

Abstract
A chronic high fat Western diet (WD) promotes a variety of morbidity factors although experimental evidence for short-term WD mediating brain dysfunction remains to be elucidated. The amyloid precursor protein and presenilin-1 (APP × PS1) knock-in mouse model has been demonstrated to recapitulate some key features of Alzheimer’s disease pathology, including amyloid-β (Aβ) pathogenesis. In this study, we placed 1-month-old APP × PS1 mice and non-transgenic littermates on a WD for 4 weeks. The WD resulted in a significant elevation in protein oxidation and lipid peroxidation in the brain of APP × PS1 mice relative to non-transgenic littermates, which occurred in the absence of increased Aβ levels. Altered adipokine levels were also observed in APP × PS1 mice placed on a short-term WD, relative to non-transgenic littermates. Taken together, these data indicate that short-term WD is sufficient to selectively promote cerebral oxidative stress and metabolic disturbances in APP × PS1 knock-in mice, with increased oxidative stress preceding alterations in Aβ. These data have important implications for understanding how WD may potentially contribute to brain dysfunction and the development of neurodegenerative disorders such as Alzheimer’s disease.
Keywords: adipokine, Alzheimer’s disease, amyloid-β, protein oxidation, Western diet
A high fat Western diet (WD) promotes a number of well-established metabolic disturbances and increased morbidity (Weisburger 1997; Litchtenstein et al. 1998; Shaw et al. 2005). The best-documented effects of long-term WD consumption include an increased incidence of obesity, hyperinsulemia, hyperglycermia, hypertension, and cardiovascular disease. Consumption of a WD is linked to altered levels of circulating adipose derived peptides known as adipokines (Ronti et al. 2006), which are known to potentially promote a number of deleterious effects in different organ systems. Accompanying each of these systemic effects of a WD, studies have identified the ability of a WD to promote numerous potentially deleterious biochemical alterations in various tissues. In particular, studies have linked consumption of a WD to increased levels of oxidative stress (Diniz et al. 2004; Ghosh et al. 2006; Milagro et al. 2006; Roberts et al. 2006; Bullo et al. 2007), including increased levels of protein oxidation and lipid peroxidation. While these effects of long-term (≥ 4 months) WD are well established, considerably less is currently known with regards to the initial effects of a WD. Identifying early biochemical events that occur following initial WD exposure may be particularly important in identifying the key substrates which are principle mediators of the deleterious effects of prolonged WD consumption.
In contrast to tissues such as the muscle and the liver, relatively little is known with regards to the effects a WD on the brain, and even less with regards to the effects of short-term WD on the brain. Recent clinical studies however have suggested that a WD may contribute to the development of Alzheimer’s disease (AD) (Morris et al. 2003; Pasinetti et al. 2007). Additionally, studies using rodent models of AD pathology have demonstrated that long-term administration of diets high in fat, cholesterol, or sucrose are sufficient to increase the levels of amyloid-β (Aβ) pathology (Rofolo et al. 2000; Oksman et al. 2006; Cao et al. 2007; Hoojimas et al. 2007). Currently the effect of a WD on the initial phase of Aβ pathogenesis is not clear. Similarly, it is unknown whether a WD modulates the levels of oxidative stress in experimental models relevant to AD. Clarifying experimentally the relationship between the potential effects of a WD on Aβ pathology and oxidative stress in the brain is particularly important given the intense interest in understanding the apparent reciprocal relationship between Aβ pathogenesis and the development of oxidative stress in AD (Markesbery 1997; Mattson 2004; Sung et al. 2004; Swerdlow 2007; Zana et al. 2007; Zhu et al. 2007; Reddy and Beal 2008; Tamagno et al. 2008).
Previous studies have demonstrated that amyloid precursor protein and presenilin-1 knock-in mice (APP × PS1) recapitulate several pathological features associated with AD, including age-related increases in Aβ insolubility and deposition (Anantharaman et al. 2006; Murphy et al. 2007). Additionally, these mice exhibit many physiological alterations associated with AD (Siman et al. 2000; Chang et al. 2006), and exhibit a time-dependent increase in the levels of oxidative stress (Anantharaman et al. 2006). In this study, we placed young (1-month old) APP × PS1 mice, and their non-transgenic littermates, on a WD for 1 month to determine if this relatively short-term WD altered the levels of oxidative stress or accelerated the development of Aβ pathology. Together, these studies demonstrate that a WD preferentially increases cerebral protein oxidation and lipid peroxidation in APP × PS1 mice in a manner apparently independent of increased levels of Aβ.
Materials and methods
Mice and Western diet studies
The APP × PS1 mouse model has been described previously by our laboratory and others (Siman et al. 2000; Anantharaman et al. 2006; Chang et al. 2006; Murphy et al. 2007). Briefly, mice were generated using the Cre-lox knock-in technology. The endogenous APP and PS1 genes were replaced with mutated ‘humanized’ APP and PS1 genes (ΔNL and P264L, respectively), which remain under the control of their endogenous promoters. Diets for the proposed studies were purchased from Research Diets Incorporated (New Brunswick, NJ, USA). At 1 month of age, 14 APP × PS1 (eight males and six females) and 14 non-transgenic littermates (eight males and six females) were placed for 4 weeks on a standard chow containing 10% fat (Diet #98052602) or a typical WD mix containing 40% fat (Diet #D12079B).
