

Abstract
Evidence shows that drug-drug interactions can occur at the level of drug transporters such as the organic anion transporting polypeptides (OATPs), a group of membrane solute carriers that mediate the sodium-independent transport of a wide range of amphipathic organic compounds. The polyspecific OATP1B1 is exclusively expressed at the basolateral membrane of hepatocytes and mediates uptake of amphipathic organic compounds from blood into hepatocytes. Nuclear receptors are ligand-activated transcription factors that play an important role in xenobiotic disposition and human diseases. Quite a few nuclear receptor ligands interact with transport proteins.
A high-resolution three-dimensional structure is critical to understand the polyspecificity of OATP1B1 to predict and prevent adverse drug-drug interactions. Unfortunately there are no crystal structures of OATPs/Oatps available to date. Therefore, in this study we attempted to elucidate the characteristics of the substrate binding site of OATP1B1 based on small molecules interacting with it. First, we identified inhibitors of the OATP1B1 model substrate estradiol-17β-glucuronide from about forty nuclear receptor ligands. Among them, GW1929, paclitaxel and troglitazone were strong inhibitors, while 5α-androstane, 5α-androstane-3β, 17β-diol-17-hexahydrobenzoate and estradiol-3-benzoate were weak inhibitors. Then, we selected 25 compounds and performed inhibition kinetic studies to identify competitive inhibitors and determine their Ki values which ranged from submicromolar to submillimolar. Finally, we performed CoMFA analysis on the identified competitive inhibitors. The CoMFA results indicate that the substrate binding site of OATP1B1 consists of a large hydrophobic middle part with basic residues at both ends that could be very important for substrate binding.
Keywords: OATP, CoMFA, drug-drug interaction, nuclear receptor
1. Introduction
Severe adverse drug-drug interactions represent an increasing risk for patients taking several drugs at the same time. There is convincing evidence that besides at the level of drug metabolizing enzymes, drug-drug interactions also occur at the level of drug transporters. The human liver is the major organ for the uptake, metabolism and excretion of numerous drugs and other xenobiotics. The first step in hepatic clearance of such chemicals is the uptake of these substances from blood into liver mediated by transporters located on the basolateral membrane of hepatocytes. Organic anion transporting polypeptides (humans: OATPs; rodents: Oatps) are a group of membrane solute carriers that mediate the sodium-independent transport of a wide range of amphipathic organic compounds including numerous drugs and other xenobiotics [1-3]. So far, eleven human OATPs have been identified [2,3]. The polyspecific OATP1B1 is exclusively expressed at the basolateral membrane of hepatocytes [4-7] and is important for drug uptake.
Nuclear receptors are important ligand-activated transcription factors that strongly affect xenobiotic disposition. Furthermore, they are important drug targets for human diseases like diabetes, obesity and cancer [8-11]. Nuclear receptors play important roles in the regulation of gene expression for metabolism, conjungation and transport of endogenous and exogenous compounds [12-14]. To exert their physiological effects, the ligands of nuclear receptors need to enter their target cells and transport proteins play a role during this process. For instance, the pregnane X receptor (PXR) ligand rifampicin [15,16] and the farnesoid X receptor (FXR) ligand cholic acid [17,18] are transported by OATP1B1 and 1B3; the thyroid hormones triiodothyronine (T3) and L-thyroxine (T4) are transported by OATP1 and OATP4 family members [4,19-23]. Besides being transported directly, interactions of nuclear receptor ligands with transporters could result in unexpected potentially adverse effects. Previous studies showed that the peroxisome proliferator-activated receptor (PPAR) ligand troglitazone caused intracellular accumulation of bile salts and subsequent liver damage due to its inhibition of the canalicular bile salt export pump (BSEP) [24], and that the glucocorticoid receptor ligand dexamethasone inhibited OATP1A2-mediated dehydroepiandrosterone sulfate transport [25]. We recently demonstrated that the PXR ligand clotrimazole inhibited OATP1B1-mediated estradiol-17β-glucuronide transport, but stimulated OATP1B3-mediated estradiol-17β-glucuronide transport [26].
Given that adverse drug-drug interactions can be a result of inhibition at the uptake transporter level, there is a critical need to understand the polyspecificity of OATP1B1 to predict and prevent such adverse effects. Due to the difficulty of isolating and crystallizing membrane proteins, researchers attempted to elucidate the characteristics of the substrate binding sites of OATPs/Oatps based on the information of interacting small molecules. Chang et al. [27] used computational pharmacophore modeling to illuminate the key features for substrate interaction with rat Oatp1a1 and human OATP1B1 transporters. Yarim et al. [28] applied a three-dimensional quantitative structure-activity relationship (3D-QSAR) technique to obtain structural information of the substrate binding site of Oatp1a5. In these studies, the substrates used to derive theoretical models might bind to different sites as experimental evidence shows that OATPs/Oatps [26,29,30] as well as other transporters [31,32] might have multiple substrate/ligand recognition sites. A computational model derived from compounds binding at the same site should be more reasonable and reliable. Therefore we decided to use competitive inhibitors of the OATP1B1 model substrate estradiol-17β-glucuronide which bind to the same site as the substrate to derive a model. From about forty nuclear receptor ligands we identified inhibitors of OATP1B1-mediated transport, carried out kinetic studies to identify the competitive inhibitors and calculated their Ki values. Finally, we performed comparative molecular field analysis (CoMFA) on the identified competitive inhibitors to elucidate possible binding mechanism between OATP1B1 and its ligands.
