody conjugated to fluorescein isothiocyanate (FITC) to each cell marker. The slides were mounted with 1 drop of Vectashield Mounting Media (Vector Laboratories, Burlingame, CA) cover slipped, and immediately viewed under a fluorescent microscope at 200× magnification.
Combined TUNEL and immunoflourescent labelings
In order to detect cell-specific death in spinal cord tissues, the terminal deoxynucleotidyl transferase (TdT)-mediated dUTP nick-end labeling (TUNEL) assay was combined with cell-specific marker labeling, as previously described (Ray et al. 2000). Briefly, spinal cord tissues were sectioned, fixed as described above, and then post-fixed in 4% methanol-free formaldehyde (in PBS) for 15 min. The slides were saturated with TdT buffer (50 μL/slide) for 5 min and then replaced with TUNEL reaction mixture (50 μL/slide) containing 10× polymerase chain reaction (PCR) mixture containing digoxigenin (DIG)-11-dUTP (2.5 μL) and terminal TdT (25 Units) in buffer (Promega, Madison, WI). Also, cell-specific staining was performed with an antibody for detecting oligodendrocytes (O4, 1:100) or neurons (NeuN, 1:100).
Detection of internucleosomal DNA fragmentation
We recently reported a method for examining internucleosomal DNA fragmentation by gel electrophoresis in spinal cord tissues (Das et al. 2008). Briefly, spinal cord segments were homogenized in 10 mmol/L Tris-HCl, pH 8.0, 150 mmol/L NaCl, 50 mmol/L EDTA in a 1.5-mL Eppendorf tube. The homogenates were digested in 10 mmol/L Tris-HCl, pH 8.0, 50 mmol/L NaCl, 10 mmol/L EDTA, 0.5% SDS, 250 ng/μL, proteinase K at 37°C for at least 12 h, extracted twice with an equal volume of a (1:1) mixture of phenol (pH 8.0) and chloroform, and then chloroform. A solution of MgCl2 to a final concentration of 10 nmol/L was then added and DNA was precipitated by adding 100% ethanol. Centrifugation was performed to obtain the pellet, which was washed with 70% ethanol, air dried, and dissolved in TE buffer (10 mmol/L Tris-HCl, pH 8.0, 1 mmol/L EDTA) containing RNase A (50 ng/μL). After 1-h incubation at 37°C, the samples were loaded onto a 1.6% agarose gel and electrophoresed in 1xTAE (40 mmol/L Tris-acetate, 1 mmol/L EDTA, pH 8.3) buffer. The gel was stained with ethidium bromide (1 μg/mL) and photographed on an ultraviolet (303 nm) transilluminator using Polaroid Type 667 (positive/negative) film and the negative was used to detect internucleosomal DNA fragmentation.
Statistical analyses
All statistical tests were chosen on the basis of well-recognized recommendations (Fleming et al. 2005) for analyzing data from EAE studies and performed using the SAS statistics software (SAS Institute, Cary, NC). Overall statistical significance for various data including median clinical score, day of onset, and number of relapses were performed using the Kruskal-Wallis test followed by the Mann-Whitney-U tests for pair-wise comparisons. Overall significant differences in protein expression were analyzed using the non-parametric Kruskal-Wallis test followed by Mann-Whitney-U tests for pair-wise comparisons. The null hypothesis for each analysis was rejected at p ≤ 0.05.
Results
Calpain inhibition decreased the encephalitogenicity of MBP-specific T cells
To understand the effect of calpain inhibition on encephalitogenicity of MBP-specific T cells, lymph node cells (LNC) isolated from MBP-immunized donor mice were incubated with MBP in the presence of calpain inhibitor SJA6017 for 4 days followed by transfer of MBP-specific T cells to naïve female SJL/J mice via tail vein. The progression of EAE was monitored by recording clinical scores (CS) of paralysis during the acute phase of RR-EAE (5–21 dpt) and late RR phases (61–94 dpt). The SJL/J mice that received MBP-specific T cells, which were incubated with vehicle (DMSO) alone (ES-0), developed severe symptoms with a clinical score reaching 2.5, and recurring relapse with lower recovery between episodes (Fig. 1), as explained previously (Dasgupta et al. 2003). In contrast, mice that received MBP-specific T cells, which were exposed to 10 μmol/L SJA6017 (ES-10) or 100 μmol/L SJA6017 (ES-100) exhibited a dose-dependent decrease in CS during the initial acute phase and during each relapse. Disease development was delayed until 11 dpt in the ES-100 group and the relapse rate was reduced from 3 relapses in the chronic phase in the ES-0.
Fig. 1.

Adoptive transfer (AT) of MBP-specific T cells following exposure to the calpain inhibitor SJA6017 reduced clinical symptoms of RR-EAE in SJL/J mice. RR-EAE was induced in naïve SJL/J mice via AT of MBP-specific T cells following exposure to 0 (vehicle only), 10, 50, and 100 μmol/L SJA6017 (ES-0, ES-10, ES-50, and ES-100). Although the animals were monitored for clinical signs daily, we only reported clinical scores (CS) during the acute phase (days 1–21 post-AT) and RR phase (days 61–94 post-AT) of the disease. Data were presented as mean score ± SEM (n = 3 to 5 in acute phase and n = 3 in RR phase). Significant difference was indicated by * p ≤ 0.05 compared with ES-0 and # p ≤ 0.05 compared with ES-10.
In second set of studies, a more complete dose-response of calpain inhibition in MBP-specific T cells before AT were performed to characterize inflammatory and neurodegenerative events during the acute phase of RR-EAE by including 50 μmol/L SJA6017 (ES-50). The peak CS in the ES-50 group during the acute phase reached 0.5, and the day of onset was delayed until day 7 dpt. These data indicated that treatment of MBP-specific T cells with SJA6017 before AT resulted in a dose-dependent reduction in CS and delay in onset of the acute episode in RR-EAE animals. The focus of the remainder of our investigation was on evaluating the effects of incubating MBP-specific T cells with SJA6017 before AT on pathophysiological changes in spinal cord of mice during the initial acute phase of RR-EAE.