Analysis of oxidative stress
The levels of protein oxidation were determined by western blot and immunohistochemical analysis of the frontal cortex as described previously (Keller et al. 1998; Ding et al. 2003). Specifically, protein oxidation was quantified by the immunodetection of derivatized protein carbonyls with anti-dinitrophenylhydrazine or antibodies which recognize proteins possessing nitrotyrosine or HNE (4-hydroxynonenal) conjugated proteins. Lipid hydroperoxides were isolated by direct extraction from frozen brain into chloroform, with the amount of lipid hydroperoxides quantified using a standard curve of lipid hydroperoxides, all in accordance with the methodologies outlined in the manufactures guidelines (Caymen Chemical; Ann Arbor, MI, USA).
Analysis of amyloid-β
The levels of DEA (diethylamine) soluble and sodium dodecyl sulfate (SDS)-soluble Aβ40 and Aβ42 were analyzed in the frontal cortex using methods as described previously (Das et al. 2003; Kukar et al. 2005; McGowan et al. 2005; Murphy et al. 2007). Briefly, the tissue was polytron homogenized in 1 mL of DEA buffer (50 mM NaCl, 0.2% diethylamine, and protease inhibitor cocktail with EDTA). The supernatant was collected after centrifugation for 30 min at 100 000 g and 4°C. The pellet was re-extracted by sonication (10 × 0.5 s microtip pulses at 100 W) in 2% SDS, centrifuged (20 000 g, 30 min) and the supernatant again collected. Prior to use, the DEA samples were neutralized by adding 10% of 0.5 M Tris-HCl, pH 7.5, and the SDS samples were diluted 1 : 20 in antigen capture buffer (0.02 M sodium phosphate buffer, 0.4 M NaCl, 2 mM EDTA, 0.4% Block Ace, 0.2% bovine serum albumin, 0.05% 3-[(3-Cholamidopropyl)di-methylammonio]-1-propanesulfonate (CHAPS), and 0.05% NaN3). The sandwich ELISA was conducted using antibodies Ab9 (human sequence Aβ1-16), horseradish peroxidase-conjugated 13.1.1 (end specific for Aβ40), and horseradish peroxidase-conjugated 2.1.3 (end specific for Aβ42).
Analysis of plasma adipokines and insulin levels
The levels of adiponectin, leptin, insulin, and resistin were quantified in the plasma from mice using ELISA based assays (Millipore, St Charles, MI, USA). After an overnight fast, plasma from all mice was collected immediately prior to the killing of animals, which occurred prior to 12 pm. Assays were conducted as per the manufactures instructions. Fasting glucose and ketones were measured in whole blood at the time of killing using the precision Xtra system with glucose and ketone sticks (Abbott Laboratories, Almeda, CA, USA).
Results
Effects of a Western diet on metabolic indices
To elucidate the effects of a short-term WD, we placed APP × PS1 mice and their non-transgenic littermates (wild type; WT) on a 4-week regimen consisting of either normal chow or a high fat WD chow. At the end of 4 weeks, there was a significant increase in the body weight and fasting glucose levels in both experimental groups of animals feeding on a WD (Fig. 1). No significant alteration in ketones was observed in any experimental groups (Fig. 1), although APP × PS1 mice trended to have decreased ketone levels. Because a WD is linked to altered levels of adipokines (peptides of predominatly adipocyte origin) we analyzed the plasma levels of a number of key adipokines in our rodents. No significant alteration in the level of insulin or adiponectin was observed in any experimental groups (Table 1). An increase in resistin was observed in both experimental groups of animals on a WD (Table 1), with WT animals demonstrating a greater increase in plasma resistin levels when compared with APP × PS1 animals. Although not statistically significant, an elevation in plasma leptin levels was also observed in WT mice on a WD, which interestingly was not observed in APP × PS1 animals (Table 1).
Fig. 1.

Effects of a Western diet on metabolic indices. Mice were placed for 4 weeks on a normal chow diet or Western diet (WD) and analyzed for their percent weight gain, fasting glucose levels, or circulating ketone levels. Data are expressed as the mean and SEM of results from seven animals in each experimental group. White bars indicate animals on control diet while black bars indicate animals on a WD; *p < 0.02 compared with animals not on a WD.
Table 1.
