

Abstract
Mycobacterium tuberculosis and other members of the Actinomycetes family produce mycothiol (MSH or Acetylcysteine-Glucosamine-Inositol, AcCys-GlcN-Ins) to protect the organism against oxidative and antibiotic stress. The biosynthesis of MSH proceeds via a five-step process that involves four unique enzymes, MshA-D, which represent specific targets for inhibitor design. Recombinant Mycobacterium smegmatis MshC catalyzes the ATP-dependent condensation of glucosamine-inositol (GlcN-Ins) and cysteine to form Cys-GlcN-Ins. The 1.6 Å three-dimensional structure of MshC in complex with a tight binding bisubstrate analog, 5'-O-[N-(L-cysteinyl)-sulfamonyl]adenosine (CSA), has suggested specific roles for T46, H55, T83, W227 and D251. In addition, a catalytic role for H55 has been proposed based on studies of related aminoacyl-tRNA synthetases. Site-directed mutagenesis was carried out to evaluate the functional roles of these highly conserved residues. All mutants exhibited significantly decreased kcat values, with the exception of T83V for which less than a seven-fold decrease was observed compared to WT. For the T46V, H55A, W227F, and D251N mutants, the rate of cysteine activation decreased 100 - 1,400 fold compared to WT, consistent with the important roles of these residues in the first half reaction. The ~2,000 fold decrease in kcat/Km as well as the ~20 fold decrease in Km for cysteine suggested a significant role for T46 in cysteine binding. Kinetic studies also indicate a function for W227 in cysteine binding but not in substrate discrimination against serine. H55 was also observed to play a significant role in ATP binding as well as cysteine adenylation. The activity of H55A was partially rescued with exogenous imidazole at acidic pH values, suggesting that the protonated form of histidine is exerting a catalytic role. The pH dependence of the kinetic parameters with the WT enzyme suggests an additional requirement for a catalytic base in cysteinyl ligation.
Keywords: Mycobacterium, mycothiol, cysteine ligase, active site residues, catalytic mechanism, quench flow, substrate inhibition, imidazole rescue
Mycothiol (MSH, AcCys-GlcN-Ins) is biosynthesized via a series of enzymatic reactions (1-4) and is the predominant low molecular weight thiol that protects actinomycetes against oxidative stress and cellular electrophilic toxins (5-8). Mycobacteria generate the highest intracellular levels of MSH among actinomycetes (9). Studies have shown that Mycobacterium smegmatis mutants lacking MSH become more sensitive towards oxidizing agents, electrophiles, and antibiotics (5-7), suggesting a critical role of MSH in the survival and pathogenicity of mycobacteria (5). MshC, which catalyzes the ATP-dependent condensation of cysteine and GlcN-Ins (Scheme 1) in the penultimate step of MSH biosynthesis, was shown to be essential for production of MSH in M. smegmatis (7). For Mycobacteria tuberculosis, the mshC gene has been demonstrated to be essential for in vitro growth (10, 11). MshC is hence a potential target for drugs to treat tuberculosis.
Scheme 1.

Reaction catalyzed by MshC
In our laboratory, MshC from M. smegmatis has been recombinantly expressed, purified and characterized (12). The steady state kinetics along with positional isotope exchange (PIX) experiments demonstrated that the reaction catalyzed by MshC follows a Bi Uni Uni Bi Ping Pong mechanism (Scheme 2), with the random binding of ATP and L-cysteine, release of pyrophosphate, binding of GlcN-Ins and finally the release of Cys-GlcN-Ins and AMP (12, 13). On the basis of this kinetic mechanism, the overall reaction can be divided into two steps, cysteine adenylation, and the subsequent ligation of cysteine to GlcN-Ins. Single-turnover reactions of the first and second half reactions and PIX studies supported the cysteinyl adenylate as a kinetically competent intermediate in the reaction by MshC (12, 13).
Scheme 2.

MshC has been suggested to share functional and, presumably, structural similarities with cysteinyl-tRNA synthetase (CysRS) based on 37.6% sequence identity (14) . CysSR is a member of the class I aminoacyl-tRNA synthetase (AaRS) family, which are characterized by the Rossmann dinucleotide binding active site along with two highly conserved signature motifs: HIGH and KMSKS (15-26). In MshC, the HIGH motif is replaced by HLGH. Both are fingerprint sequences for ATP-binding in the cytidylyltransferase enzyme superfamily (27). Crystallization efforts on MshC failed to produce protein crystals. This was overcome by incubation of the enzyme with a cysteinyl adenylate analogue 5'-O-[N-(L-cysteinyl)-sulfamonyl]adenosine (CSA, with an inhibition constant of ~300 nM versus ATP), followed by a 24-hour limited proteolysis. The resulting enzyme preparation was successfully crystallized, yielding the three-dimensional structure of MshC with CSA bound in the active site at 1.6 Å resolution (28). The Rossman catalytic domain revealed that CSA binding is stabilized by a set of completely conserved residues in the MshC active site (Figure 1). The OG1 sidechain oxygens of T46 and T83 form hydrogen bonds with the cysteine moiety of CSA. The 2'-oxygen of the ATP moiety makes a 2.7 Å hydrogen bond with the D251 side chain carboxylate group. The conserved H55 on the HxGH motif is 3.4 Å away from the ribose oxygen atom in the MshC-CSA complex, but it is likely that a hydrogen bond would form in the structure with the cysteinyl adenylate intermediate. The indole ring nitrogen of W227, proposed to be responsible for amino acid discrimination and positioning in CysRS (15), is 3.3 Å away from the thiol group of cysteine, and is likely to form a hydrogen bond with the cysteine substrate in the adenylation reaction. To assess the functional roles of these residues in substrate binding and catalysis, we have constructed the following mutant forms of the enzyme: T46V, H55A, T83V, D251A, D251N, W227F and W227H. We have studied the pH effects on wild type (WT) MshC, and examined the catalytic effects of these mutants via biochemical and kinetic approaches.
Figure 1.

The active site residues of MshC from the three-dimensional structure resolved at 1.6 Å (PDB code 3D8Z).