Exposure to SJA6017 did not affect viability of MBP-specific T cells
In order to determine if exposure of MBP-specific T cells to SJA6017 before AT reduced encephalogenecity by killing T cells, we assessed changes in cell viability using the MTT assay (Fig. 2). After MBP-specific T cells were exposed to MBP ± SJA6017 (10 or 50 μmol/L) for 3 days, no significant differences in cell viability were detected in any of the MBP-specific T cell groups (Fig. 2a). In order to confirm these results, also LDH assay was employed in another set of experiments. No significant increases in LDH were noted in the MBP-specific T cells esposed to SJA6017, when compared with vehicle-treated MBP-specific T cells (Fig. 2b). These data demonstrated that calpain inhibition in MBP-specific T cells did not reduce EAE clinical signs by reducing the viability of these cells before AT.
Fig. 2.

Exposure to SJA6017 did not reduce cell viability of MBP-specific T cells. Cell viability was determined using colorometric (a) MTT assay and (b) LDH release assay. Data were presented as mean score ± SEM (n = 4 for MTT assay, n = 3 for LDH release assay, * p ≤ 0.05 for vehicle vs. SJA6017 treated MBP-specific T cells).
Calpain inhibition in MBP-specific T cells before AT decreased calpain:calpastatin ratio and calpain activity in RR-EAE mice
Previous studies in our laboratory demonstrated that SJA6017 therapy attenuated pathophysiological changes in spinal cord from Lewis rats with acute EAE (Guyton et al. 2006). The goal of the present study was to determine if blocking calpain activity in MBP-specific T cells before AT into naïve SJl/J mice was sufficient to reduce inflammatory and neurodegenerative events during the initial acute phase of RR-EAE (21 dpt). First, Western blot analysis of spinal cord tissues on 21 dpt was performed to determine calpain:calpastatin ratio and calpain activity (Fig. 3). Calpain:calpastatin ratios were significantly decreased to 0.70 ± 0.18% and 0.68 ± 0.09% in the ES-10 and ES-50 groups, respectively, when compared with calpain:calpastatin ratio of 1.54 ± 0.33% in ES-0 group (Fig. 3a, 3b). Calpain activity was also determined using an antibody, which recognized calpain-cleaved 145 kD spectrin break down product (SBDP) (Fig. 3a, 3c). In correlation with decrease in calpain:calpastatin ratios, calpain activities were also significantly decreased by 37.5 ± 3.9% and 48.7 ± 5.3% in spinal cords from ES-10 and ES-50 groups, respectively.
Fig. 3.

AT of MBP-specific T cells following exposure to SJA6017 reduced calpain:calpastatin ratio and calpain activity during the acute phase of RR-EAE in SJL/J mice. Calpain and calpastatin expression and calpain activity were examined in RR-EAE animals via Western blotting of spinal cord homogenates on day 21 post-AT. (a) Representative Western blots of 42 kD β-actin, 80 mcalpain, 100 kD calpastatin, and calpain-cleaved 145 kD SBDP. Expression of β-actin was used to monitor loading of equal amount of protein in each lane. (b) Determination of calpain:calpastatin ratio. (c) Determination of calpain activity presented as % change compared with ES-0. Results were presented as box-plot of inter-quartile data with the white line denoting the median expression. Diamonds represent mean protein expression (n = 3 per group, * p ≤ 0.05 compared with ES-0).
Calpain inhibition in MBP-specific T cells before AT decreased inflammatory events in RR-EAE mice
Microgliosis and astrogliosis are well-documented inflammatory events that occur during EAE and the effects of exposure to SJA6017 before AT of MBP-specific T cells on these events were examined (Fig. 4). As expected, microgliosis (ED-2 immunoreactivity) and astrogliosis (GFAP immunoreactivity) were increased in spinal cords from the ES-0 group (Fig. 4). In line with decrease in CS of SJL/J mice that received MBP-specific T cells following exposure to SJA6017, both microgliosis and astrogliosis were substantially attenuated in spinal cord tissues from the ES-10 and ES-50 groups.
Fig. 4.
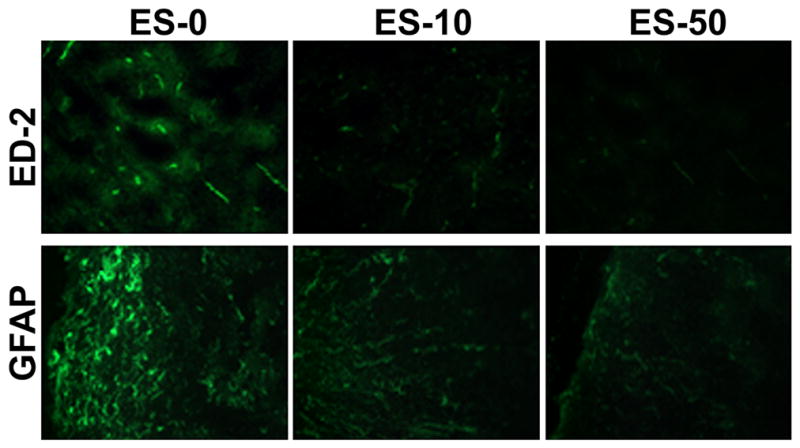
AT of MBP-specific T cells following exposure to SJA6017 reduced inflammatory events during the acute phase of RR-EAE in SJL/J mice. Representative images of spinal cord tissue sections collected from RR-EEAE mice at day 21-post-AT. Tissue sections were stained for microgliosis and astrogliosis using the antibodies against ED-2 and GFAP, respectively (n = 3, magnification 200×).
The effects of calpain inhibition in MBP-specific T cells on expression of signaling proteins involved in pro-inflammatory events were also examined via Western blot analyses of spinal cord homogenates (Fig. 5). While significant changes were not achieved in the ES-10 group, exposure of MBP-specific T cells to SJA6017 resulted in a significant decrease in the expression of NF-κB RelA from 91.7 ± 12.0% in ES-0 group of mice to 33.3 ± 2.4 in ES-50 group of mice (Fig. 5a, 5b). The levels of COX-2 expression were decreased by 68.3 ± 20.9% and 78.2 ± 4.1% in mice that received MBP-specific T cells following exposure to 10 and 50 μmol/L SJA6017, respectively (Fig. 5a, 5c). Further, the levels of expression of iNOS, the enzyme involved in nitric oxide production, were also significantly decreased to 68.5 ± 19.2% and 35.1 ± 6.4% in spinal cord tissues as a result of exposing MBP-specific T cells to 10 and 50 μmol/L SJA6017, respectively (Fig. 5a, 5d).