Effects of a Western diet on adipokine levels
| Adiponectin | Leptin | Insulin | Resistin | |
|---|---|---|---|---|
| WT normal diet | 11.0 ± 1.0 | 2.2 ± 0.38 | 0.5 ± 0.05 | 7.2 ± 1.08 |
| WT Western diet | 9.8 ± 0.7 | 4.8 ± 2.00 | 0.6 ± 0.11 | 12.2 ± 0.77** |
| APP × PS1 normal diet | 8.1 ± 0.7 | 2.2 ± 0.37 | 0.6 ± 0.05 | 7.1 ± 0.63 |
| APP × PS1 Western diet | 9.1 ± 0.8 | 2.1 ± 0.65 | 0.5 ± 0.05 | 8.9 ± 0.81* |
WT, wild type; APP × PS1, amyloid precursor protein and presenilin-1 knock-in mice. All values are expressed as the mean and SEM of results from seven animals for each condition. Data are expressed as ng/mL plasma.
p < 0.01 compared with normal diet
p < 0.01 compared with APP × PS1 with same treatment.
Effects of Western diet on indices of oxidative stress
To elucidate the effects of a WD on the levels of oxidative stress in the brain we conducted western blot analysis to quantify the level of protein oxidation in brain homogenates from WT and APP × PS1 mice on a WD. We observed that the levels of protein carbonyls (anti-dinitrophenylhydrazine) and protein nitrosylation were significantly elevated in APP × PS1 on a WD (Fig. 2), when compared with all other experimental groups. Although more variable than APP × PS1, WT mice on a WD exhibited some elevation in both protein carbonyls and protein nitrosylation (Fig. 2). Immunoreactivity was observed across a wide range of molecular weights suggesting that a broad spectrum of proteins in the brain possessed increased levels of oxidative damage following a WD (Fig. 2). No significant difference in HNE-protein conjugation was observed in WT or APP × PS1 mice on a WD (Fig. 2), when compared with animals fed a normal chow diet.
Fig. 2.
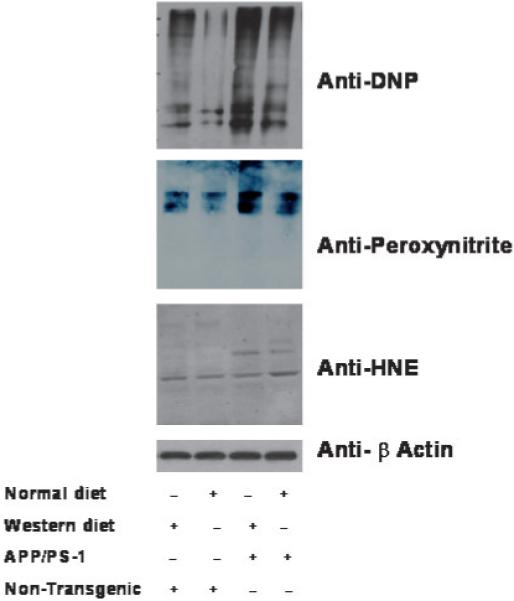
Western diet increases the levels of protein oxidation in APP × PS1 mice. The level of protein carbonyls [anti-dinitrophenylhydrazine (DNP)], nitrotyrosine, and HNE-protein conjugates in the frontal cortex were analyzed by Western blotting. Molecular weights are indicated at 131 and 85 kDa. Data represent the results from at least six mice in each experimental condition.
To determine which cell types possessed the greatest amount of protein oxidation following a WD, we conducted immunohistochemistry within the frontal cortex. In this analysis, we observed that the immunoreactivity appeared to be primarily of neuronal origin for both protein carbonyls (Fig. 3) and protein nitrosylation (Fig. 4). To expand our analysis of oxidative stress in the brain beyond protein oxidation, we conducted an additional analysis to quantify the gross levels of lipid hydroperoxides in the brain, with lipid hydroperoxides being a rapid and sensitive marker for increased levels of oxidative stressors in a variety of tissues (Esterbauer 1993; Girotti 1998). In these studies, we observed a significant elevation in lipid hydroperoxides in brain homogenates of both WT and APP × PS1 mice on a WD (Fig. 5). However, it is important to note that the level of lipid hydroperoxides were significantly higher in APP × PS1 mice on a WD when compared with all other experimental groups (Fig. 5).
Fig. 3.
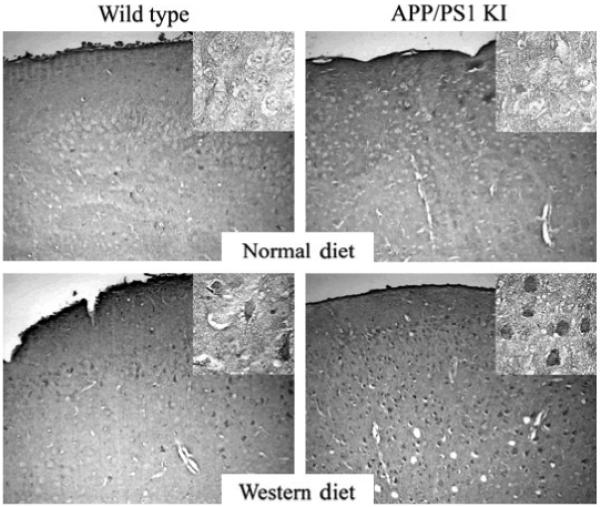
Increased levels of protein carbonyls are observed in neurons following a Western diet. The level of protein carbonyl immunoreactivity appears to be increased in individual neurons within the frontal cortex following a WD, with APP × PS1 mice exhibiting enhanced numbers of neurons with carbonyl immunoreactivity when compared with non-transgenic littermates on a WD. Data represent the results from at least six mice in each experimental condition.