2. Materials and methods
2.1. Chemicals and reagents
Radiolabeled [3H]estradiol-17β-glucuronide (39.8 Ci/mmol) was purchased from PerkinElmer Life Sciences (Boston, MA). Nuclear receptor ligands were obtained from Sigma-Aldrich (St. Louis, MO) and Steraloids Inc. (Newport, RI). Cell culture reagents were from Invitrogen (Carlsbad, CA), fetal bovine serum from Hyclone (Logan, UT). The BCA protein assay kit was from Pierce (Rockford, IL).
2.2. Cell culture and uptake experiments
The Chinese Hamster Ovary (CHO) stable cell line expressing human OATP1B1 was described previously [26]. Wild-type CHO cells served as control. Cells were grown at 37 °C in a humidified 5% CO2 atmosphere in Dulbecco’s Modified Eagle Medium (DMEM), containing 1 g/l D-glucose, 2 mM L-glutamine, 25 mM Hepes buffer and 110 mg/l sodium pyruvate, supplemented with 10% FBS, 50 μg/ml L-proline, 100 U/ml penicillin and 100 μg/ml streptomycin without (for wild-type cells) and with (for OATP1B1-expressing cells) 500 μg/ml G418.
For uptake experiments CHO wild-type and OATP-expressing cells were plated at 40,000 cells/well on 24-well plates and 48 h later medium was replaced with medium containing 5 mM Na-butyrate to induce nonspecific gene expression [33]. After another 24 h in culture, cells were used for uptake experiments. Cells were washed three times with 1 ml of pre-warmed uptake buffer (116.4 mM NaCl, 5.3 mM KCl, 1 mM NaH2PO4, 0.8 mM MgSO4, 5.5 mM D-glucose and 20 mM Hepes, pH adjusted to 7.4 with Trizma base) and uptake was started by adding 200 μl of uptake buffer containing 0.3 to 0.6 μCi/ml of the radiolabeled substrate in the presence or absence of inhibitors. After 20 s, at previously determined initial linear rate conditions, uptake was stopped by removing the uptake solution and washing the cells four times with 1 ml of ice-cold uptake buffer. The cells were then solubilized with 500 μl of 1% Triton X-100 and 300 μl of the lysate were used for liquid scintillation counting. The protein concentration was determined from the rest using the BCA assay with bovine serum albumin as a standard.
2.3. Identification of competitive inhibitors and calculation of Ki values
First, inhibition screening experiments were performed with 1 μM estradiol-17β-glucuronide (containing 0.4 μCi/ml [3H]estradiol-17β-glucuronide) in the absence and presence of two different concentrations for each compound. To analyze whether the groups were different from the control, one-way ANOVA was performed followed by the Bonferroni t-test with SigmaStat 3.5 (Systat Software, Inc., San Jose, CA). The p value for statistical significance was set to be < 0.05. Inhibitors were selected based on their structures and inhibition effects and then kinetic studies were carried out using four different substrate concentrations in the absence or presence of two inhibitor concentrations that were selected based on the results of the first screening experiment. Inhibition models for each inhibitor were compared by corrected Akaike’s Information Criterion (AICc) and the Ki values of competitive inhibitors were determined by nonlinear regression fitting with the Enzyme Kinetics Module of SigmaPlot 9.01 (Systat Software, Inc., San Jose, CA).
2.4. Computational methods
2.4.1. Determination of substrate and inhibitor conformation and their structural alignment
All molecules were constructed using Sybyl7.1 [34] and the initial conformations were obtained by molecular mechanics optimization using the Tripos force field and Gasteiger-Hückel charges [35], with an energy gradient convergence criterion of 0.001 kcal/mol and a distance-dependent dielectric constant of 1. First, systematic conformational search was performed for the model substrate estradiol-17β-glucuronide. The conformation with lowest energy was selected and subjected to energy minimization until convergence. The resulting conformation was chosen as the binding conformation for estradiol-17β-glucuronide. The distance between the negative charge and the hydrophobic center was measured and was employed as a distance constraint during the conformational search of OATP1B1 inhibitors. After that, systematic conformational search was carried out on the structures of the 21 competitive inhibitors of estradiol-17β-glucuronide with the distance constraint mentioned above. The conformation with lowest energy for each inhibitor was chosen for molecular alignment.