Fig. 5.

AT of MBP-specific T cells following exposure to SJA6017 reduced expression of proteins involved in inflammation in spinal cord during the acute phase of RR-EAE in SJL/J mice. (a) Representative Western blots of 42 kD β-actin, 65 kD NF-κB, 70 kD COX-2, and 130 kD iNOS using the spinal cord homogenates on day 21-post-AT. (b) Determination of changes in NF-κB expression. (c) Determination of changes in COX-2 expression. (c) Determination of changes in iNOS expression. Results were presented as box-plot of inter-quartile data with the white line denoting the median expression. Diamonds represent mean protein expression (n = 3 per group, * p ≤ 0.05 compared with ES-0, and # p ≤ 0.05 for ES-10 vs. ES-50).
Calpain inhibition in MBP-specific T cells before AT decreased demyelination in RR-EAE mice
Demyelination is a hallmark of the MS and EAE pathology and can lead to axonal damage and neurodegeneration. Therefore, the effects of blocking calpain in MBP-specific T cells before AT on axonal damage were assessed in RR-EAE mice using LFB staining and Western blot analysis of MBP, a major component of myelin, and de-NFP in spinal cord tissues (Fig. 6). The LFB staining indicated myelin was more diffuse and patchy (black arrows) in the spinal cords from ES-0 group of mice (Fig. 6a). In contrast, spinal cord tissues from ES-10 and ES-50 groups exhibited reduced myelin loss, when compared with the ES-0 group. The MBP degradation was also reduced due to calpain inhibiton, as noted by significant 109.2 ± 8.9% and 145.4 ± 35.6% increases in MBP expression in spinal cord tissues from the ES-10 and ES-50 groups, respectively (Fig. 6b, 6c). Similarly, Western blot analysis of total spinal cord homogenates indicated dose-dependent significant decrease in de-NFP to 47.8 ± 6.7% and 27.4 ± 10.9% in the ES-10 and ES-50 groups, respectively, when compared with the ES-0 group (Fig. 6b, 6d).
Fig. 6.

AT of MBP-specific T cells following exposure to SJA6017 reduced demyelination and axonal damage during the acute phase of RR-EAE in SJL/J mice. (a) Demyelination in spinal cord tissue was assessed using the LFB staining. Arrows indicate demyelinated plagues (magnification 200×). (b) Representative Western blots of 42 kD β-actin, 18 kD MBP, and 140 kD de-NFP using the spinal cord homogenates on day 21-post-AT. (c) Determination of percent change in MBP. (d) Determination of percent change in de-NFP. Results were presented as box-plot of inter-quartile data with the white line denoting the median expression. Diamonds represent mean protein expression (n = 3 per group, * p ≤ 0.05 compared with ES-0, and # p ≤ 0.05 for ES-10 vs. ES-50).
Calpain inhibition of MBP-specific T cells before AT decreased oligodendrocyte and neuron death in RR-EAE mice
In order to determine if blocking calpain activity in MBP-specific T cells before AT would reduce cell death in spinal cord tissues from RR-EAE mice, tissue sections were stained with oligodendrocyte-specific marker (O4) or neuron-specific marker (NeuN) and TUNEL (Fig. 7). Many cells were TUNEL positive in white matter from the ES-0 group, with some cells co-stained for O4 (Fig. 7a). While very few O4 positive cells appeared to co-stain with TUNEL in the ES-10 and ES-50 groups, the effects of calpain inhibitor on oligodendrocyte death was not clear, due to the relatively small number of O4 positive cells in the ES-0 group. In contrast to oligodendrocytes, many neurons in spinal cord grey matter also stained TUNEL positive, with multiple cells co-stained for NeuN (Fig. 7b). Interestingly, TUNEL staining was reduced in NeuN positive cells within the grey matter from the ES-10 and ES-50 groups. Thus, in order to characterize whether the cell death identified in spinal cord tissues from animals that received the MBP-specific T cells following exposure to SJA6017 before AT was apoptotic in nature, we examined occurrence of internucleosomal DNA fragmentation, the hallmark of apoptosis (Fig. 8). Genomic DNA from spinal cord tissue from the ES-0 group displayed the 180 bp DNA laddering indicating characteristic apoptotic death, which was almost completely abolished in spinal cord tissues from the mice that received the MBP-specific T cells exposed to the calpain inhibitor SJA6017 (Fig. 8).
Fig. 7.

AT of MBP-specific T cells following exposure to SJA6017 reduced apoptosis of oligodendrocytes and neurons during the acute phase of RR-EAE in SJL/J mice. (a) Representative images of oligodendrocyte [using O4 antibody (green) for detecting mature oligodendrocytes] death [using TUNEL staining (red) for detecting fragmented DNA] in spinal cords. (b) Representative images of neuron [using NeuN antibody (green)] death [using TUNEL staining (red) for detecting fragmented DNA] in spinal cords. Arrows indicate co-staining in merged images (n =3, magnification 200×).
Fig. 8.

AT of MBP-specific T cells following exposure to SJA6017 reduced internucleosomal DNA fragmentation during the acute phase of RR-EAE in SJL/J mice. Detection of internucleosomal DNA fragmentation (180 bp laddering), a hallmark of apoptosis, by agarose gel electrophoresis of genomic DNA isolated from spinal cord tissues (n = 3).