MATERIALS AND METHODS
Materials
Pfu DNA polymerase was from Stratagene. pET28a(+) and the Escherichia coli strains, Rosetta(DE3)2 and Top10, were obtained from Novagen. T4 DNA ligase and the restriction enzymes NdeI and EcoRI were from New England Biolabs. The oligonucleotide primers were synthesized by Invitrogen. Luria-Bertani broth was purchased from Fisher. All chromatographic materials were from Pharmacia. Protease inhibitor cocktail tablet (Complete™, EDTA free) was obtained from Roche. 5'-O-[N-(L-cysteinyl)sulfamonyl]adenosine was from Integrated DNA Technologies. [14C]-L-cysteine and [α-33P]-ATP were purchased from PerkinElmer Life Sciences. PEI-F-TLC and Silica-TLC plates were obtained from EMD Chemicals Inc. and Whatman Ltd., respectively. All other chemicals were purchased from Sigma or Aldrich.
Site-directed mutagenesis
The mutations were generated using the QuikChange site-directed mutagenesis kit (Stratagene) according to the manufacturer's instructions, with the template pET28amshC plasmid (12), as well as the forward and reverse primers to introduce the desired mutation. DNA was sequenced at the DNA Sequencing Facility at the Albert Einstein College of Medicine. The verified plasmid was then transformed into to Escherichia coli Rosetta(DE3)pLysS competent cells.
Expression and purification mutants of MshC
The sequence verified plasmids were transformed into Rosetta(DE3)2 for protein expression. A 10 ml pre-culture was used to inoculate 1L of Luria-Bertani broth media containing ampicillin and chloramphenicol at final concentrations of 50 and 34 μg/ml, respectively. Cells were grown at 37 °C. When the optical density at 600 nm of the culture reached 0.8, IPTG was added into the culture to a final concentration of 0.3 mM and the temperature of the culture was lowered to 20 °C for overnight growth. Cells were harvested by centrifugation at ~ 3,000 × g for 30 min at 4 °C and stored at -20 °C. The mutant enzymes were purified to homogeneity using the same protocol used for the wild type MshC (12). The concentration of enzyme was determined using the Bio-Rad protein assay kit with bovine serum albumin as the standard.
Enzyme Activity Assay
Initial velocities of the MshC reaction were assayed spectrophotometrically by coupling the formation of AMP to the reaction of myokinase, pyruvate kinase, and lactate dehydrogenase as described previously (29). The decrease in absorbance of NADH at 340 nm (ε340 = 6,220 M-1 cm-1) was measured at 25 °C using a UVIKON 943 spectrophotometer with a circulating water bath and thermospacers. The standard reaction contains 100 mM HEPES, pH 7.8, 10 mM MgCl2, 10 mM ATP, 1 mM L-cysteine, 2 mM DTT, 100 μM GlcN-Ins, 1 mM potassium PEP, 300 μM NADH, 18 units of myokinase, 18 units of pyruvate kinase and 18 units of lactate dehydrogenase in a total volume of 1 ml. After incubation for 5 min at 25 °C, reactions were initiated by the addition of MshC (≤ 10 μl to a final concentration of ~ 25 nM). MshC enzymatic activities were corrected for the background activity, i.e., the decrease of absorbance at 340 nm caused by ATP hydrolysis. The rate of cys-GlcN-Ins formation is proportional to the rate of NADH oxidation where two molecules of NADH are oxidized for each molecule of cys-GlcN-Ins formed.
Steady state kinetics
Initial velocity experiments were carried out at various concentrations of one substrate in the presence of different fixed levels of a second substrate, and with the concentration of the third substrate kept saturating and constant as described previously (12). Product inhibition studies with pyrophosphate were performed at varying concentrations of ATP, 500 μM GlcN-Ins, and 40 μM or 2 mM cysteine in a total volume of 1 ml. The bi-substrate analog CSA was tested as an inhibitor, versus ATP (at 50 μM cysteine and 100 μM GlcN-Ins) and cysteine (at 1 mM ATP and 200 μM GlcN-Ins), respectively.
The pH dependence of the kinetic parameters exhibited by MshC were determined for kcat, kcat/KATP and kcat/KGlcN-Ins. Reactions were performed by varying concentrations of ATP and at saturating concentrations of cysteine and GlcN-Ins to determine kcat and kcat/KATP. For the determination of kcat/KGlcN-Ins, GlcN-Ins was the variable substrates, with ATP and cysteine kept at saturating concentrations. MshC activity was monitored over the pH range from 5.5 to pH 10 using bis-Tris propane.
The effect of imidazole on the turnover number of MshC H55A was determined by measuring the enzymatic activity with 2 mM cysteine, 5 mM ATP, 200 μM GlcN-Ins in the presence of varying concentrations of imidazole in the range from 0 -100 mM in 100 mM HEPES, pH 7.8 or 100 mM Bis-Tris propane at other pH values.
Pre-steady state kinetics of cysteine adenylation
Single turnover experiments were performed at 25 °C using a KinTek rapid quench apparatus (Model RQF-3) equipped with a constant temperature circulating water bath. The rates of the first half reaction (cysteine adenylation) were determined by rapidly mixing MshC containing 1 mM L-cysteine, 10 mM DTT, in 200 mM HEPES, pH 7.8, with a solution containing 0.1 - 4 mM [α-33P]-ATP (20 μCi/μmol), 10 mM MgCl2 in 100 mM HEPES, pH 7.8 in a total volume of 20 μl. Control reactions excluded MshC from the reaction. The reaction was incubated for a given time interval and then quenched with 110 μl of 150 mM EDTA. The enzyme was denatured by heat treatment in a boiling water bath for 1 min after quenching. After centrifugation, 1 μl of the reactions was spotted onto PEI TLC plates, and radiolabeled ATP and AMP were resolved using 0.9 M guanidine hydrochloride as mobile phase.
Fluorescence studies
Emission fluorescence spectra were recorded on a Jobin Yvon Horiba spectrofluorimeter Model Fluoro Max-3 at 25 °C with a 1 cm light path quartz cuvette and 5 nm slits. The protein was dissolved in 50 mM HEPES buffer, 5 mM Mg2+, pH 7.8. The tryptophan intrinsic fluorescence was measured on a 0.1 μM solution of the protein using an excitation wavelength of 295 nM and recording the emission spectrum between 300-400 nm. To measure the affinity of CSA to MshC, the protein solution was titrated with aliquots of 100 μM CSA to a final concentration of 8 -250 nM.