For molecular alignment, estradiol-17β-glucuronide was used as a reference and the initial alignment was obtained by aligning the molecules according to their pharmacophoric centers. An initial CoMFA model was developed (method described in section 2.4.2) and outliers (leave-one-out cross-validated prediction error of greater than 0.5 log unit) were subjected to conformational adjustment by rotating the rotatable bonds. However, all these rotations were confined within 10 kcal/mol to its lowest energy. The obtained alignment was further iteratively refined by the CoMFA field-fit repositioning tool (all settings were left at their default value; no external perturbations or scalings were employed in the field specification; iteration terminated when subsequent field-fitting of outliers failed to further improve their cross-validated prediction errors).
2.4.2. CoMFA
Steric and electrostatic field energies were probed by an sp3 carbon atom and a +1 net charge atom, respectively. Steric and electrostatic interactions were calculated using a Tripos force field with a distance-dependent dielectric constant at all intersections in a regularly spaced (2 Å) grid. The minimum σ (column filtering) was set to 1.0 kcal/mol to improve the signal-to-noise ratio by omitting those lattice points whose energy variation was below this threshold. A cutoff of 30 kcal/mol was adopted, and the regression analysis was carried out using the partial least-squares (PLS) method with region focusing to improve the cross-validation correlation coefficient (q2). The final model was developed with the optimum number of components equal to that yielding the highest q2.
3. Results and discussion
3.1. Identification of inhibitors of the model substrate estradiol-17β-glucuronide for OATP1B1
Estradiol-17β-glucuronide is one of the typical and well characterized OATP1B1 substrates which are amphipathic organic compounds containing hydrophobic and negatively charged groups. To identify potential inhibitors of estradiol-17β-glucuronide, inhibition experiments were performed for 39 compounds including 37 nuclear receptor ligands, the HMG-CoA reductase inhibitor pravastatin and the anti-fungal agent fluconazole (Fig. 1). As shown in Fig. 1, estradiol-17β-glucuronide was strongly inhibited by PPAR ligands GW1929, WY-14643 and troglitazone, PXR ligands rifampicin and paclitaxel, and high concentration FXR ligand chenodeoxycholic acid. Most bile acids (cholic acid, deoxycholic acid, dehydrocholic acid, glycocholic acid, lithocholic acid and taurocholic acid) and fatty acids (9-cis retinoic acid, 13-cis retinoic acid and all-trans retinoic acid) showed moderate inhibition, while some highly hydrophobic compounds such as 5α-androstane, 5α-androstane-3β, 17β-diol-17-hexahydrobenzoate and estradiol-3-benzoate were very weak inhibitors.
Fig. 1.
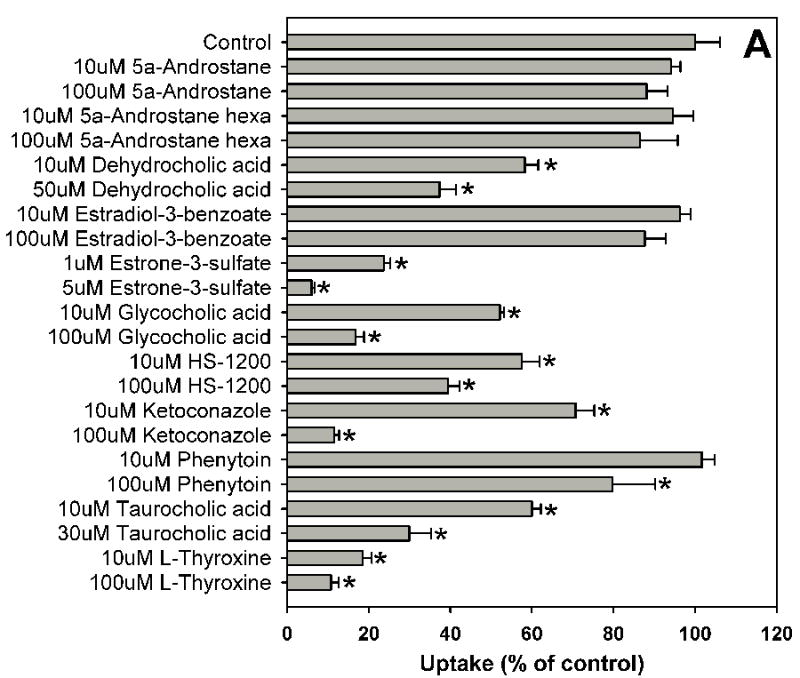



Inhibition of OATP1B1-mediated estradiol-17β-glucuronide uptake by 37 nuclear receptor ligands, the HMG-CoA reductase inhibitor pravastatin and the anti-fungal agent fluconazole. (A) Androgen receptor (AR), constitutive androstane receptor (CAR) and glucocorticoid receptor (GR) ligands. (B) Peroxisome proliferator-activated receptor (PPAR) ligands. (C) Pregnane X receptor (PXR) ligands. (D) Farnesoid X receptor (FXR), retinoic acid receptor (RAR) and retinoid X receptor (RXR) ligands together with pravastatin and fluconazole. Uptake of 1 μM estradiol-17β-glucuronide (containing 0.4 μCi/ml [3H]estradiol-17β-glucuronide) was measured at 37 °C for 20 s with OATP1B1-expressing and wild-type CHO cells in the absence or presence of inhibitors. Values obtained with wild-type CHO cells were subtracted from values obtained with OATP1B1-expressing CHO cells and are given as percent of the control. Values are given as the mean ± SD of triplicate determinations; * p < 0.05. Abbreviations: 5α-Androstane hexa: 5α-androstane-3β, 17β-diol 17-hexahydrobenzoate; HETE: 8(S)-hydroxy-(5Z,9E,11Z,14Z)-eicosatetraenoic acid.