Fig. 4.
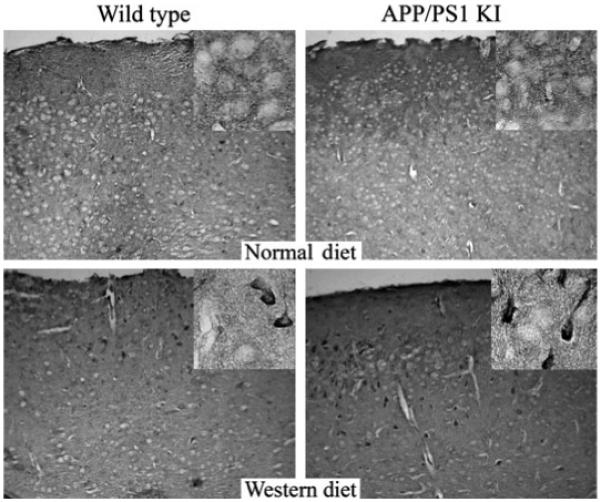
Increased levels of nitrotyrosine immunoreactivity are observed in neurons following a Western diet. The level of nitrotyrosine immunoreactivity appears to be increased in individual neurons within the frontal cortex following a WD, with APP × PS1 mice exhibiting enhanced numbers of neurons with carbonyl immunoreactivity when compared with non-transgenic littermates on a WD. Data represent the results from at least six mice in each experimental condition.
Fig. 5.
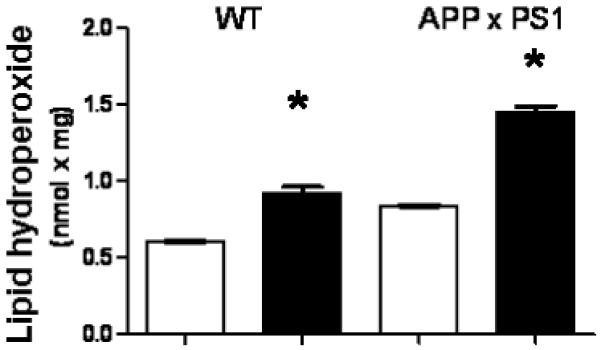
Western diet increases the levels of lipid hydroperoxides in the brain. The level of lipid hydroperoxides were analyzed in frozen brain tissue from the frontal cortex of APP × PS1 mice and their non-transgenic littermates following control or WD. Graphs represent the mean and SEM from seven animals for each treatment group. White bars indicate animals on control diet while black bars indicate animals on a WD; *p < 0.02 compared with animals not on a WD.
Short-term Western diet consumption does not increase soluble or insoluble Aβ levels in APP × PS1 mice
In our last set of experiments we sought to elucidate the effects of a 4-week WD on the levels of Aβ. After measuring the levels of Aβ40 and Aβ42 in cortical extracts, we did not observe any elevation the soluble (DEA) or detergent soluble (SDS) pools of Aβ following a short-term WD (Fig. 6). Additionally, immunohistochemical analysis did not detect any Aβ deposition in any of the mice (data not shown), consistent with previous reports from our group that APP × PS1 mice do not possess detectible levels of Aβ deposition until 12 weeks of age (Anantharaman et al. 2006; Murphy et al. 2007). These data are important because they suggest that the elevation in oxidative stress of APP × PS1 mice following a short-term WD is not mediated by increased levels of Aβ.
Fig. 6.

A Western diet does not elevate the level of Aβ in the brain. Brain homogenates were analyzed by ELISA for the level of soluble Aβ40 or Aβ42 (DEA) and detergent soluble Aβ40 or Aβ42 (SDS). Graphs represent the mean and SEM from seven animals for the APP × PS1 control diet (white bars) and the WD (black bars). White bars indicate animals on control diet while black bars indicate animals on a WD. Table 1
Discussion
The data in the current study demonstrate that consumption of a WD for 1 month is sufficient to increase the level of highly reactive lipid hydroperoxides in the brains of young adult mice. Because the brain is a highly vascularized and highly metabolic tissue rich in lipids and polyunsaturated fatty acids, it is extremely susceptible to undergo rapid elevations in oxidative damage. Consumption of a WD may promote lipid oxidation in the brain by elevating the levels of reactive oxygen species, as has been reported in non-CNS tissues following long-term consumption of a WD (Diniz et al. 2004; Ghosh et al. 2006; Milagro et al. 2006; Roberts et al. 2006; Bullo et al. 2007; Vincent et al. 2007). While the initial products of polyunsaturated fatty acid oxidation are almost exclusively monohydroperoxides, these products are often rapidly put through a progression of secondary and tertiary oxidative modifications that involve the genesis of lipid peroxyl and lipid alkoxyl radicals (Esterbauer 1993; Girotti 1998). These increasingly toxic species are capable of rapidly inducing a milieu of deleterious signaling cascades, promoting disturbances in the plasma membranes, and inactivating vital cellular enzymatic activities (Esterbauer 1993; Girotti 1998). It is interesting to note that in this study we did not observe a significant increase in the amount of HNE-protein conjugation, suggesting that HNE may not be a principle lipid peroxidation product generated in response to a WD in the brain. However, because we utilized whole brain extracts in the current studies we may have missed the presence of brain region specific elevations in HNE-protein conjugation. For example, previous studies have demonstrated that a 4-month diet of a highly palatable diet selectively increases oxidative stress in the rat frontal cortex (Souza et al. 2007). Regardless, the presence of elevated lipid peroxidation in this study suggests that oxidative stress, via the elevation of potentially toxic products such as lipid hydroperoxides, may promote cellular stress and toxicity in the brain following even the short-term consumption of a WD.