Data analysis
Parallel initial velocity patterns were fitted to eq 1 and intersecting patterns were fit to eq 2, where Ka and Kb are the Michaelis constants for the varied substrates A and B. The pH dependences of steady state kinetic parameters were determined by fitting initial rate data to eqs 3 and 4, which describe a curve with slope of +1 and a plateau region at high pH, and a bell shape curve with slope of +1 at low pH and a slope of -1 at high pH, respectively. Inhibition data were fitted to eq 5 for competitive inhibition, where P represents the concentration of inhibitor (5'-O-[N-(L-cysteinyl)sulfamonyl]adenosine or pyrophosphate), and Kis is the inhibition constant for the slope term. The apparent substrate inhibition constant for ATP (Kai) was determined by fitting the data to eq 6.
| (1) |
| (2) |
| (3) |
| (4) |
| (5) |
| (6) |
Single-turnover progress curve data were fitted to eq 7:
| (7) |
For a two-step binding mechanism, the dependence of the single-exponential rate constant, kobs, as a function of substrate concentration is given by eq 8. For both half reactions by MshC, the intercept of kobs versus substrate concentration curves appeared to be approximately zero. The data were thus fitted to eq 9, where koff = 0.
| (8) |
| (9) |
Fluorescence data were fit to an equation describing binding to a single site (eq 10), where ΔF is the fluorescence quenching (change in fluorescence to the initial value), following addition of a ligand at a concentration L, and Kd is the dissociation constant for binding to the protein.
| (10) |
RESULTS
pH-Dependent studies with WT-MshC
The kinetic parameters of WT-MshC were measured as a function of pH to probe the ionization state of groups responsible for catalysis and binding. Since the enzyme activity was monitored by coupled enzyme assays, the pH dependence of the coupling enzymes was considered. At the pH extremes, concentrations of coupling enzymes and substrate were increased to ensure that the pH dependence of the MshC reaction is the observed reaction. For the overall reaction, kinetic parameters were determined at varying concentrations of ATP and fixed saturating concentrations of cysteine and GlcN-Ins. The kcat increases with increasing pH values, depending on the ionization group of one group with pKa value of 6.8 ± 0.1 (Figure 2A). The kcat/KATP pH profile exhibits a bell shape curve (Figure 2B), that is depends on the ionization of two groups exhibiting pKa values of 6.8 ± 0.1, and 10.1 ± 0.1, respectively. The pH dependence was also determined at varying concentrations of GlcN-Ins and fixed saturating concentrations of ATP and cysteine. In this case, kcat/KGlcN-Ins (Figure 2C) values increase with increases pH and reached a plateau above pH 8, due to a single ionizable group exhibiting a pKa value of 7.9 ± 0.1.
Figure 2.

pH Dependence of the kcat (panel A), kcat/Km for ATP (panel B) and kcat/Km for GlcN-Ins (panel C). MshC activity was assayed at saturating concentrations GlcN-Ins and cysteine, and varying concentrations of ATP at 25 °C as described in the Experimental Procedures section. The lines are fits of the data to eqs 3 and 4.
Mutagenesis, expression and purification
Mutagenesis studies were preformed to probe the possible identity of the ionizable residues observed in the pH profiles. Both the structure of the MshC-CSA complex as well as previous studies with CysRS (15) suggested important roles for five completely conserved active site residues: T46, H55, T83, W227, and D251. Alanine was substituted for H55. Valine substitutions were introduced for both T46 and T83. W227 was substituted with both phenylalanine and histidine. D251 was changed to both alanine and asparagine. All mutations were confirmed by nucleotide sequencing, subsequently expressed in Rosetta(DE3) cells and purified to homogeneity in a yield of ~ 25 mg from 1 L of cell culture as previously described (12). The subunit molecular mass of purified MshC mutant forms were determined SDS-PAGE to be ~ 47 kDa.
Steady state kinetic studies
The kinetic parameters of WT and mutant forms of MshC were determined by initial velocity experiments using a coupled spectrophotometric assay. As expected, the best fits of the initial data support a sequential steady-state kinetic mechanism for all mutant enzymes with the exception of H55A, where parallel lines were observed by varying concentrations of both ATP and cysteine (data not shown). These are consistent with the formation of a ternary MshC-ATP-cysteine complex. The kcat values of these mutants were determined to be 0.08 - 15% that of the WT enzyme (Table 1). The highest activity was observed with T83V, with Km values for both ATP and cysteine similar to those of WT, suggesting that the importance of the T83 interaction with the cysteinyl α-amino group is modest. The nature and strength of inhibition by CSA was also determined for mutant MshC enzymes. Competitive inhibition patterns were observed with all mutant enzymes against ATP (data not shown). The highest inhibition constants were observed for the H55A, D251N and D251A mutants, with values ranging from ~500 - 900 nM. Surprisingly, the Ki values are decreased to ~ 30 and 56 nM for W227F and W227H mutants. In addition, similar to WT enzyme, no activity was observed when serine was tested as substrate up to 5 mM with both W227 mutants.
Table 1.