3.2. Inhibition type and Ki determination of OATP1B1 inhibitors by kinetic studies
Based on the result of the inhibition experiment, we selected 25 compounds that share some structural similarity to the model substrate estradiol-17β-glucuronide. They all have hydrophobic and charge/hydrogen bonding groups and their inhibition activities cover a wide range from strong to weak inhibition. The chemical structures of these 25 compounds together with the model substrate estradiol-17β-glucuronide are shown in Fig. 2.
Fig. 2.

Chemical structures of the model substrate estradiol-17β-glucuronide and 25 inhibitors. These 25 inhibitors were selected based on their structures which contain a hydrophobic center and a negative charge/hydrogen bonding center as the model substrate, and their inhibition activities cover a wide range from strong to weak inhibition.
Inhibition kinetic studies were carried out for the selected 25 compounds. Their inhibition types were determined by AICc method and the Ki values of competitive inhibitors were calculated by nonlinear regression fitting with the Enzyme Kinetics Module of SigmaPlot. The results are listed in Table 1. The Ki values for 5α-androstane-3β, 17β-diol-17-hexahydrobenzoate and estradiol-3-benzoate could not be determined due to their low water solubility and extremely weak inhibition effect. Ketoconazole and HS-1200 are noncompetitive and uncompetitive inhibitors for estradiol-17β-glucuronide respectively, indicating that they bind at a different site than the substrate. The remaining 21 compounds are competitive inhibitors for the model substrate estradiol-17β-glucuronide and their Ki values range from submicromolar to submillimolar. Most strikingly, estrone-3-sulfate (Ki = 0.2 μM) and estradiol (Ki = 3.3 μM) were much stronger inhibitors than estradiol-3-benzoate (its inhibition effect being very weak, Fig. 1A), indicating that a polar or charge group at position C3 of the steroid ring (estradiol-17β-glucuronide, Fig. 2) is very important for their activity. Cholic acid has three hydroxyl groups at positions C3, C7 and C12; chenodeoxycholic acid and deoxycholic acid have two hydroxyl groups at positions C3, C7 and C3, C12, respectively, while lithocholic acid has only one hydroxyl group at position C3 (Fig. 2). The inhibition potency for these bile acids increased with decreasing number of hydroxyl groups which is reflected by Ki values for cholic acid of 25.0 μM, for chenodeoxycholic acid of 3.4 μM, for deoxycholic acid of 3.2 μM and for lithocholic acid of 1.2 μM. Dehydrocholic acid which contains three carbonyl groups instead of three hydroxyl groups at positions C3, C7 and C12 showed similar inhibition activity (Ki = 29.7 μM) as cholic acid. Therefore we conclude that polar groups at positions C7 and C12 are unfavorable for bile acids in the interaction with OATP1B1. The inhibition activity of glycocholic acid (Ki = 22.2 μM) was very close to that of cholic acid, suggesting that the chain length at position C17 is not very critical for their interaction with the protein. However, when the carboxyl group of glycocholic acid was replaced by a much stronger negatively charged sulfonate group, the activity increased two-fold (taurocholic acid, Ki = 11.4 μM).
Table 1.
Ki values of competitive inhibitors of OATP1B1-mediated estradiol-17β-glucuronide transport.