Amyloid precursor protein and presenilin-1 knock-in mice exhibit increased susceptibility to oxidative stress induced by WD. The APP × PS1 mice exhibited an elevation in lipid peroxidation, and in protein carbonyls and protein nitrosylation that was not observed in non-transgenic littermates. Because of the broad spectrum of proteins that exhibited elevations in protein oxidation, it is likely that this observed increase in carbonylated and nitrosylated protein, represents a potentially deleterious alteration in the proteome. Previous studies have identified a potential role for protein oxidation as a potential mediator of neuropathology and cognitive disturbances observed in AD, and experimental models relevant to AD (Markesbery 1997; Sung et al. 2004; Zana et al. 2007; Zhu et al. 2007). These data suggest that genetic modifiers associated with familial forms of AD predispose the brain to undergo rapid increases in oxidative stress in response to consumption of a WD. Our immunohistochemistry revealed that neurons, and likely even specific neuron subtypes, selectively exhibit elevated levels of protein oxidation. Preferential elevation in oxidative stress within neurons may have important implications for potentially deleterious effects of a WD on both cognition and the development of AD related neuropathology. In this scenario, the increased susceptibility of the brain to undergo oxidative stress following a WD may be an important component for the ‘second hit hypothesis’ (Mattson 2004; Zhu et al. 2004, 2007; Zana et al. 2007), and thereby link diet and oxidative stress to the potential development of AD. Defining whether this WD-induced increase in oxidative stress is mediated by elevations in pro-reactive oxygen species reactions and/or via decreased levels of antioxidants is a critical issue to be resolved experimentally. Previous studies have suggested the potential contribution of each of these factors to WD-induced oxidative stress (Diniz et al. 2004; Ghosh et al. 2006; Milagro et al. 2006; Roberts et al. 2006), with the prooxidative stress environment also initiated by the presence of AD related mutations (Mattson 2004).
Previous studies have demonstrated that dietary manipulations can accelerate the development and severity of Aβ pathogenesis. These previous studies have however typically used longer periods of dietary manipulation (up to 12 months), and more severe models of forced Aβ expression (Rofolo et al. 2000; Oksman et al. 2006; Cao et al. 2007; Hoojimas et al. 2007). The APP × PS1 mice utilized in the current study have the endogenous mouse promotersand do not drive ectopic expression of APP × PS1 and recapitulate key features of Aβ insolubility and deposition observed in the progression of AD (Anantharaman et al. 2006; Murphy et al. 2007). Our findings in these mice suggest that a relatively short exposure to a WD (4 weeks) results in an elevation in oxidative stress in APP × PS1 mice that is not correlated with an increase in Aβ or detectible levels of Aβ deposition. This suggests that a mechanism other than increased levels of insoluble Aβ or Aβ deposition is capable of increasing the levels of oxidative stress in the brain following a WD. However, while the role of intraneuronal Aβ remains controversial, our data do not exclude the possibility that a WD specifically elevates the levels of intraneuronal Aβ, which then promotes the increased levels of neuronal oxidative stress observed in this study. These data may be particularly important to AD, where the reciprocal relationship between Aβ and the development of oxidative stress in the AD brain has proven difficult to clarify experimentally. For example, while the ability of Aβ to promote oxidative stress is well established (Markesbery 1997; Mattson 2004; Zhu et al. 2004, 2007), studies have also demonstrated that oxidative stress appears to be critical for the initiating the development of Aβ pathogenesis (Sung et al. 2004), as well as initiating a number of aspects relevant to brain aging (Pratico 2002). Additionally, while our short-term studies in young mice did not result in the development of diabetes, we did observe a significant alteration in blood glucose regulation. These studies raise the possibility of hyperglycemia contributing to brain pathogenesis and altered brain function (Farr et al. 2008; Stranahan et al. 2008), via the generation of cerebral oxidative stress. Similarly, our data raise the possibility that circulating levels of adipokines may become altered in models relevant to AD, with a neuroprotective role for adipokines emerging in a number of paradigms relevant to AD (Zhang et al. 2007; Guo et al. 2008). Analysis of superoxide dismutase activity and catalase in the brains of mice from this study revealed that there was no significant change between the different experimental groups (data not shown). These findings suggest that while changes in antioxidant enzyme activity may be involved in promoting oxidative stress, it does not appear that the modulation of antioxidant enzyme activity plays a significant role in the earliest elevations in oxidative damage following WD consumption.