Calpain inhibition in MBP-specific T cells before AT decreased receptor and mitochondria mediated pathways of apoptosis in RR-EAE mice
We performed Western blotting to determine the effects of blocking calpain activation in MBP-specific T cells before AT on proteins involved in regulating receptor mediated events (e.g., caspase-8, tBid) and mitochondrial mediated events (e.g., Bax:Bcl-2) of apoptosis in spinal cord tissues of animals (Fig. 9). Receptor mediated caspase-8 activation, the formation of active 42 kD caspase-8 fragment, was significantly reduced in ES-10 (15.1 ± 10.9%) and ES-50 (6.2 ± 4.6%) animals, when compared with the ES-0 group (Fig. 9a, 9b). Caspase-8 activation causes proteotytic cleavage of Bid to truncated Bid (tBid), which is then translocated to mitochondria for triggering apoptosis. Production of tBid due to caspase-8 activity was significantly decreased in spinal cord tissues from the animals that received the MBP-specific T cells following exposure to SJA6017 (10 and 50 μmol/L) before AT (Fig. 9a, 9c). Changes in the levels of pro-apoptotic Bax protein and anti-apoptotic Bcl-2 protein may result in an increase in Bax:Bcl-2 ratio for making a commitment of cells to apoptotic death via mitochondria mediated pathway. While a trend in a decrease in Bax:Bcl-2 ratio was noted in the ES-10 group, compared with the ES-0 group, Bax:Bcl-2 ratio were significantly decreased in spinal cord tissue from the ES-50 group (Fig. 9a, 9d).
Fig. 9.

AT of MBP-specific T cells following exposure to SJA6017 reduced expression and activation of apoptotic proteins during the acute phase of RR-EAE in SJL/J mice. (a) Representative Western blots of specific proteins of receptor and mitochondria mediated pathways of apoptosis using the spinal cord homogenates on day 21-post-AT. Determination of percent changes in active 42 kD caspase-8 (b), production of 15 kD tBid expression (c), Bax:Bcl-2 ratio (d), active 20 kD caspase-3 (e), and formation of the caspase-3-cleaved 120 kD SBDP (f). Data were presented as box-plot of inter-quartile data with the white line denoting the median expression. Diamonds represent mean protein expression (n = 3 per group, * p ≤ 0.05 compared with ES-0).
The effects of calpain inhibition on activation and activity of caspase-3, the final mediator of apoptosis, were also assessed in the formation of active 20 kD caspase-3 fragment (Fig. 9a, 9e) and production of the caspase-3-cleaved 120 kD SBDP (Fig. 9a, 9f). Blocking calpain activity before AT of MBP-specific T cells resulted in reduction in formation of the active 20 kD caspase-3 fragment to 56.1 ± 2.8% and 52.13 ± 11.5% in the ES-10 and ES-50 groups, respectively, when compared with 85.6 ± 15.9% in the ES-0 group (Fig. 9e). Similarly, decreases were found in the production of 120 kD SBDP in the ES-10 and ES-50 groups (Fig. 9f).
Discussion
The focus of the present study was to examine the effects of calpain inhibition on encephalitogenicity of MBP-specific T cells by incubating these cells with the calpain inhibitor SJA6017 before AT in the SJL/J RR-EAE mice. CS and relapse rates were decreased dose-dependently in mice that received MBP-specific T cells following exposure to SJA6017 before AT. Cell viability was not altered by incubation of MBP-specific T cells with SJA6017 before AT, indicating that decreased encephalogenecity was not due to loss of viability of these cells. Changes in pathophysiological events were then examined in spinal cord tissue collected at 21 dpt, which corresponds with the peak of clinical symptoms during the initial acute phase of RR-EAE in the SJL/J mouse model. Calpain:calpastatin ratio and calpain activity were decreased, correlating with attenuation of inflammatory events including microgliosis and astrogliosis and also reduction in expression of proteins associated with inflammation (NF-κB, COX-2, iNOS). Calpain inhibition prevented neurodegenerative events including myelin degradation, axonal damage, and loss of oligodendrocytes and neurons. Decreases in internucleosomal DNA fragmentation and pro-apoptotic proteins indicated that attenuation of cell death was due largely to blocking apoptosis. The major findings of this investigation are summarized in Table 1.
Table 1.
Summary of the effects of calpain inhibition on encephalogenicity of MBP-specific T cells
| Features of EAE | Effects of calpain inhibition |
|---|---|
| Signs of disease |
|
| Inflammation |
|
| Neurodegeneration |
|
Previous studies in our laboratory and others have indicated that the reduction in disease severity following calpain inhibition was due to decreases in both inflammatory and neurodegenerative events in both acute model and chronic progressive model of EAE (Guyton et al. 2006; Hassen et al. 2006). The current study expands previous findings by determining whether blocking auto-reactive T cells alone is sufficient to attenuate disease severity in the more clinically relevant RR-EAE mouse model. The exact mechanisms by which calpain inhibition attenuated the encepalogenicity of MBP-specific T cells is unclear. However, since the viability of the T cells in each group was >95%, calpain inhibition did not appear to decrease CS of disease by killing MBP-specfic T cells before AT. Reducing T cell activity with IL-27, which reduces IL-2 production in TH17 cells (Villarino et al. 2006), before AT reduced CS in EAE mice (Fitzgerald et al. 2007). Compounds such as gemfibrozil and cinnamon metabolite sodium benzoate also reduced clinical symptoms of disease when incubated with MBP-specific T cells before AT by switching the T cells from a pro-inflammatory Th1 to an anti-inflammatory Th2 mode (Brahmachari and Pahan 2007; Dasgupta et al. 2007). In the current study, both CS of disease and relapse rate were reduced in a dose-dependent manner following AT of SJA6017 exposed MBP-specific T cells, further suggesting that modulating T cell activation through calpain inhibition and/or through switching T cell profiles from Th1 to Th2 profiles was sufficient to lower disease severity. Whether or not SJA6017 exposure had a similar effect in switching the MBP-specific T cells to a Th2 mode has not been examined in the present study, although we think that calpain inhibitors may reduce the encephalogenicity of MBP-specific T cells in a manner similar to gemfibrozil and sodium benzoate. However, lower calpain level correlated with higher production of Th2 cytokines in MS patients during remission, as opposed to higher calpain level and increased Th1 cytokine levels in relapse patients (Imam et al. 2007). One important limitation of the current study is that RR-EAE, along with most other EAE models, is an artificially induced model of MS that takes advantage of the host immune system to induce EAE. Whether or not auto-reactive T cell activation precedes axonal damage in MS is still unclear (Peterson et al. 2001); however, the spontaneous EAE models available are the result of altering T cells (Bettelli 2007; Lee et al. 2007).