| No. | Compound | Ki (μM) |
|---|---|---|
| 1 | Estradiol-17β-glucuronide | 5.9 ± 1.2* |
| 2 | 5α-Androstane | 374 ± 123 |
| 3 | 5α-Androstane-3β, 17β-diol-17-hexahydrobenzoate | Undeterminable |
| 4 | Bezafibrate | 45.2 ± 5.9 |
| 5 | Chenodeoxycholic acid | 3.4 ± 0.5 |
| 6 | Cholic acid | 25.0 ± 3.3 |
| 7 | Clotrimazole | 7.6 ± 1.9 |
| 8 | Dehydrocholic acid | 29.7 ± 5.7 |
| 9 | Deoxycholic acid | 3.2 ± 0.5 |
| 10 | Estradiol | 3.3 ± 0.6 |
| 11 | Estradiol-3-benzoate | Undeterminable |
| 12 | Estrone-3-sulfate | 0.2 ± 0.02 |
| 13 | Fenofibrate | 105.2 ± 26.7 |
| 14 | Glycocholic acid | 22.2 ± 3.9 |
| 15 | GW1929 | 0.3 ± 0.05 |
| 16 | GW4064 | 32.4 ± 6.7 |
| 17 | HS-1200 | Uncompetitive |
| 18 | Ketoconazole | Noncompetitive |
| 19 | Lithocholic acid | 1.2 ± 0.2 |
| 20 | Mevinolin | 12.7 ± 1.9 |
| 21 | Mifepristone | 2.2 ± 0.3 |
| 22 | Pravastatin | 21.8 ± 3.6 |
| 23 | Taurocholic acid | 11.4 ± 3.2 |
| 24 | Thyroxine | 3.5 ± 0.7 |
| 25 | Troglitazone | 1.0 ± 0.18 |
| 26 | WY-14643 | 1.6 ± 0.11 |
For estradiol-17β-glucuronide the Km value is given. Values are given as the mean ± SD.
The inhibition activities of compounds are governed by their chemical structures. Some properties of the ligand binding site of a protein could be estimated based on the structures and activities of the ligands. Therefore, we went on to carry out a 3D-QSAR study for OATP1B1 with the 21 competitive inhibitors of the model substrate estradiol-17β-glucuronide.
3.3. CoMFA model
Seventeen of the 21 competitive inhibitors together with the substrate estradiol-17β-glucuronide were used as training set for constructing a CoMFA model. Fig. 3 shows the structural alignment of the 18 compounds in the training set. The remaining four compounds, namely clotrimazole, fenofibrate, mevinolin and troglitazone, were used as test set for model validation. They were weak (fenofibrate), moderate (clotrimazole and mevinolin) and strong (troglitazone) OATP1B1 inhibitors.
Fig. 3.

Structural alignment of the 18 compounds in training set. Hydrogen atoms were omitted for clarity.
PLS analysis was carried out and the result is summarized in Table 2. The cross-validated value q2 is 0.615 with an optimal number of components of 3. The noncross-validation PLS analysis with the optimal components of 3 revealed a conventional r2 of 0.966. The F value is 133.7 and the standard error is 0.164. The steric fields contribute 70.2% to the model’s information, while the electrostatic fields represent the other 29.8%. The correlation between the predicted activities and experimental activities for the 18 compounds in the training set is depicted in Fig. 4. These results demonstrate that the predicted activities are in good agreement with the experimental data and suggest that our CoMFA model is reliable.
Table 2.
Statistical indexes of CoMFA model based on seventeen competitive inhibitors and estradiol-17β-glucuronide.
| CoMFA | |
|---|---|
| PLS statistics | |
| q2 (leave-one-out) | 0.615 |
| q2 (five-fold) | 0.578 |
| r2 | 0.966 |
| S | 0.164 |
| F | 133.7 |
| Optimal components | 3 |
| Field contribution (%) | |
| Steric | 70.2 |
| Electrostatic | 29.8 |
Fig. 4.

Predicted activities versus experimental activities of 22 OATP1B1 compounds. ●, compounds in the training set; ▲, compounds in the test set.
The stability and predictive ability of the CoMFA model were further verified using the 4 test compounds clotrimazole, fenofibrate, mevinolin and troglitazone. The correlation between the predicted and experimental results for these 4 compounds is depicted in Fig. 4 (shown as ▲). The predictions were in good agreement with the experimental data with an r2 of 0.917.
Fig. 5 shows the CoMFA contour plots of steric and electrostatic fields. The model substrate estradiol-17β-glucuronide and the inhibitor cholic acid are displayed to aid in visualization. Detrimental and beneficial steric interactions are displayed in yellow and green contours, while blue and red contours illustrate the regions of desirable positive and negative electrostatic interactions. The steric contours are mainly located in the area of the hydrophobic steroid ring (Fig. 5A). It seems that a bulky group in the C7 and C15 positions of the four-fused ring of the steroids would enhance their inhibition activities. Due to the conjugation effect, the orientation of the aromatic ring A of estrogen is different from that of the saturated ring A of bile acids. In the area between these two rings, there is a sterically unfavorable region. We consider this is the main reason why estradiol-3-benzoate shows very weak inhibition effect because the bulky benzoyl group of estradiol-3-benzoate is supposed to be located in this sterically unfavorable region. The negative CoMFA region is mainly located in the area where most tested compounds contain a negatively charged group (Fig. 5B). Most strikingly, there is another negative region near the C3 position of the steroid ring which indicates that a negatively charged group in C3 position is favorable. This might explain why estrone-3-sulfate which contains a negatively charged sulfate group in the C3 position, showed a very high activity. On the contrary, QSAR analysis done by Yarim et al. [28] showed that the sulfate group of estrone-3-sulfate diminished its affinity for Oatp1a5 because the region where the sulfate group was located favored a positive charge. This might reflect the difference of the substrate binding pockets of these two transporters as OATP1B1 shows 100-fold higher affinity for estrone-3-sulfate (Km = 2.4 μM) [26] than Oatp1a5 (Km = 268 μM) [36].