Taken together, our studies raise the possibility of diet contributing to brain pathogenesis, even in young adults, via the generation of cerebral oxidative stress. These data have important implications for understanding how diet and oxidative stress may ultimately contribute to the development of age-related neurodegenerative disorders such as AD. These data suggest that neuronal oxidative stress is one of the earliest factors in the initiation of the development of brain pathology, with oxidative stress potentially preceding the development of Aβ insolubility and deposition. Clarifying the cellular mechanisms responsible for increased levels of oxidative stress, and identifying the peripheral factors by which diet modulates cerebral oxidative stress, will likely be key to the development of effective interventions to reverse or prevent the deleterious effects of a WD on the brain.
Acknowledgements
This work was supported by funds from the NIA (MPM, AJBK, WRM, JNK), NCRR (RR020171 to MPM), NINDS (NS058382 to MPM), Alzheimer’s Association (JNK), R. C Durr Endowed Chair (JNK), and a fellowship from the Myositis Association (CMS). The authors thank Dr Daret St Clair (University of Kentucky) for assistance with maintaining the mouse colony and assistance genotyping the animals.
Abbreviation used
- AD
Alzheimer’s disease
- APP × PS1
amyloid precursor protein and presenilin-1 knock-in mice
- APP
amyloid precursor protein
- Aβ
amyloid-β
- DEA
diethylamine
- HNE
4-hydroxynonenal
- PS
presenilin-1
- SDS
sodium dodecyl sulfate
- WD
Western diet
- WT
wild type
References
- Anantharaman M, Tongpong J, Keller JN, Murphy MP, Markesbery WR, Kiningham KK, St Clair DK. β-amyloid mediated nitration of manganese superoxide dismutase: implication for oxidative stress in a APPNLh/NLh × PS-1P264L/P264L double knock-in mouse model of Alzheimer’s disease. Am. J. Pathol. 2006;168:1608–1618. doi: 10.2353/ajpath.2006.051223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bullo M, Casas-Agustench P, Amigo-Correigm P, Arancetam J, Salas-Salvadom J. Inflammation, obesity and comorbidities: the role of diet. Public Health Nutr. 2007;10:1164–1172. doi: 10.1017/S1368980007000663. [DOI] [PubMed] [Google Scholar]
- Cao D, Lu H, Li L. Intake of sucrose-sweetened water induces insulin resistance and exacerbates memory deficits and amyloidosis in a transgenic mouse model of Alzheimer’s disease. J. Biol. Chem. 2007;282:36275–36282. doi: 10.1074/jbc.M703561200. [DOI] [PubMed] [Google Scholar]
- Chang EH, Savage MJ, Flood DG, Thomas JM, Levy RB, Mahadomrongkul V, Shirao T, Aoki C, Huerta PT. AMPA receptor downscaling at the onset of Alzheimer’s disease pathology in double knock-in mice. Proc. Natl Acad. Sci. USA. 2006;103:3410–3415. doi: 10.1073/pnas.0507313103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das P, Howard V, Loosbrock N, Dickson D, Murphy MP, Golde T. Amyloid-beta immunization effectively reduces amyloid deposition in FcR gamma-/- knock out mice. J. Neurosci. 2003;23:8532–8538. doi: 10.1523/JNEUROSCI.23-24-08532.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding Q, Reinacker K, Dimayuga E, Nukala V, Drake J, Butterfield DA, Dunn JC, Martin S, Bruce-Keller AJ, Keller JN. Role of the proteasome in protein oxidation and neural viability following low-level oxidative stress. FEBS Lett. 2003;546:228–232. doi: 10.1016/s0014-5793(03)00582-9. [DOI] [PubMed] [Google Scholar]
- Diniz YS, Cicogna AC, Padovani CR, Santana LS, Faine LA, Novelli EL. Diets rich in saturated and polyunsaturated fatty acids: metabolic shifting and cardiac health. Nutrition. 2004;20:230–234. doi: 10.1016/j.nut.2003.10.012. [DOI] [PubMed] [Google Scholar]
- Esterbauer H. Cytotoxicity and genotoxicity of lipid-oxidation products. Am. J. Clin. Nutr. 1993;57:779S–789S. doi: 10.1093/ajcn/57.5.779S. [DOI] [PubMed] [Google Scholar]
