

Abstract
Depression is one of the most prevalent and debilitating of the psychiatric disorders. Studies have shown that cognitive therapy is as efficacious as antidepressant medications at treating depression, and it seems to reduce the risk of relapse even after its discontinuation. Cognitive therapy and antidepressant medication probably engage some similar neural mechanisms, as well as mechanisms that are distinctive to each. A precise specification of these mechanisms might one day be used to guide treatment selection and improve outcomes.
Major depressive disorder is a serious illness that in the United States alone is estimated to affect 13 to 14 million adults each year. The lifetime prevalence rate (16%) is even higher, with an estimated 32 to 35 million US residents expected to develop the disorder at some point during their lifetime. Not only can depression be debilitating on its own, it also has a high rate of co-morbidity with other mental illnesses. Indeed, nearly three-quarters of people who meet the criteria for depression at some point during their lifetime will also meet the criteria for another psychiatric disorder: approximately three-fifths will be co-morbid for one of the anxiety disorders, one-quarter for substance use disorders and one-third for impulse-control disorders1.
Substantial impairment in social and occupational functioning is also frequently observed in depressed individuals2. Not surprisingly, the economic burden of depression is enormous. Workplace-related costs in the United States have been estimated at more than fifty billion dollars annually.3 Although most episodes of depression resolve without treatment, over one-quarter of all patients suffer from chronic depression, and the vast majority of those who do recover from a depressive episode will experience recurrences. Owing to its prevalence, its chronic and recurrent nature and its frequent co-morbity with other chronic illnesses - both as a contributing factor and as a consequence – depression is considered to be the condition that is most responsible for health decrements worldwide. It is therefore a global health priority to understand, prevent and treat depression.4
The nature of depression
Depression can be defined as both a syndrome and a disorder. As a syndrome it involves episodes of sadness, loss of interest, pessimism, negative beliefs about the self, decreased motivation, behavioural passivity, changes in sleep, appetite and sexual interest, and suicidal thoughts and impulses. As a disorder it comes in two forms. The unipolar type, which affects approximately 10% of men and 20% of women, includes only episodes of depression.1 Heritability estimates for this unipolar type have ranged from approximately 25% in less-severe samples up to 50% in more-severe samples.5
In the bipolar form, which is commonly known as manic depression, patients also (or exclusively) experience episodes of mania or hypomania that are in many ways the opposite of depression. Manic episodes are marked by euphoria or irritability, sleeplessness, grandiosity, recklessness and uncontrollable impulses that can lead to buying sprees and sexual promiscuity.6 This Perspective concentrates on unipolar depression, as the phenomenology differs from bipolar depression, as do the medication and psychological treatments. In addition, more is known about the neural mechanisms that underlie unipolar disorder and its treatment, and it is unclear how these mechanisms are related to those of bipolar disorder.
Current treatment practices
The goal of acute treatment for unipolar depression with antidepressant medication (ADM), the current standard of treatment, is generally to provide symptom relief. In this context, response is defined as a noticeable improvement, and remission is defined by the near absence of symptoms.7 Even when remission is achieved, patients retain a high risk of relapse (that is, the return of symptoms within the same episode). For this reason, patients who have been treated to the point of remission with ADM are advised to continue treatment for at least six months (the period of greatest risk of relapse). Patients whose remission lasts for six months are said to have recovered. However, those who recover from depression with ADM but then discontinue the treatment have a risk of experiencing a new episode of depression (recurrence) that is three to five times the risk of a member of the general population experiencing a first episode of depression. Consequently, practice guidelines emphasize the benefits of maintaining recovered patients on ADMs indefinitely, especially for patients who have a history of recurrent (or chronic) depression.