Fig. 5.


CoMFA contour maps in combination with estradiol-17β-glucuronide (cyan) and cholic acid (white). Heteroatoms were rendered according to their default CPK colors (O = red). (A) Steric field distribution. Regions where increasing the molecular volume increases activity are in green and regions where increasing the volume decreases the activity are in yellow. (B) Electrostatic field distribution. Regions where increasing the positive charges increases activity are in blue and regions where increasing the negative charges increases the activity are in red.
3.4. Potential interaction mode between OATP1B1 and its ligands
Based on the structures and activities of the OATP1B1 inhibitors and the CoMFA result, we propose a model for their interactions with the protein as shown in Fig. 6. A typical inhibitor/substrate of OATP1B1 contains a hydrophobic center and a negatively charged center which form hydrophobic and electrostatic interactions with the protein, respectively. Another negatively charged or polar group on the hydrophobic center in the opposite direction of the negatively charged center is important for ligand binding probably by forming a salt bridge or a hydrogen bond with the protein. The hydrophobic center area is a large space where bulky substitutions might be beneficial for inhibition activity.
Fig. 6.

Schematic representation of potential interaction mode between OATP1B1 and its inhibitors.
4. Conclusion
In the present study, we determined the Ki values for 21 competitive inhibitors of the model substrate estradiol-17β-glucuronide for OATP1B1 ranging from submicromolar to submillimolar. Most of these compounds are nuclear receptor ligands and contain a hydrophobic center and a negatively charged center. CoMFA analysis was carried out on the substrate and competitive inhibitors to explore the structural requirement for inhibitors that interact with OATP1B1 at the same site as the model substrate estradiol-17β-glucuronide. CoMFA results show that besides the hydrophobic and negatively charged centers, another negatively charged or polar group at the C3 position is very important for strong inhibition. This suggests that both ends of this substrate binding site of OATP1B1 have positively charged residues. The middle of the binding pocket is relatively large as it is a sterically favorable region. The obtained information could be helpful to better understand the characteristics of the substrate binding site of OATP1B1 and to predict novel inhibitors and thus potential new substrates.
Acknowledgments
This work was supported by National Institute of Health grants RR021940, GM077336 and the KUMC Biomedical Research Training Program 2007-2008.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Hagenbuch B, Meier PJ. Organic anion transporting polypeptides of the OATP/ SLC21 family: phylogenetic classification as OATP/ SLCO superfamily, new nomenclature and molecular/functional properties. Pflugers Arch. 2004;447:653–65. doi: 10.1007/s00424-003-1168-y. [DOI] [PubMed] [Google Scholar]
- 2.Hagenbuch B, Meier PJ. The superfamily of organic anion transporting polypeptides. Biochim Biophys Acta. 2003;1609:1–18. doi: 10.1016/s0005-2736(02)00633-8. [DOI] [PubMed] [Google Scholar]
- 3.König J, Seithel A, Gradhand U, Fromm MF. Pharmacogenomics of human OATP transporters. Naunyn Schmiedebergs Arch Pharmacol. 2006;372:432–43. doi: 10.1007/s00210-006-0040-y. [DOI] [PubMed] [Google Scholar]
- 4.Abe T, Kakyo M, Tokui T, Nakagomi R, Nishio T, Nakai D, et al. Identification of a novel gene family encoding human liver-specific organic anion transporter LST-1. J Biol Chem. 1999;274:17159–63. doi: 10.1074/jbc.274.24.17159. [DOI] [PubMed] [Google Scholar]
- 5.Abe T, Unno M, Onogawa T, Tokui T, Kondo TN, Nakagomi R, et al. LST-2, a human liver-specific organic anion transporter, determines methotrexate sensitivity in gastrointestinal cancers. Gastroenterology. 2001;120:1689–99. doi: 10.1053/gast.2001.24804. [DOI] [PubMed] [Google Scholar]