- Farr SA, Yamada KA, Butterfield DA, Abdul HM, Miller NE, Banks WA, Morley JE. Obesity and hypertrigly-ceridemia produce cognitive impairment. Endocrinology. 2008;149:28–36. doi: 10.1210/en.2007-1722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh S, Kewairamani G, Yuen G, Pulinilkunnil T, An D, Innis SM, Allard MF, Wambolt RB, Abrahani A, Rodrigues B. Induction of mitochondrial nitrative damage and cardiac dysfunction by chronic provision of dietary omega-6 polyunsaturated fatty acids. Free Radic. Biol. Med. 2006;41:1413–1424. doi: 10.1016/j.freeradbiomed.2006.07.021. [DOI] [PubMed] [Google Scholar]
- Girotti AW. Lipid hydroperoxide generation, turnover, and effector action in biological systems. J. Lipid Res. 1998;39:1529–1542. [PubMed] [Google Scholar]
- Guo Z, Jiang H, Xu X, Duan W, Mattson MP. Leptin-mediated cell survival signaling in hippocampal neurons mediated by JAK STAT3 and mitochondrial stabilization. J. Biol. Chem. 2008;283:1754–1763. doi: 10.1074/jbc.M703753200. [DOI] [PubMed] [Google Scholar]
- Hoojimas CR, Rutters F, Dederen PJ, et al. Changes in cerebral blood volume and amyloid pathology in aged Alzheimer APP/PS1 mice on a docohexaenoic acid (DHA) diet or cholesterol enriched typical Western diet (TWD) Neurobiol. Dis. 2007;28:16–29. doi: 10.1016/j.nbd.2007.06.007. [DOI] [PubMed] [Google Scholar]
- Keller JN, Kindy MS, Holtsberg FW, St Clair DK, Yen HC, Germeyer A, Steiner SM, Bruce-Keller AJ, Hutchins JB, Mattson MP. Mitochondrial manganese superoxide dismutase prevents neural apoptosis and reduces ischemic brain injury: suppression of peroxynitrite production, lipid peroxidation, and mitochondrial dysfunction. J. Neurosci. 1998;18:687–697. doi: 10.1523/JNEUROSCI.18-02-00687.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kukar T, Murphy MP, Eriksen JL, et al. Diverse compounds mimic Alzheimer’s disease-causing mutations by augmenting Aβ42 production. Nat. Med. 2005;11:545–550. doi: 10.1038/nm1235. [DOI] [PubMed] [Google Scholar]
- Litchtenstein AH, Kennedy E, Barrier P, Danford D, Ernst ND, Grundy SM, Leveille GA, Van Horn L, Williams CL, Booth SL. Dietary fat consumption and health. Nutr. Rev. 1998;56:S3–S19. doi: 10.1111/j.1753-4887.1998.tb01728.x. [DOI] [PubMed] [Google Scholar]
- Markesbery WR. Oxidative stress hypothesis in Alzheimer’s disease. Free Radic. Biol. Med. 1997;23:134–147. doi: 10.1016/s0891-5849(96)00629-6. [DOI] [PubMed] [Google Scholar]
- Mattson MP. Pathways towards and away from Alzheimer’s disease. Nature. 2004;430:631–639. doi: 10.1038/nature02621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGowan E, Pickford F, Kim J, et al. Aβ42 is essential for parenchymal and vascular amyloid deposition in mice. Neuron. 2005;47:191–199. doi: 10.1016/j.neuron.2005.06.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milagro FI, Campion J, Martinez JA. Weight gain induced by high-fat feeding involves increased liver oxidative stress. Obesity. 2006;14:1118–1123. doi: 10.1038/oby.2006.128. [DOI] [PubMed] [Google Scholar]
- Morris MC, Evans DA, Bienias JL, Tangney CC, Bennett DA, Aggarwal N, Schneider J, Wilson RS. Dietary fats and the risk of incident Alzheimer’s disease. Arch. Neurol. 2003;60:194–200. doi: 10.1001/archneur.60.2.194. [DOI] [PubMed] [Google Scholar]
- Murphy MP, Beckett TL, Ding Q, Patel E, Markesbery WR, St Clair DK, LeVine H, Keller JN. Aβ solubility and deposition during AD progression and in APP × PS1 knock-in mice. Neurobiol. Dis. 2007;27:301–311. doi: 10.1016/j.nbd.2007.06.002. [DOI] [PubMed] [Google Scholar]
- Oksman M, Livonen H, Hogyes E, Atmul Z, Penke B, Leenders I, Broersen L, Lutjohann D, Hartmann T, Tanila H. Impact of different saturated fatty acid, polyunsaturated fatty acid and cholesterol containing diets on beta-amyloid accumulation in APP/PS1 transgenic mice. Neurobiol. Dis. 2006;23:563–572. doi: 10.1016/j.nbd.2006.04.013. [DOI] [PubMed] [Google Scholar]
- Pasinetti GM, Zhao Z, Qin W, et al. Caloric intake and Alzheimer’s disease. Experimental approaches and therapeutic implications. Interdiscip. Top. Gerontol. 2007;35:159–175. doi: 10.1159/000096561. [DOI] [PubMed] [Google Scholar]
- Pratico D. Lipid peroxidation and the aging process. Sci. Aging Knowledge Env. 2002;18(50):re5. doi: 10.1126/sageke.2002.50.re5. [DOI] [PubMed] [Google Scholar]