Steady State Kinetics Parameters of WT and Mutants MshC from M.Smegmatisa
| Protein | kcat, min-1 | Km-ATP, mM | Km-cys, mM | Km-GlcN-Ins, mM |
|---|---|---|---|---|
| WT | 189 ± 11 | 1.8 ± 0.1 | 0.1 ± 0.01 | 0.16 ± 0.05 |
| H55A | 4.4 ± 0.5 | 5.2 ± 0.1 | 1.1 ± 0.2 | 0.01 ± 10-3 |
| W227F | 1.0 ± 0.1 | 0.20 ± 0.05 | 0.4 ± 0.1 | 0.15 ± 0.06b |
| W227H | 1.9 ± 0.1 | 0.12 ± 0.01 | 0.12 ± 0.04 | 0.6 ± 0.01b |
| T46V | 1.8 ± 0.5 | 1.1 ± 0.03 | 1.9 ± 0.2 | 0.15 ± 0.08b |
| T83V | 29 ± 1 | 0.32 ± 0.06 | 0.02 ± 0.001 | 0.17 ± 0.05b |
| D251N | 0.45 ± 0.05 | 8.9 ± 1.6 | 0.48 ± 0.15 | 0.10 ± 0.06b |
| D251A | 0.16 ± 0.02 | 0.36 ± 0.011 | 0.17 ± 0.02 | 0.16 ± 0.01b |
Substrate inhibition studies
Substrate inhibition is commonly observed in Ping Pong and steady state ordered mechanisms in a bireactant mechanism (30). For WT MshC, no substrate inhibition was observed up to 10 mM ATP (12). In contrast, the enzyme activity decreases at concentrations of ATP above ~1.1 mM with the D251A mutant as shown in Figure 3 and the substrate inhibition constant was determined to be ~ 8.1 mM. Substrate inhibition is not observed for the D251N mutant at up to 20 mM ATP. This type of inhibition can be observed when two molecules of substrate bind to the enzyme, however, we believe that the loss of the hydrogen bonding between D251 and the 2'-hydroxyl may result in the incorrect binding of ATP at high concentrations.
Figure 3.
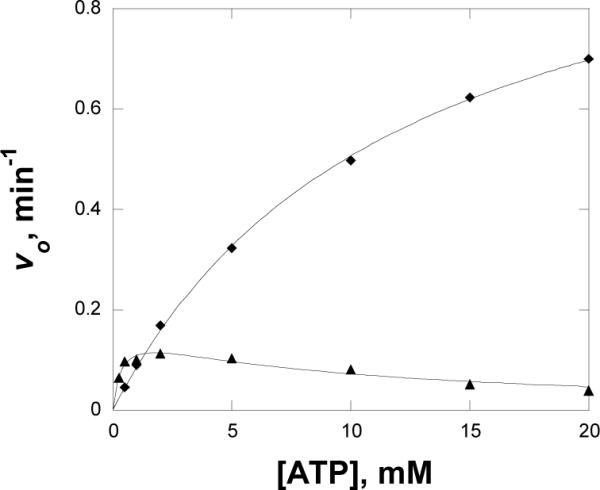
Substrate inhibition of D251A (▲) with ATP, with D251N (◆) as a control. MshC activity was assayed at 2 mM cysteine, 200 μM GlcN-Ins, and varying concentration of ATP at 25 °C as described in the Experimental Procedures section. The line of D251A is a fit of the data to eq 6.
Chemical Rescue
The histidine to alanine mutation in the active site is equivalent to the removal of an imidazole moiety from the side chain. In principle, both the kinetic and biochemical properties that have been affected by such a mutation should at least be partially restored in the enzyme in the presence of exogenous imidazole. No significant activating or inhibitory effect of added imidazole was observed with the WT enzyme. At pH 7.8 and pH 9, no changes were observed in enzyme activity upon addition of imidazole to the H55A mutant, whereas significant increases of activity were seen at 6.75. A seven-fold increase was observed on the kcat value of H55A at 100 mM added imidazole (Figure 4), indicating that the protonated form of imidazole is the catalytically relevant species responsible for the partial rescue of enzyme activity in MshC.
Figure 4.
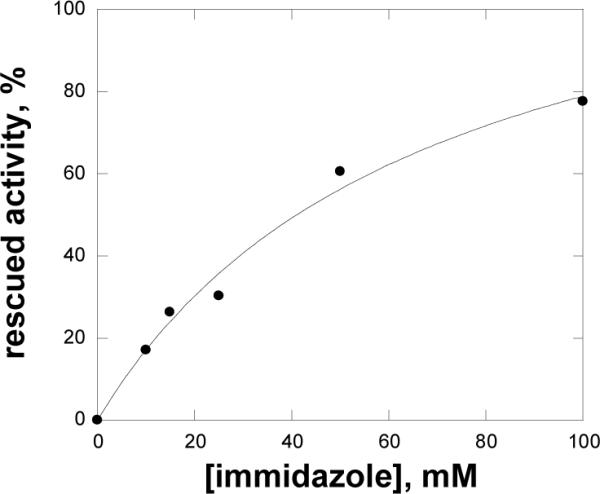
Activity of H55A mutant rescued by imidazole. Assays were carried out as described in Materials and Methods.
Fluorescence studies of inhibitor binding
The WT and mutant forms of MshC were investigated for binding of CSA by fluorescence spectrophotometry. The WT enzyme exhibits an emission peak at 340 nm (Figure 5). The normalized fluorescence intensity for W227H change 0.9 compared to WT, indicating that W227 is not the only tryptophan whose fluorescence changes with CSA binding to the WT and mutant forms of MshC. The tryptophan fluorescence of WT and mutant forms of MshC decreases with the addition of CSA (Figure 5B), with the Kd value for WT MshC determined to be 110 ± 10 nM. Significant increases in the Kd values of CSA were observed for the H55A, D251N, and D251A mutants, whereas the W227 (H or F) mutants showed decreased Kd values for CSA, following the trend observed in steady-state inhibition studies (Table 2).
Figure 5.

Fluorescence studies with CSA as inhibitor. Panel A, fluorescence titration of WT MshC with 0 (solid line), 91 (broken line), and 410 (dotted line) nM CSA; Panel B, fluorescence spectra of WT (solid line), W227F (big broken line), D251N (small broken line) and H55A (dotted line) forms of free MshC. In all experiments, the concentration of enzyme utilized is 0.1 μM.
Table 2.
Inhibition Constants of WT and Mutants MshC with Acylsulfamate Analogue (CSA) versus ATP.
Pre-steady state kinetic analysis
The rate of cysteine-adenylate formation in the first half reaction was determined by single turnover experiments using rapid quench flow techniques with [α-33P]-ATP in the absence of GlcN-Ins. Plots of [α-33P]-AMP formation were obtained using different concentrations of [α-33P]-ATP. In experiments using the T46V and W227F mutants, cysteine was varied. The data were fit to eq 6 to obtain rate constants for adenylate formation at each concentration of substrate used. These rate constants were plotted as a function of ATP or cysteine (Figure 6). The data were fit to eq 7 to obtain the maximum value of the adenylate formation. The maximal rates of adenylate formation for the H55A, T46V, W227F and D251A mutants are lower than that of WT by 100 - 1,400 fold (Table 3). The D251N has the lowest activity among the mutants tested. Both H55A and D251N exhibited 5-10 fold higher Kd values for ATP, while for the T46V and W227F mutants, a significantly increased Kd value for cysteine was observed.