Studies in peripheral blood mononuclear cells (PBMCs) from normal and MS patients have indicated that calpain is involved in receptor mediated activation of T cells (Schaecher et al. 2002; Imam et al. 2007). The increase in T cell activity correlates with increase in nuclear translocation of active NF-κB heterodimers since calpain degrades IκB (Schaecher et al. 2004). Upon nuclear translocation, NF-κB increases the transcription of pro-inflammatory mediators COX-2 and iNOS (Surh et al. 2001). In the present study, levels of NF-κB p65, COX-2, and iNOS were decreased in spinal cords from RR-EAE mice that received SJA6017 exposed MBP-specific T cells.
Recently, we and others have shown involvement of calpain in immune cell migration and calpain inhibitors have been found to block migration of immune cells in vitro and in vivo in EAE (Guyton et al. 2006; Butler et al. 2009). Although infiltration of activated inflammatory cells, T cells, and macrophages are thought to enter the CNS through a compromised blood-brain barrier (BBB), the mechanisms by which the BBB is broken are not clearly known. Nonetheless, inflammatory molecules, including nitric oxide that is formed by iNOS, have been implicated in this process (Thiel and Audus 2001) and blocking iNOS expression in T cells before AT may decrease the ability of these immune cells to cross the BBB. Decreases in COX-2 and iNOS expression following calpain inhibition may be directly related to the decrease in NF-κB activity as promoters of both COX-2 and iNOS genes harbor several consensus NF-κB binding sites. However, recent studies have also linked calpain to the proteolytic cleavage and activity of COX-2 (Mancini et al. 2007), suggesting both direct and indirect modulation of this pro-inflammatory mediator. A previous study (Kone et al. 2003) suggests that decreases in iNOS expression may occur indirectly through NF-κB modulation because calpain degrades iNOS as part of a negative feedback mechanism that helps to lessen out-of-control inflammation.
Gliosis involving microglia and astrocytes is commonly associated with CNS injuries and diseases including MS (Ray et al. 2000) and EAE (Guyton et al. 2005); however, whether or not these inflammatory events are detrimental or reparative is still under debate. Treatment with a compound that specifically blocked activation of astrocytes and microglia reduced clinical symptoms of EAE (Guo et al. 2007). In the present study, blocking calpain in MBP-specific T cells before AT also decreased microgliosis and astrogliosis in spinal cords from RR-EAE mice. The decreases in astrogliosis and microgliosis were possibly due to a reduction in signals from injured neurons that are associated with gliosis (Streit 2000). Thus, one could speculate that reducing MBP-specific T cell activity in the periphery decreased migration of these auto-immune cells into the CNS and was sufficient to reduce the inflammatory and neurodengenerative events that occurred in the local CNS environment.
Another consequence of decreased activity of MBP-specific T cells would be a reduction in the release of detrimental mediators that degrade myelin proteins including MBP. Previous study demonstrated that calpain released from activated PBMCs degraded myelin in vitro (Deshpande et al. 1995a). Calpain released from activated T cells would thus degrade myelin in the CNS of EAE animals and MS patients, resulting in axonal damage and production of immunogenic peptides from myelin proteins that could lead to epitope spreading (Banik et al. 1994). Evidence from the present study indicated that myelin loss and MBP degradation were attenuated in female SJL/J mice that received the MBP-specific T cells exposed to SJA6017 before AT. These animals also demonstrated reduction in axonal damage in spinal cord during the acute phase of RR-EAE.
The extent of axonal damage in the CNS from MS patients directly correlates with disease severity (Trapp et al. 1998). Thus, axonal damage can be reduced either directly through inhibiting proteases such as calpain that degrade axonal proteins or by blocking the activity of the inflammatory cells that produce calpain as well as cytokines such as TNF-α and IL-6, which are also known to directly damage axons during pathogenesis of MS and EAE (Aarli 2003). Previous studies have indicated that calpain inhibition reduces axonal injury in neurodegenerative disorders such as EAE (Guyton et al. 2006; Hassen et al. 2008) and SCI (Sribnick et al. 2007). While the AT of auto-reactive T cells is necessary for induction of RR-EAE, earlier research suggests that axonal injury occurs early in MS pathology and continues even in the absence of inflammation in later disease states (Peterson et al. 2001). Thus, the decrease in MBP degradation and axonal damage in the current study is most likely a result of inhibition of T cell activation and subsequent production of calpain, cytokines, NO, and other mediators associated with inflammation.
In addition to axonal damage, mediators produced by activated T cells also lead to destruction of oligodendrocytes and neurons in neurodegenerative diseases. Oligodendrocyte and neuronal death are increased in MS and EAE and contribute to disease pathology (Hisahara et al. 2003; Guyton et al. 2005). Thus, treatments that reduce oligodendrocyte and neuronal death should correlate with reduced disease process. In the present study oligodendrocyte and neuronal death was attenuated by calpain inhibitor. These findings suggest that blocking MBP-specific T cell activity with calpain inhibitor is sufficient to reduce loss of oligodendrocytes and neurons in the RR-EAE mouse model. The loss of oligodendrocytes and neurons is presumably due to decrease in the induction of apoptotic cascades. The effects of calpain inhibition on oligodendrocyte death was unclear from this study, since very few oligodendrocytes were TUNEL positive in the group that did not receive MBP-specific T cells exposed to SJA6017. These studies have been performed using tissues from RR-EAE mice during the acute phase of disease; whether or not more oligodendrocytes are killed in the chronic phase and the effects of calpain inhibition will be determined in future studies.
The production of cytokines and other mediators by activated T cells and other immune cells leads to induction of multiple apoptotic signaling cascades. Thus, blocking T cell activity in MBP-specific T cells through calpain inhibition was hypothesized to decrease activity of proteins associated with apoptosis in RR-EAE spinal cord during the acute phase. In the present study, caspase-8, a protease involved in receptor mediated apoptosis (Frisch 2008), was decreased in animals that received MBP-specific T cells exposed to SJA6017. The formation of tBid and increase in Bax:Bcl-2 ratio were also attenuated, indicating that mitochondria mediated apoptosis was also diminished. Finally, the activity of caspase-3 was reduced in spinal cord tissue from mice that received MBP-specific T cells exposed to SJA6017 before AT. Our previous study has linked these molecular events to apoptosis in EAE animals (Das et al. 2008), and current data indicate that blocking auto-reactive T cell activity before AT results in a reduction in the induction of multiple pathways of apoptosis.