- Reddy PH, Beal MF. Amyloid beta, mitochondrial dysfunction and synaptic damage: implications for cognitive decline in aging and Alzheimer’s disease. Trends Mol. Med. 2008;14:45–53. doi: 10.1016/j.molmed.2007.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts CK, Bernard RJ, Sindhu RK, Jurczak M, Ehdale A, Vaziri ND. Oxidative stress and dysregulation of NAD(P)H oxidase and antioxidant enzymes in diet induced metabolic syndrome. Metabolism. 2006;55:928–934. doi: 10.1016/j.metabol.2006.02.022. [DOI] [PubMed] [Google Scholar]
- Rofolo LM, Malester B, LaFrancois J, Bryant-Thomas T, Wang R, Tint GS, Sambamurti K, Duff K, Pappolla MA. Hypercholesterolemia accelerates the Alzheimer’s amyloid pathology in a transgenic mouse model. Neurobiol. Dis. 2000;7:321–331. doi: 10.1006/nbdi.2000.0304. [DOI] [PubMed] [Google Scholar]
- Ronti T, Lupattelli G, Mannarino E. The endocrine function of adipose tissue: an update. Clin. Endocrinol. 2006;64:355–365. doi: 10.1111/j.1365-2265.2006.02474.x. [DOI] [PubMed] [Google Scholar]
- Shaw DI, Hall WL, Williams CM. Metabolic syndrome: what is it and what are the implications? Proc. Nutr. Soc. 2005;64:349–357. doi: 10.1079/pns2005442. [DOI] [PubMed] [Google Scholar]
- Siman R, Reaume AG, Savage MJ, Trusko S, Lin YG, Scott RW, Flood DG. Presenilin-1 P264L knock-in mutation: differential effects on Aβ production, amyloid deposition, and neuronal vulnerability. J. Neurosci. 2000;20:8717–8726. doi: 10.1523/JNEUROSCI.20-23-08717.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Souza CG, Moreira JD, Siqueira IR, Pereira AG, Rieger DK, Souza DO, Souza TM, Portela LV, Perry ML. Highly palatable diet consumption increases protein oxidation in rat frontal cortex and anxiety-like behavior. Life Sci. 2007;81:198–203. doi: 10.1016/j.lfs.2007.05.001. [DOI] [PubMed] [Google Scholar]
- Stranahan AM, Arumugam TV, Cutler RG, Lee K, Egan JM, Mattson MP. Diabetes impairs hippocampal function through glucocorticoid-mediated effects on new and mature neurons. Nat. Neurosci. 2008;11:309–317. doi: 10.1038/nn2055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sung S, Yao Y, Uryu K, Yang H, Lee VM, Trojanowski JQ, Pratico D. Early vitamin E supplementation in young but not aged mice reduces Aβ levels and amyloid deposition in a transgenic model of Alzheimer’s disease. FASEB J. 2004;18:323–325. doi: 10.1096/fj.03-0961fje. [DOI] [PubMed] [Google Scholar]
- Swerdlow RH. Pathogenesis of Alzheimer’s disease. Clin. Interv. Aging. 2007;2:347–359. [PMC free article] [PubMed] [Google Scholar]
- Tamagno E, Guglielmotto M, Aragno M, et al. Oxidative stress activates a positive feedback between the gamma- and beta-secretase cleavages of the beta-amyloid precursor protein. J. Neurochem. 2008;104:683–695. doi: 10.1111/j.1471-4159.2007.05072.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vincent HK, Innes KE, Vincent KR. Oxidative stress and potential interventions to reduce oxidative stress in overweight and obesity. Diabetes Obes. Metab. 2007;9:813–839. doi: 10.1111/j.1463-1326.2007.00692.x. [DOI] [PubMed] [Google Scholar]
- Weisburger JH. Dietary fat and risk of chronic disease: mechanistic insights from experimental studies. J. Am. Diet. Assoc. 1997;97:S16–S23. doi: 10.1016/s0002-8223(97)00725-6. [DOI] [PubMed] [Google Scholar]
- Zana M, Janka Z, Kalman J. Oxidative stress: a bridge between Down’s syndrome and Alzheimer’s disease. Neurobiol. Aging. 2007;28:648–676. doi: 10.1016/j.neurobiolaging.2006.03.008. [DOI] [PubMed] [Google Scholar]
- Zhang F, Wang S, Signore AP, Chen J. Neuroprotective effects of leptin against ischemic injury induced by oxygen-glucose deprivation. Stroke. 2007;38:2329–2336. doi: 10.1161/STROKEAHA.107.482786. [DOI] [PubMed] [Google Scholar]
- Zhu X, Raina AK, Perry G, Smith MA. Alzheimer’s disease: the two-hit hypothesis. Lancet Neurol. 2004;3:219–226. doi: 10.1016/S1474-4422(04)00707-0. [DOI] [PubMed] [Google Scholar]
- Zhu X, Lee HG, Perry G, Smith MA. Alzheimer’s disease, the two-hit hypothesis: an update. Biochem. Biophys. Acta. 2007;1772:494–502. doi: 10.1016/j.bbadis.2006.10.014. [DOI] [PubMed] [Google Scholar]