Figure 6.

Pre-steady state kinetics of AMP formation in the first half reaction of MshC. Panel A and B, plots of the determined rate constants as a function of the ATP concentration used with D251N and H55A mutants; Panels C and D, plots of the determined rate constants as a function of the cysteine concentration used with T46V and W227F mutant, respectively. Assays were carried out as described in Materials and Methods. The curves are fits of the data to eq 9.
Table 3.
Pre-Steady State Kinetics Parameters of WT and Mutants MshC
| Protein | kobs, min-1 | Kd-ATP, mM | Kd-cys, HIM |
|---|---|---|---|
| WT | 560 ± 40 | 1.7 ± 0.2 | 0.16 ± 0.03 |
| H55A | 6 ± 1 | 15 ± 3 | |
| W227F | 2.6 ± 0.2 | 0.6 ± 0.1 | |
| T46V | 3.2 ± 0.2 | 0.8 ± 0.1 | |
| D251N | 0.41 ± 0.06 | 10.2 ± 2.8 |
a The first half reaction, data were fit to eq 9.
DISCUSSION
The 1.6 Å structure of MshC in complex with CSA (28) revealed a number of active site residues that interact with CSA as shown in Figure 1. Among these residues, T46 and T83 interact with the α-amino group of the cysteine moiety of CSA, while D251 interacts with the 2'-ribose hydroxyl group of the ATP moiety. We have also selected two other highly conserved active site residues, H55 and W227, for mechanistic characterization. Histidine 55 is the first histidine residue on the HxGH signature motif, and is likely to form a hydrogen bond with the cysteinyl adenylate intermediate. In the structurally and functional related CysRS, the highly conserved tryptophan residue W205 (corresponding to W227 in MshC) was suggested to be responsible for cysteine binding and for the discrimination against serine as a substrate (15). We have constructed the T46V, H55A, T83V, D251A, D251N, W227F and W227H mutant forms of MshC. Decreased but measurable MshC activities (~ 0.1 - 15%) were observed for all mutant enzyme forms. We have examined the catalytic effects of these mutants via biochemical and kinetic approaches.
ATP binding residue - D251
The three-dimensional structure of MshC revealed that the carboxylate oxygen atom of D251 is 2.7 Å from the 2'-oxygen on the ribose ring of CSA, suggesting its role in ATP binding. The alanine and asparagine mutations of D251 were therefore introduced for kinetic characterization. As expected, D251N has higher activity than D251A, but both exhibit steady state turnover numbers that are ~ 400 and ~ 1,200 fold lower in comparison with WT MshC, respectively. The pre-steady state kinetic analysis of D251N showed a 1,200 fold decrease in the rate of the first half reaction compared to WT, confirming that the effect of substitution of D251 primarily lies within the first, adenylation reaction (12). The observation that D251N exhibits a ~ 400 fold decrease in kcat yet a ~ 2,000 fold decrease in kcat/Km for ATP favors the interpretation that D251 is primarily involved in ATP binding rather than the chemical adenylation reaction. The observation of the substrate inhibition with the D251A mutant is also consistent with this. Differences in the steady-state affinity for the D251 mutants are mirrored in the Kd value for ATP determined in pre-steady state kinetics. Finally, the side chain carboxyl group of D251 also influences the affinity of the enzyme for CSA, measured both by steady state inhibition and inhibitor affinity changes upon CSA binding based on fluorescence measurements with the WT and the D251 mutant enzyme forms.
Cysteine binding residues
The side chain hydroxyl groups of T46 and T83 are both observed in the MshC-CSA complex 2.9 Å from the α-amino group of the cysteine moiety of CSA, suggesting roles in cysteine binding. In addition, a highly conserved tryptophan W227 was observed 3.3 Å from the zinc coordinated thiol, which has been proposed to be responsible for the amino acid discrimination and positioning in CysRS (15).
T46V exhibited an ~ 100 fold decrease in kcat compared to WT, while T83V showed less than a 7 fold decrease. When compared to WT, the kcat/Km values of T46V decrease ~ 60 and ~2,000 fold for ATP and cysteine, respectively. This is consistent with a role for T46V in cysteine rather than ATP binding. The modest decrease in the kcat/Km value for ATP in T46V is presumably due to the random binding order of ATP and cysteine to free MshC (31). The observation that T83V exhibits a similar kcat value to WT, as well as the kcat/Km values for both ATP and cysteine, argues against any significant role of T83 in binding and catalysis, despite its proximity to the cysteine moiety. Pre-steady state kinetics were therefore only carried out with T46V, where a ~ 180 fold decrease was observed in the adenylation rate compared to WT.
Phenylalanine and histidine were substituted for W227 and characterized. The phenylalanine (W227F) and histidine (W227H) mutants exhibited similar kinetic parameters, with an ~ 100 fold decrease in kcat, a ~ 800 fold decrease in kcat/Km value for cysteine and a ~ 4 fold increase in the Kd value for cysteine, suggesting that W227 plays a role in cysteine binding. Interestingly, the Kd values for CSA determined via fluorescence and the Ki values determined by inhibition studies for these mutants are decreased compared to the WT enzyme. Whether the substitution of these smaller aromatic side chains allows for enhanced thiolate-metal interaction is unclear, but under investigation. Finally, there is no detectable MshC activity with serine as substrate for either the phenylalanine or histidine mutant, which argues that W227 does not function in MshC to discriminate between cysteine and serine, as proposed for CysRS (15). The pre-steady state rate of the first adenylation reaction with W227F exhibited a ~ 220 fold decrease compared to WT, confirming its role in this half reaction.