- 6.Hsiang B, Zhu Y, Wang Z, Wu Y, Sasseville V, Yang WP, et al. A novel human hepatic organic anion transporting polypeptide (OATP2). Identification of a liver-specific human organic anion transporting polypeptide and identification of rat and human hydroxymethylglutaryl-CoA reductase inhibitor transporters. J Biol Chem. 1999;274:37161–8. doi: 10.1074/jbc.274.52.37161. [DOI] [PubMed] [Google Scholar]
- 7.König J, Cui Y, Nies AT, Keppler D. A novel human organic anion transporting polypeptide localized to the basolateral hepatocyte membrane. Am J Physiol. 2000;278:G156–G64. doi: 10.1152/ajpgi.2000.278.1.G156. [DOI] [PubMed] [Google Scholar]
- 8.Robinson-Rechavi M, Escriva Garcia H, Laudet V. The nuclear receptor superfamily. J Cell Sci. 2003;116:585–6. doi: 10.1242/jcs.00247. [DOI] [PubMed] [Google Scholar]
- 9.Giguere V. Orphan nuclear receptors: from gene to function. Endocrin Rev. 1999;20:689–725. doi: 10.1210/edrv.20.5.0378. [DOI] [PubMed] [Google Scholar]
- 10.Wang H, LeCluyse EL. Role of orphan nuclear receptors in the regulation of drug-metabolising enzymes. Clin Pharmacokinet. 2003;42:1331–57. doi: 10.2165/00003088-200342150-00003. [DOI] [PubMed] [Google Scholar]
- 11.Xie W, Uppal H, Saini SPS, Mu Y, Little JM, Radominska-Pandya A, et al. Orphan nuclear receptor-mediated xenobiotic regulation in drug metabolism. Drug Discov Today. 2004;9:442–9. doi: 10.1016/S1359-6446(04)03061-2. [DOI] [PubMed] [Google Scholar]
- 12.Chawla A, Repa JJ, Evans RM, Mangelsdorf DJ. Nuclear receptors and lipid physiology: opening the X-files. Science. 2001;294:1866–70. doi: 10.1126/science.294.5548.1866. [DOI] [PubMed] [Google Scholar]
- 13.Honkakoski P, Sueyoshi T, Negishi M. Drug-activated nuclear receptors CAR and PXR. Ann Med. 2003;35:172–82. doi: 10.1080/07853890310008224. [DOI] [PubMed] [Google Scholar]
- 14.Teng S, Piquette-Miller M. Regulation of transporters by nuclear hormone receptors: implications during inflammation. Mol Pharm. 2008;5:67–76. doi: 10.1021/mp700102q. [DOI] [PubMed] [Google Scholar]
- 15.Vavricka SR, Van Montfoort J, Ha HR, Meier PJ, Fattinger K. Interactions of rifamycin SV and rifampicin with organic anion uptake systems of human liver. Hepatology. 2002;36:164–72. doi: 10.1053/jhep.2002.34133. [DOI] [PubMed] [Google Scholar]
- 16.Tirona RG, Leake BF, Wolkoff AW, Kim RB. Human organic anion transporting polypeptide-C (SLC21A6) is a major determinant of rifampin-mediated pregnane X receptor activation. J Pharmacol Exp Ther. 2003;304:223–8. doi: 10.1124/jpet.102.043026. [DOI] [PubMed] [Google Scholar]
- 17.Cui Y, Konig J, Leier I, Buchholz U, Keppler D. Hepatic uptake of bilirubin and its conjugates by the human organic anion transporter SLC21A6. J Biol Chem. 2001;276:9626–30. doi: 10.1074/jbc.M004968200. [DOI] [PubMed] [Google Scholar]
- 18.Briz O, Romero MR, Martinez-Becerra P, Macias RI, Perez MJ, Jimenez F, et al. OATP8/1B3-mediated cotransport of bile acids and glutathione: an export pathway for organic anions from hepatocytes? J Biol Chem. 2006;281:30326–35. doi: 10.1074/jbc.M602048200. [DOI] [PubMed] [Google Scholar]
- 19.Fujiwara K, Adachi H, Nishio T, Unno M, Tokui T, Okabe M, et al. Identification of thyroid hormone transporters in humans: different molecules are involved in a tissue-specific manner. Endocrinology. 2001;142:2005–12. doi: 10.1210/endo.142.5.8115. [DOI] [PubMed] [Google Scholar]
- 20.Kullak-Ublick GA, Ismair MG, Stieger B, Landmann L, Huber R, Pizzagalli F, et al. Organic anion-transporting polypeptide B (OATP-B) and its functional comparison with three other OATPs of human liver. Gastroenterology. 2001;120:525–33. doi: 10.1053/gast.2001.21176. [DOI] [PubMed] [Google Scholar]
- 21.Pizzagalli F, Hagenbuch B, Stieger B, Klenk U, Folkers G, Meier PJ. Identification of a novel human organic anion transporting polypeptide as a high affinity thyroxine transporter. Mol Endocrinol. 2002;16:2283–96. doi: 10.1210/me.2001-0309. [DOI] [PubMed] [Google Scholar]