In conclusion, our current study adds strong support to the notion that blocking auto-reactive T cell activity by inhibition of calpain reduces inflammatory and neurodegenerative events in EAE and possibly in MS patients. Although, by the time MS patients are diagnosed, neurodegenerative events are progressive; however, calpain remains an attractive target for treating MS because this protease is involved in not only T cell activation but also migration of immune cells, myelin degradation, axonal damage, and cell death. The beneficial effects of calpain inhibition of MBP-specific T cells before AT include reduction of clinical disease signs. Therefore, future studies for treating RR-EAE mice with calpain inhibitor at various time-points following AT of MBP-specific T cells would be helpful to dissect the timing of therapy that would result in the greatest reduction in clinical symptoms and relapse rates.
Acknowledgments
This work was supported in part by the R01 grants (NS-41088, NS-45967, NS-57811, and CA-91460) from the National Institutes of Health, and also a Spinal Cord Injury Research Fund grant (SCIRF-0803) from the state of South Carolina.
Footnotes
Disclosures
The authors have no financial conflicts of interest.
References
- Aarli JA. Role of cytokines in neurological disorders. Curr Med Chem. 2003;10:1931–1937. doi: 10.2174/0929867033456918. [DOI] [PubMed] [Google Scholar]
- Banik NL, Mauldin LB, Hogan EL. Activity of 2′,3′-cyclic nucleotide 3′-phosphohydrolase in human cerebrospinal fluid. Ann Neurol. 1979;5:539–541. doi: 10.1002/ana.410050608. [DOI] [PubMed] [Google Scholar]
- Banik NL, McAlhaney WW, Hogan EL. Calcium-stimulated proteolysis in myelin: evidence for a Ca2+-activated neutral proteinase associated with purified myelin of rat CNS. J Neurochem. 1985;45:581–588. doi: 10.1111/j.1471-4159.1985.tb04026.x. [DOI] [PubMed] [Google Scholar]
- Banik NL, Chou CH, Deibler GE, Krutzch HC, Hogan EL. Peptide bond specificity of calpain: proteolysis of human myelin basic protein. J Neurosci Res. 1994;37:489–496. doi: 10.1002/jnr.490370408. [DOI] [PubMed] [Google Scholar]
- Bettelli E. Building different mouse models for human MS. Ann N Y Acad Sci. 2007;1103:11–18. doi: 10.1196/annals.1394.021. [DOI] [PubMed] [Google Scholar]
- Brahmachari S, Pahan K. Sodium benzoate, a food additive and a metabolite of cinnamon, modifies T cells at multiple steps and inhibits adoptive transfer of experimental allergic encephalomyelitis. J Immunol. 2007;179:275–283. doi: 10.4049/jimmunol.179.1.275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler JT, Samantaray S, Beeson CC, Ray SK, Banik NL. Involvement of calpain in the process of Jurkat T cell chemotaxis. J Neurosci Res. 2009;87:626–635. doi: 10.1002/jnr.21882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerghet M, Skoff RP, Bessert D, Zhang Z, Mullins C, Ghandour MS. Proliferation and death of oligodendrocytes and myelin proteins are differentially regulated in male and female rodents. J Neurosci. 2006;26:1439–1447. doi: 10.1523/JNEUROSCI.2219-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuzner ML, McDonald WI, Rudge P, Smith M, Borshell N, Davison AN. Leucocyte proteinase activity and acute multiple sclerosis. J Neurol Sci. 1975;26:107–111. doi: 10.1016/0022-510x(75)90118-5. [DOI] [PubMed] [Google Scholar]
- Das A, Guyton MK, Matzelle DD, Ray SK, Banik NL. Time-dependent increases in protease activities for neuronal apoptosis in spinal cords of Lewis rats during development of acute experimental autoimmune encephalomyelitis. J Neurosci Res. 2008;86:2992–3001. doi: 10.1002/jnr.21737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dasgupta S, Zhou Y, Jana M, Banik NL, Pahan K. Sodium phenylacetate inhibits adoptive transfer of experimental allergic encephalomyelitis in SJL/J mice at multiple steps. J Immunol. 2003;170:3874–3882. doi: 10.4049/jimmunol.170.7.3874. [DOI] [PubMed] [Google Scholar]
- Dasgupta S, Roy A, Jana M, Hartley DM, Pahan K. Gemfibrozil ameliorates relapsing-remitting experimental autoimmune encephalomyelitis independent of peroxisome proliferator-activated receptor-alpha. Mol Pharmacol. 2007;72:934–946. doi: 10.1124/mol.106.033787. [DOI] [PubMed] [Google Scholar]
- de Rosbo NK, Bernard CC. Multiple sclerosis brain immunoglobulins stimulate myelin basic protein degradation in human myelin: a new cause of demyelination. J Neurochem. 1989;53:513–518. doi: 10.1111/j.1471-4159.1989.tb07363.x. [DOI] [PubMed] [Google Scholar]
- Deshpande RV, Goust JM, Hogan EL, Banik NL. Calpain secreted by activated human lymphoid cells degrades myelin. J Neurosci Res. 1995a;42:259–265. doi: 10.1002/jnr.490420214. [DOI] [PubMed] [Google Scholar]
- Deshpande RV, Goust JM, Chakrabarti AK, Barbosa E, Hogan EL, Banik NL. Calpain expression in lymphoid cells. Increased mRNA and protein levels after cell activation. J Biol Chem. 1995b;270:2497–2505. doi: 10.1074/jbc.270.6.2497. [DOI] [PubMed] [Google Scholar]
- Diaz-Sanchez M, Williams K, DeLuca GC, Esiri MM. Protein co-expression with axonal injury in multiple sclerosis plaques. Acta Neuropathol. 2006;111:289–299. doi: 10.1007/s00401-006-0045-0. [DOI] [PubMed] [Google Scholar]