pH studies - implications to the chemical mechanism
The pH dependence of the kinetic parameters is consistent with the presence of ionizable group(s) involved in catalysis and substrate binding. The decreased enzyme stability at high pH values (>10) has prevented the examination of the kinetic parameters above pH 10. The pH dependence of kcat depends on a single ionizable group that must be unprotonated and could function as a general base in catalysis. The kcat/KATP pH profile is bell shaped, suggesting two ionizable groups must be deprotonated and protonated in catalysis. Since the release of pyrophosphate is reversible based on previous PIX studies (13), the catalytic base observed in kcat/KATP is likely the same group observed in the kcat profile. The group whose protonation affects kcat/KATP is likely a group that binds ATP. The pH-profile of kcat/KGlcN-Ins reports on the steps in the second half reaction, which is the nucleophilic attack on the cysteine adenylate (12). As shown in Figure 2C, the pH-profile of kcat/KGlcN-Ins reveals the involvement of one catalytic base. This catalytic base could be an enzyme residue that initiates the reaction by abstracting a proton from the protonated amine group of GlcN-Ins, or initially formed zwitterionic tetrahedral intermediate (Scheme 4, path a). An alternative mechanism involving the nonbridging Sp oxygen of the cysteinyl adenylate acting as the base (Scheme 4, path b) has also been proposed based on mechanistic investigations of other aminoacyl-tRNA synthetases (32, 33). This substrate-assisted mechanism was proposed for E. coli histidyl-tRNA synthetase as the aminoacyl transfer rate decreased 10,000 fold when using phosphorothioate-substituted ATP as substrate (32). Structural examination of histidyl-tRNA synthetase showed no other side chains with ionizating groups in the immediate vicinity of the α-carboxylate carbon, which supports the idea of nonbridging phosphate oxygen being the general base (32).
Scheme 4.

The involvement of a general acid in catalysis is unlikely both experimentally and mechanistically. In the adenylation reaction, the cysteinyl carboxylate makes a direct nucleophilic attack on the α-phosphate of ATP (Scheme 3), and the cysteinyl carboxylate group is likely fully ionized while pyrophosphate is an excellent leaving group for such a nucleophilic mechanism. This mechanism could also be assisted by a Mg2+-coordinated water molecule. The nucleophilic mechanism has been proposed for other aminoacyl-tRNA synthetases (33-38). Biochemical and structural studies of the E. coli glutaminyl-tRNA synthetase revealed no enzyme residues that act as acid or base groups, suggesting that adenylation occurs via a direct nucleophilic attack of the glutamine carboxylate on the ATP (33, 38). Structural studies of lysyl-tRNA synthetase from E. coli in complex with AMP-PCP, lysine, and the lysyl-adenylate intermediate also revealed no major conformational changes and no residues directly involved in catalysis other than those involved in stabilizing the transition state (36). This nucleophilic mechanism has also been proposed for tyrosyl- and histinyl-tRNA synthetases (32, 34, 35, 39, 40). The group exhibiting the pKa value of 10.1 observed in the kcat/KATP pH profile likely reflects a protonated group involved in ATP binding and orientation.
Scheme 3.

Studies of H55A
H55 resides on the HxGH signature motif, and it is likely that a hydrogen bond would form between H55 and the cysteinyl adenylate intermediate. A modeling study with tyrosyl-tRNA synthetase suggested that the first conserved histidine (H45) residue is hydrogen bonded with the γ-phosphate during the transition state of tyrosine activation (41). Indeed the role of H45 was confirmed by studies of the H45D mutant form of TyrRS, for which a dramatically decreased activation rate was observed (42). For MshC, the H55A mutant exhibited an ~ 40 fold decrease in kcat, and an ~ 120 fold decrease in kcat/Km value for ATP compared to WT MshC, suggesting that H55 plays a significant but less essential role in ATP binding than D251. The dissociation constant for CSA determined by fluorescence and the Ki value determined by inhibition studies increased significantly for the H55A compared to WT. Consistent with this role of H55 in cysteine adenylation, the rate of the half reaction decreased ~100 fold compared to WT enzyme. Intriguingly, H55A is the only mutant that exhibits a parallel initial velocity pattern with varying cysteine and ATP concentrations. This could be the result of a change in kinetic mechanism for the H55A mutant. The binding of ATP is significantly impaired in H55A, causing cysteine to bind prior to ATP in an ordered sequence. If cysteine binding is much faster than dissociation and ATP binding is at equilibrium, the observed “parallel” initial velocity pattern simply reflects the near zero value of Kia.
We propose that H55 assists in the nucleophilic attack on the α-phosphate of ATP during cysteine adenylation (Scheme 5). Evidence supporting this role comes from kinetic studies as well as the imidazole rescue effect with the H55A mutant. From the three dimensional structure of MshC, the interaction of protonated H55 could be with either the α- phosphate via a hydrogen bond (Scheme 5, path a), or with the α-carboxylate moiety of cysteine to maintain its ionization state (Scheme 5, path b). Additional characterization is required to differentiate between these two mechanisms.
Scheme 5.

ACKNOWLEDGMENT
We thank Dr. Gavin Painter and group (Gracefield Research Center, New Zealand) for the synthesis of GlcN-Ins.
This work was supported in part by a grant from the National Institute of Health ( AI33696 to J. S. B.), and a Fellowship from the Heiser Program for Research in Leprosy and Tuberculosis of The New York Community Trust to F. F.