- 22.Mikkaichi T, Suzuki T, Onogawa T, Tanemoto M, Mizutamari H, Okada M, et al. Isolation and characterization of a digoxin transporter and its rat homologue expressed in the kidney. Proc Natl Acad Sci U S A. 2004;101:3569–74. doi: 10.1073/pnas.0304987101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hagenbuch B. Cellular entry of thyroid hormones by organic anion transporting polypeptides. Best Pract Res Clin Endocrinol Metab. 2007;21:209–21. doi: 10.1016/j.beem.2007.03.004. [DOI] [PubMed] [Google Scholar]
- 24.Funk C, Ponelle C, Scheuermann G, Pantze M. Cholestatic potential of troglitazone as a possible factor contributing to troglitazone-induced hepatotoxicity: in vivo and in vitro interaction at the canalicular bile salt export pump (Bsep) in the rat. Mol Pharmacol. 2001;59:627–35. [PubMed] [Google Scholar]
- 25.Kullak-Ublick GA, Fisch T, Oswald M, Hagenbuch B, Meier PJ, Beuers U, et al. Dehydroepiandrosterone sulfate (DHEAS): identification of a carrier protein in human liver and brain. FEBS Lett. 1998;424:173–6. doi: 10.1016/s0014-5793(98)00168-9. [DOI] [PubMed] [Google Scholar]
- 26.Gui C, Miao Y, Thompson L, Wahlgren B, Mock M, Stieger B, et al. Effect of pregnane X receptor ligands on transport mediated by human OATP1B1 and OATP1B3. Eur J Pharmacol. 2008;584:57–65. doi: 10.1016/j.ejphar.2008.01.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chang C, Pang KS, Swaan PW, Ekins S. Comparative Pharmacophore Modeling of Organic Anion Transporting Polypeptides: A Meta-analysis of Rat Oatp1a1 and Human OATP1B1. J Pharmacol Exp Ther. 2005 doi: 10.1124/jpet.104.082370. [DOI] [PubMed] [Google Scholar]
- 28.Yarim M, Moro S, Huber R, Meier PJ, Kaseda C, Kashima T, et al. Application of QSAR analysis to organic anion transporting polypeptide 1a5 (Oatp1a5) substrates. Bioorg Med Chem. 2005;13:463–71. doi: 10.1016/j.bmc.2004.10.009. [DOI] [PubMed] [Google Scholar]
- 29.Sugiyama D, Kusuhara H, Shitara Y, Abe T, Sugiyama Y. Effect of 17β-estradiol-D-17β-glucuronide on the rat organic anion transporting polypeptide 2-mediated transport differs depending on substrates. Drug Metab Dispos. 2002;30:220–3. doi: 10.1124/dmd.30.2.220. [DOI] [PubMed] [Google Scholar]
- 30.Noe J, Portmann R, Brun ME, Funk C. Substrate-dependent drug-drug interactions between gemfibrozil, fluvastatin and other organic anion-transporting peptide (OATP) substrates on OATP1B1, OATP2B1, and OATP1B3. Drug Metab Dispos. 2007;35:1308–14. doi: 10.1124/dmd.106.012930. [DOI] [PubMed] [Google Scholar]
- 31.Zelcer N, Huisman MT, Reid G, Wielinga P, Breedveld P, Kuil A, et al. Evidence for two interacting ligand binding sites in human multidrug resistance protein 2 (ATP binding cassette C2) J Biol Chem. 2003;278:23538–44. doi: 10.1074/jbc.M303504200. [DOI] [PubMed] [Google Scholar]
- 32.Van Aubel RA, Smeets PH, van den Heuvel JJ, Russel FG. Human organic anion transporter MRP4 (ABCC4) is an efflux pump for the purine end metabolite urate with multiple allosteric substrate binding sites. Am J Physiol Renal Physiol. 2005;288:F327–33. doi: 10.1152/ajprenal.00133.2004. [DOI] [PubMed] [Google Scholar]
- 33.Palermo DP, DeGraaf ME, Marotti DR, Rehberg E, Post LE. Production of analytical quantities of recombinant proteins in chinese hamster ovary cells using sodium butyrate to elevate gene expression. J Biotechnol. 1991;19:35–48. doi: 10.1016/0168-1656(91)90073-5. [DOI] [PubMed] [Google Scholar]
- 34.Tripos. Sybyl [molecular modeling package], Version 7.1. St Louis (MO): [Google Scholar]
- 35.Gasteiger J, Marsili M. Iterative partial equalization of orbital electronegativity: a rapid access to atomic charges. Tetrahedron. 1980:3219–28. [Google Scholar]
- 36.Cattori V, van Montfoort JE, Stieger B, Landmann L, Meijer DK, Winterhalter KH, et al. Localization of organic anion transporting polypeptide 4 (Oatp4) in rat liver and comparison of its substrate specificity with Oatp1, Oatp2 and Oatp3. Pflugers Arch. 2001;443:188–95. doi: 10.1007/s004240100697. [DOI] [PubMed] [Google Scholar]