- Einstein ER, Csejtey J, Dalal KB, Adams CW, Bayliss OB, Hallpike JF. Proteolytic activity and basic protein loss in and around multiple sclerosis plaques: combined biochemical and histochemical observations. J Neurochem. 1972;19:653–662. doi: 10.1111/j.1471-4159.1972.tb01382.x. [DOI] [PubMed] [Google Scholar]
- Fitzgerald DC, Ciric B, Touil T, Harle H, Grammatikopolou J, Das Sarma J, Gran B, Zhang GX, Rostami A. Suppressive effect of IL-27 on encephalitogenic Th17 cells and the effector phase of experimental autoimmune encephalomyelitis. J Immunol. 2007;179:3268–3275. doi: 10.4049/jimmunol.179.5.3268. [DOI] [PubMed] [Google Scholar]
- Fleming KK, Bovaird JA, Mosier MC, Emerson MR, LeVine SM, Marquis JG. Statistical analysis of data from studies on experimental autoimmune encephalomyelitis. J Neuroimmunol. 2005;170:71–84. doi: 10.1016/j.jneuroim.2005.08.020. [DOI] [PubMed] [Google Scholar]
- Frisch SM. Caspase-8: fly or die. Cancer Res. 2008;68:4491–4493. doi: 10.1158/0008-5472.CAN-08-0952. [DOI] [PubMed] [Google Scholar]
- Govindarajan KR, Rauch HC, Clausen J, Einstein ER. Changes in cathepsins B-1 and D, neutral proteinase and 2′,3′-cyclic nucleotide-3′-phosphohydrolase activities in monkey brain with experimental allergic encephalomyelitis. J Neurol Sci. 1974;23:295–306. doi: 10.1016/0022-510x(74)90232-9. [DOI] [PubMed] [Google Scholar]
- Guo X, Nakamura K, Kohyama K, Harada C, Behanna HA, Watterson DM, Matsumoto Y, Harada T. Inhibition of glial cell activation ameliorates the severity of experimental autoimmune encephalomyelitis. Neurosci Res. 2007;59:457–466. doi: 10.1016/j.neures.2007.08.014. [DOI] [PubMed] [Google Scholar]
- Guyton MK, Das A, Matzelle DD, Samantaray S, Azuma M, Inoue J, Ray SK, Banik NL. SJA6017 attenuates immune cell infiltration and neurodegeneration in EAE. In: Tabisa T, Yamamora T, Kira J, editors. 8th International Congress of Neuroimmunology; Nagoya, Japan: Medimond International Proceedings; 2006. pp. 107–112. [Google Scholar]
- Guyton MK, Wingrave JM, Yallapragada AV, Wilford GG, Sribnick EA, Matzelle DD, Tyor WR, Ray SK, Banik NL. Upregulation of calpain correlates with increased neurodegeneration in acute experimental auto-immune encephalomyelitis. J Neurosci Res. 2005;81:53–61. doi: 10.1002/jnr.20470. [DOI] [PubMed] [Google Scholar]
- Hassen GW, Feliberti J, Kesner L, Stracher A, Mokhtarian F. A novel calpain inhibitor for the treatment of acute experimental autoimmune encephalomyelitis. J Neuroimmunol. 2006;180:135–146. doi: 10.1016/j.jneuroim.2006.08.005. [DOI] [PubMed] [Google Scholar]
- Hassen GW, Feliberti J, Kesner L, Stracher A, Mokhtarian F. Prevention of axonal injury using calpain inhibitor in chronic progressive experimental autoimmune encephalomyelitis. Brain Res. 2008;1236:206–215. doi: 10.1016/j.brainres.2008.07.124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higuchi M, Tomioka M, Takano J, Shirotani K, Iwata N, Masumoto H, Maki M, Itohara S, Saido TC. Distinct mechanistic roles of calpain and caspase activation in neurodegeneration as revealed in mice overexpressing their specific inhibitors. J Biol Chem. 2005;280:15229–15237. doi: 10.1074/jbc.M500939200. [DOI] [PubMed] [Google Scholar]
- Hisahara S, Okano H, Miura M. Caspase-mediated oligodendrocyte cell death in the pathogenesis of autoimmune demyelination. Neurosci Res. 2003;46:387–397. doi: 10.1016/s0168-0102(03)00127-5. [DOI] [PubMed] [Google Scholar]
- Imam SA, Guyton MK, Haque A, Vandenbark A, Tyor WR, Ray SK, Banik NL. Increased calpain correlates with Th1 cytokine profile in PBMCs from MS patients. J Neuroimmunol. 2007;190:139–145. doi: 10.1016/j.jneuroim.2007.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keegan BM, Noseworthy JH. Multiple sclerosis. Annu Rev Med. 2002;53:285–302. doi: 10.1146/annurev.med.53.082901.103909. [DOI] [PubMed] [Google Scholar]
- Kone BC, Kuncewicz T, Zhang W, Yu ZY. Protein interactions with nitric oxide synthases: controlling the right time, the right place, and the right amount of nitric oxide. Am J Physiol Renal Physiol. 2003;285:F178–190. doi: 10.1152/ajprenal.00048.2003. [DOI] [PubMed] [Google Scholar]
- Lee SU, Grigorian A, Pawling J, Chen IJ, Gao G, Mozaffar T, McKerlie C, Demetriou M. N-Glycan processing deficiency promotes spontaneous inflammatory demyelination and neurodegeneration. J Biol Chem. 2007;282:33725–33734. doi: 10.1074/jbc.M704839200. [DOI] [PubMed] [Google Scholar]
- Mancini A, Jovanovic DV, He QW, Di Battista JA. Site-specific proteolysis of cyclooxygenase-2: a putative step in inflammatory prostaglandin E(2) biosynthesis. J Cell Biochem. 2007;101:425–441. doi: 10.1002/jcb.21191. [DOI] [PubMed] [Google Scholar]
- Marks N, Benuck M, Hashim G. Hydrolysis of myelin basic protein with brain acid proteinase. Biochem Biophys Res Commun. 1974;56:68–74. doi: 10.1016/s0006-291x(74)80316-5. [DOI] [PubMed] [Google Scholar]
- Noonan CW, Kathman SJ, White MC. Prevalence estimates for MS in the United States and evidence of an increasing trend for women. Neurology. 2002;58:136–138. doi: 10.1212/wnl.58.1.136. [DOI] [PubMed] [Google Scholar]