ABBREVIATIONS
- MSH
mycothiol
- CSA
5'-O-[N-(L-cysteinyl)-sulfamonyl]adenosine
- CysRS
cysteinyl tRNA synthetase
- AaRS
aminoacyl-tRNA synthetase
- PIX
positional isotope exchance
- WT
wild type
References
- (1).Anderberg SJ, Newton GL, Fahey RC. Mycothiol biosynthesis and metabolism. Cellular levels of potential intermediates in the biosynthesis and degradation of mycothiol in Mycobacterium smegmatis. J. Biol. Chem. 1998;273:30391–30397. doi: 10.1074/jbc.273.46.30391. [DOI] [PubMed] [Google Scholar]
- (2).Bornemann C, Jardine MA, Spies HS, Steenkamp DJ. Biosynthesis of mycothiol: elucidation of the sequence of steps in Mycobacterium smegmatis. Biochem. J. 1997;325(Pt 3):623–629. doi: 10.1042/bj3250623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Newton GL, Fahey RC. Mycothiol biochemistry. Arch. Microbiol. 2002;178:388–94. doi: 10.1007/s00203-002-0469-4. [DOI] [PubMed] [Google Scholar]
- (4).Newton GL, Ta P, Bzymek KP, Fahey RC. Biochemistry of the initial steps of mycothiol biosynthesis. J. Biol. Chem. 2006;281:33910–33920. doi: 10.1074/jbc.M604724200. [DOI] [PubMed] [Google Scholar]
- (5).Newton GL, Av-Gay Y, Fahey RC. A novel mycothiol-dependent detoxification pathway in mycobacteria involving mycothiol S-conjugate amidase. Biochemistry. 2000;39:10739–10746. doi: 10.1021/bi000356n. [DOI] [PubMed] [Google Scholar]
- (6).Newton GL, Unson MD, Anderberg SJ, Aguilera JA, Oh NN, delCardayre SB, Av-Gay Y, Fahey RC. Characterization of Mycobacterium smegmatis mutants defective in 1-d-myo-inosityl-2-amino-2-deoxy-alpha-d-glucopyranoside and mycothiol biosynthesis. Biochem. Biophys. Res. Commun. 1999;255:239–244. doi: 10.1006/bbrc.1999.0156. [DOI] [PubMed] [Google Scholar]
- (7).Rawat M, Newton GL, Ko M, Martinez GJ, Fahey RC, Av-Gay Y. Mycothiol-deficient Mycobacterium smegmatis mutants are hypersensitive to alkylating agents, free radicals, and antibiotics. Antimicrob. Agents. Chemother. 2002;46:3348–3355. doi: 10.1128/AAC.46.11.3348-3355.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Buchmeier NA, Newton GL, Koledin T, Fahey RC. Association of mycothiol with protection of Mycobacterium tuberculosis from toxic oxidants and antibiotics. Mol. Microbiol. 2003;47:1723–1732. doi: 10.1046/j.1365-2958.2003.03416.x. [DOI] [PubMed] [Google Scholar]
- (9).Newton GL, Arnold K, Price MS, Sherrill C, Delcardayre SB, Aharonowitz Y, Cohen G, Davies J, Fahey RC, Davis C. Distribution of thiols in microorganisms: mycothiol is a major thiol in most actinomycetes. J Bacteriol. 1996;178:1990–1995. doi: 10.1128/jb.178.7.1990-1995.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Sareen D, Newton GL, Fahey RC, Buchmeier NA. Mycothiol is essential for growth of Mycobacterium tuberculosis Erdman. J. Bacteriol. 2003;185:6736–6740. doi: 10.1128/JB.185.22.6736-6740.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Sassetti CM, Boyd DH, Rubin EJ. Genes required for mycobacterial growth defined by high density mutagenesis. Mol Microbiol. 2003;48:77–84. doi: 10.1046/j.1365-2958.2003.03425.x. [DOI] [PubMed] [Google Scholar]
- (12).Fan F, Luxenburger A, Painter GF, Blanchard JS. Steady-state and pre-steady-state kinetic analysis of Mycobacterium smegmatis cysteine ligase (MshC) Biochemistry. 2007;46:11421–11429. doi: 10.1021/bi7011492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Williams L, Fan F, Blanchard JS, Raushel FM. Positional isotope exchange analysis of the Mycobacterium smegmatis cysteine ligase (MshC) Biochemistry. 2008;47:4843–4850. doi: 10.1021/bi800327u. [DOI] [PubMed] [Google Scholar]
- (14).Sareen D, Steffek M, Newton GL, Fahey RC. ATP-dependent L-cysteine:1D-myo-inosityl 2-amino-2-deoxy-alpha-D-glucopyranoside ligase, mycothiol biosynthesis enzyme MshC, is related to class I cysteinyl-tRNA synthetases. Biochemistry. 2002;41:6885–6890. doi: 10.1021/bi012212u. [DOI] [PubMed] [Google Scholar]
- (15).Newberry KJ, Hou YM, Perona JJ. Structural origins of amino acid selection without editing by cysteinyl-tRNA synthetase. Embo J. 2002;21:2778–2787. doi: 10.1093/emboj/21.11.2778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Cavarelli J, Delagoutte B, Eriani G, Gangloff J, Moras D. L-arginine recognition by yeast arginyl-tRNA synthetase. Embo J. 1998;17:5438–5448. doi: 10.1093/emboj/17.18.5438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Cusack S. Aminoacyl-tRNA synthetases. Curr Opin Struct Biol. 1997;7:881–889. doi: 10.1016/s0959-440x(97)80161-3. [DOI] [PubMed] [Google Scholar]
- (18).Cusack S, Yaremchuk A, Tukalo M. The 2 A crystal structure of leucyl-tRNA synthetase and its complex with a leucyl-adenylate analogue. Embo J. 2000;19:2351–2361. doi: 10.1093/emboj/19.10.2351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Fukai S, Nureki O, Sekine S, Shimada A, Tao J, Vassylyev DG, Yokoyama S. Structural basis for double-sieve discrimination of L-valine from L-isoleucine and L-threonine by the complex of tRNA(Val) and valyl-tRNA synthetase. Cell. 2000;103:793–803. doi: 10.1016/s0092-8674(00)00182-3. [DOI] [PubMed] [Google Scholar]
- (20).Fukai S, Nureki O, Sekine S, Shimada A, Vassylyev DG, Yokoyama S. Mechanism of molecular interactions for tRNA(Val) recognition by valyl-tRNA synthetase. Rna. 2003;9:100–111. doi: 10.1261/rna.2760703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Mechulam Y, Schmitt E, Maveyraud L, Zelwer C, Nureki O, Yokoyama S, Konno M, Blanquet S. Crystal structure of Escherichia coli methionyl-tRNA synthetase highlights species-specific features. J Mol Biol. 1999;294:1287–1297. doi: 10.1006/jmbi.1999.3339. [DOI] [PubMed] [Google Scholar]