- Peterson JW, Bo L, Mork S, Chang A, Trapp BD. Transected neurites, apoptotic neurons, and reduced inflammation in cortical multiple sclerosis lesions. Ann Neurol. 2001;50:389–400. doi: 10.1002/ana.1123. [DOI] [PubMed] [Google Scholar]
- Pitt D, Werner P, Raine CS. Glutamate excitotoxicity in a model of multiple sclerosis. Nat Med. 2000;6:67–70. doi: 10.1038/71555. [DOI] [PubMed] [Google Scholar]
- Ray SK, Banik NL. Calpain and its involvement in the pathophysiology of CNS injuries and diseases: therapeutic potential of calpain inhibitors for prevention of neurodegeneration. Curr Drug Targets CNS Neurol Disord. 2003;2:173–189. doi: 10.2174/1568007033482887. [DOI] [PubMed] [Google Scholar]
- Ray SK, Schaecher KE, Shields DC, Hogan EL, Banik NL. Combined TUNEL and double immunofluorescent labeling for detection of apoptotic mononuclear phagocytes in autoimmune demyelinating disease. Brain Res Protoc. 2000;5:305–311. doi: 10.1016/s1385-299x(00)00027-1. [DOI] [PubMed] [Google Scholar]
- Schaecher K, Goust JM, Banik NL. The effects of calpain inhibition on IkB alpha degradation after activation of PBMCs: identification of the calpain cleavage sites. Neurochem Res. 2004;29:1443–1451. doi: 10.1023/b:nere.0000026410.56000.dd. [DOI] [PubMed] [Google Scholar]
- Schaecher K, Rocchini A, Dinkins J, Matzelle DD, Banik NL. Calpain expression and infiltration of activated T cells in experimental allergic encephalomyelitis over time: increased calpain activity begins with onset of disease. J Neuroimmunol. 2002;129:1–9. doi: 10.1016/s0165-5728(02)00142-x. [DOI] [PubMed] [Google Scholar]
- Schaecher KE, Shields DC, Banik NL. Mechanism of myelin breakdown in experimental demyelination: a putative role for calpain. Neurochem Res. 2001a;26:731–737. doi: 10.1023/a:1010903823668. [DOI] [PubMed] [Google Scholar]
- Schaecher KE, Goust JM, Banik NL. The effects of calpain inhibition upon IL-2 and CD25 expression in human peripheral blood mononuclear cells. J Neuroimmunol. 2001b;119:333–342. doi: 10.1016/s0165-5728(01)00367-8. [DOI] [PubMed] [Google Scholar]
- Shields DC, Banik NL. Pathophysiological role of calpain in experimental demyelination. J Neurosci Res. 1999;55:533–541. doi: 10.1002/(SICI)1097-4547(19990301)55:5<533::AID-JNR1>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- Shields DC, Tyor WR, Deibler GE, Banik NL. Increased calpain expression in experimental demyelinating optic neuritis: an immunocytochemical study. Brain Res. 1998a;784:299–304. doi: 10.1016/s0006-8993(97)01381-4. [DOI] [PubMed] [Google Scholar]
- Shields DC, Schaecher KE, Saido TC, Banik NL. A putative mechanism of demyelination in multiple sclerosis by a proteolytic enzyme, calpain. Proc Natl Acad Sci U S A. 1999;96:11486–11491. doi: 10.1073/pnas.96.20.11486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shields DC, Tyor WR, Deibler GE, Hogan EL, Banik NL. Increased calpain expression in activated glial and inflammatory cells in experimental allergic encephalomyelitis. Proc Natl Acad Sci U S A. 1998b;95:5768–5772. doi: 10.1073/pnas.95.10.5768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sribnick EA, Matzelle DD, Banik NL, Ray SK. Direct evidence for calpain involvement in apoptotic death of neurons in spinal cord injury in rats and neuroprotection with calpain inhibitor. Neurochem Res. 2007;32:2210–2216. doi: 10.1007/s11064-007-9433-7. [DOI] [PubMed] [Google Scholar]
- Sribnick EA, Wingrave JM, Matzelle DD, Wilford GG, Ray SK, Banik NL. Estrogen attenuated markers of inflammation and decreased lesion volume in acute spinal cord injury in rats. J Neurosci Res. 2005;82:283–293. doi: 10.1002/jnr.20622. [DOI] [PubMed] [Google Scholar]
- Streit WJ. Microglial response to brain injury: a brief synopsis. Toxicol Pathol. 2000;28:28–30. doi: 10.1177/019262330002800104. [DOI] [PubMed] [Google Scholar]
- Surh YJ, Chun KS, Cha HH, Han SS, Keum YS, Park KK, Lee SS. Molecular mechanisms underlying chemopreventive activities of anti-inflammatory phytochemicals: down-regulation of COX-2 and iNOS through suppression of NF-kappa B activation. Mutat Res. 2001:480–481. 243–268. doi: 10.1016/s0027-5107(01)00183-x. [DOI] [PubMed] [Google Scholar]
- Thiel VE, Audus KL. Nitric oxide and blood-brain barrier integrity. Antioxid Redox Signal. 2001;3:273–278. doi: 10.1089/152308601300185223. [DOI] [PubMed] [Google Scholar]
- Trapp BD, Ransohoff R, Rudick R. Axonal pathology in multiple sclerosis: relationship to neurologic disability. Curr Opin Neurol. 1999;12:295–302. doi: 10.1097/00019052-199906000-00008. [DOI] [PubMed] [Google Scholar]
- Trapp BD, Peterson J, Ransohoff RM, Rudick R, Mork S, Bo L. Axonal transection in the lesions of multiple sclerosis. N Engl J Med. 1998;338:278–285. doi: 10.1056/NEJM199801293380502. [DOI] [PubMed] [Google Scholar]
- Tyor WR, Avgeropoulos N, Ohlandt G, Hogan EL. Treatment of spinal cord impact injury in the rat with transforming growth factor-beta. J Neurol Sci. 2002;200:33–41. doi: 10.1016/s0022-510x(02)00113-2. [DOI] [PubMed] [Google Scholar]
- Villarino AV, Stumhofer JS, Saris CJ, Kastelein RA, de Sauvage FJ, Hunter CA. IL-27 limits IL-2 production during Th1 differentiation. J Immunol. 2006;176:237–247. doi: 10.4049/jimmunol.176.1.237. [DOI] [PubMed] [Google Scholar]