- (22).Nureki O, Fukai S, Sekine S, Shimada A, Terada T, Nakama T, Shirouzu M, Vassylyev DG, Yokoyama S. Structural basis for amino acid and tRNA recognition by class I aminoacyl-tRNA synthetases. Cold Spring Harb Symp Quant Biol. 2001;66:167–173. doi: 10.1101/sqb.2001.66.167. [DOI] [PubMed] [Google Scholar]
- (23).Nureki O, Vassylyev DG, Katayanagi K, Shimizu T, Sekine S, Kigawa T, Miyazawa T, Yokoyama S, Morikawa K. Architectures of class-defining and specific domains of glutamyl-tRNA synthetase. Science. 1995;267:1958–1965. doi: 10.1126/science.7701318. [DOI] [PubMed] [Google Scholar]
- (24).Silvian LF, Wang J, Steitz TA. Insights into editing from an ile-tRNA synthetase structure with tRNAile and mupirocin. Science. 1999;285:1074–1077. [PubMed] [Google Scholar]
- (25).Sugiura I, Nureki O, Ugaji-Yoshikawa Y, Kuwabara S, Shimada A, Tateno M, Lorber B, Giege R, Moras D, Yokoyama S, Konno M. The 2.0 A crystal structure of Thermus thermophilus methionyl-tRNA synthetase reveals two RNA-binding modules. Structure. 2000;8:197–208. doi: 10.1016/s0969-2126(00)00095-2. [DOI] [PubMed] [Google Scholar]
- (26).Crepin T, Schmitt E, Mechulam Y, Sampson PB, Vaughan MD, Honek JF, Blanquet S. Use of analogues of methionine and methionyl adenylate to sample conformational changes during catalysis in Escherichia coli methionyl-tRNA synthetase. J Mol Biol. 2003;332:59–72. doi: 10.1016/s0022-2836(03)00917-3. [DOI] [PubMed] [Google Scholar]
- (27).von Delft F, Lewendon A, Dhanaraj V, Blundell TL, Abell C, Smith AG. The crystal structure of E. coli pantothenate synthetase confirms it as a member of the cytidylyltransferase superfamily. Structure. 2001;9:439–450. doi: 10.1016/s0969-2126(01)00604-9. [DOI] [PubMed] [Google Scholar]
- (28).Tremblay LW, Fan F, Vetting MW, Blanchard JS. The 1.6 Å Crystal Structure of Mycobacterium smegmatis MshC: The Penultimate Enzyme in the Mycothiol Biosynthetic Pathway. Biochemistry. 2008;47:11326–11335. doi: 10.1021/bi801708f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Zheng R, Blanchard JS. Steady-state and pre-steady-state kinetic analysis of Mycobacterium tuberculosis pantothenate synthetase. Biochemistry. 2001;40:12904–12912. doi: 10.1021/bi011522+. [DOI] [PubMed] [Google Scholar]
- (30).Cook PF, Cleland WW. Enzyme Kinetics and Mechanism. Taylor & Francis Group; New York: 2007. [Google Scholar]
- (31).Williams L, Zheng R, Blanchard JS, Raushel FM. Positional isotope exchange analysis of the pantothenate synthetase reaction. Biochemistry. 2003;42:5108–5013. doi: 10.1021/bi0340853. [DOI] [PubMed] [Google Scholar]
- (32).Guth E, Connolly SH, Bovee M, Francklyn CS. A substrate-assisted concerted mechanism for aminoacylation by a class II aminoacyl-tRNA synthetase. Biochemistry. 2005;44:3785–3794. doi: 10.1021/bi047923h. [DOI] [PubMed] [Google Scholar]
- (33).Perona JJ, Rould MA, Steitz TA. Structural basis for transfer RNA aminoacylation by Escherichia coli glutaminyl-tRNA synthetase. Biochemistry. 1993;32:8758–8771. doi: 10.1021/bi00085a006. [DOI] [PubMed] [Google Scholar]
- (34).Arnez JG, Augustine JG, Moras D, Francklyn CS. The first step of aminoacylation at the atomic level in histidyl-tRNA synthetase. Proc. Natl. Acad. Sci. U. S. A. 1997;94:7144–7149. doi: 10.1073/pnas.94.14.7144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Brick P, Bhat TN, Blow DM. Structure of tyrosyl-tRNA synthetase refined at 2.3 A resolution. Interaction of the enzyme with the tyrosyl adenylate intermediate. J. Mol. Biol. 1989;208:83–98. doi: 10.1016/0022-2836(89)90090-9. [DOI] [PubMed] [Google Scholar]
- (36).Desogus G, Todone F, Brick P, Onesti S. Active site of lysyl-tRNA synthetase: structural studies of the adenylation reaction. Biochemistry. 2000;39:8418–8425. doi: 10.1021/bi0006722. [DOI] [PubMed] [Google Scholar]
- (37).Leatherbarrow RJ, Fersht AR. Investigation of transition-state stabilization by residues histidine-45 and threonine-40 in the tyrosyl-tRNA synthetase. Biochemistry. 1987;26:8524–8528. doi: 10.1021/bi00400a005. [DOI] [PubMed] [Google Scholar]
- (38).Leatherbarrow RJ, Fersht AR, Winter G. Transition-state stabilization in the mechanism of tyrosyl-tRNA synthetase revealed by protein engineering. Proc. Natl. Acad. Sci. U. S. A. 1985;82:7840–7844. doi: 10.1073/pnas.82.23.7840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Fersht AR, Leatherbarrow RJ, Wells TN. Structure-activity relationships in engineered proteins: analysis of use of binding energy by linear free energy relationships. Biochemistry. 1987;26:6030–6038. doi: 10.1021/bi00393a013. [DOI] [PubMed] [Google Scholar]
- (40).Leatherbarrow RJ, Fersht AR. Protein engineering. Protein Eng. 1986;1:7–16. doi: 10.1093/protein/1.1.7. [DOI] [PubMed] [Google Scholar]
- (41).Leatherbarrow RJ, Fersht AR, Winter G. Transition-state stabilization in the mechanism of tyrosyl-tRNA synthetase revealed by protein engineering. Proc Natl Acad Sci U S A. 1985;82:7840–7844. doi: 10.1073/pnas.82.23.7840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Carter P, Bedouelle H, Winter G. Construction of heterodimer tyrosyl-tRNA synthetase shows tRNATyr interacts with both subunits. Proc Natl Acad Sci U S A. 1986;83:1189–1192. doi: 10.1073/pnas.83.5.1189. [DOI] [PMC free article] [PubMed] [Google Scholar]