In addition to ADM, other treatments are effective in alleviating depression. These include cognitive therapy (CT) and other forms of psychotherapy, such as interpersonal therapy,8 electroconvulsive therapy9 and electrical stimulation of the vagus nerve.10 Newer interventions, such as behavioural activation,11 neurofeedback,12,13 and chronic stimulation of the subgenual cingulate region (Brodmann area 25)14 have also shown promise in recent clinical trials. The fact that there are various means through which depression can be alleviated suggests that different treatments might engage common neural mechanisms, although they might affect a final common pathway through widely differing processes. This Perspective focuses on a comparison of CT and ADM because, among the common effective treatments, these two treatments have been the focus of the most intensive research efforts, both with regard to the outcomes that they produce and the mechanisms that might explain their effects. We argue that comparative investigations of CT and ADM promise to further our understanding of the nature of depression, as these treatments use very different routes of administration and because CT, but not ADM, seems to alter the subsequent course of depression.
Antidepressant medication
ADMs fall into a few main classes, including older types of medication such as monoamine oxidase inhibitors (MAOIs) and tricyclic antidepressants (TCAs). More recently developed treatments include agents that block the reuptake of serotonin, noradrenaline or dopamine – their names refer to the neurotransmitter systems that they are believed to affect the most. The selective serotonin-reuptake inhibitors (SSRIs) have been the most widely prescribed ADMs to date, although the newer serotonin/noradrenaline-reuptake inhibitors (SNRIs) are also now coming into widespread use.
The efficacy of ADM has been established in literally thousands of placebo-controlled clinical trials.15,16 Approximately one-half of all patients will respond to any given ADM irrespective of its class, and many of the other half will respond to another ADM or to a combination of ADMs.17 However, ADMs seem to be symptom-suppressive rather than curative.18 That is, although ADM is effective in the treatment of the acute depressive episode and is preventive so long as its use is maintained, no published findings to date suggest that ADMs reduce future risk of depressive episodes once their use is terminated.19 This suggests that causal mechanisms of depression are unchanged by ADM treatment, and so patients are left with an elevated risk for subsequent episodes if they stop taking their medications. Because the risk is not absolute – not all patients experience a return of depression after they stop taking ADM – it is likely that these causal mechanisms take the form of stable diatheses that interact with negative life events to determine the onset of subsequent episodes. These causal diatheses might be represented by neural processes, and research that aims to specify these processes is ongoing.
Research on humans and animals indicates that ADMs alter the regulatory processes of monoamine systems, especially the serotonin, noradrenaline and dopamine systems, in such a way as to reverse pathological patterns of functioning that arise during the depressive episode. These agents affect neurotransmitter degradation (in the case of the MAOIs) or reuptake mechanisms (in the case of the TCAs, SSRIs, and SNRIs), in areas of the limbic system that subserve emotional responses, appetite, sexual interest and sleep, all of which are notably disordered in depression. Changes in neurotransmitter availability are known to trigger various other changes in the brain, such as decreases in amygdala activity20 and decreases in neurogenesis in the hippocampus.21 They also affect higher cortical processes, through direct projections as well as indirect connections. However, the long-term effects of these changes on cognition and behaviour have not been examined. Moreover, the lack of evidence that in humans ADM treatment reduces the risk of developing subsequent depressive episodes suggests that the changes in brain function and structure that are thought to result from ADM treatment do not confer protection against the return of symptoms once ADM is discontinued.
Cognitive therapy
CT is the best-known and most widely tested of a larger family of cognitive behavioural interventions. Like ADM, it is a safe and efficacious treatment for acute episodes of major depressive disorder. CT is based on the premise that inaccurate beliefs and maladaptive information processing (forming the bases for repetitive negative thinking) have a causal role in depression. This ‘cognitive model’ posits that when maladaptive thinking is corrected, both acute distress and the risk for subsequent symptom return will be reduced. Contrasting with the lack of evidence of enduring effects of ADMs is the substantiation of claims that CT provides protection against relapse and, possibly, recurrence.18
Aaron T. Beck developed CT for depression in the 1960s. By 1979 he and his colleagues had codified the treatment in a detailed manual.22 In CT, therapists aim to help patients acquire the abilities to, first, identify the thoughts and images that accompany and precede the experience of upsetting emotions; second, distance themselves from the beliefs embedded in, or implied by, these thoughts and images; third, question, often through experiments, the validity of their beliefs (for example., What is the evidence for this belief? Are there alternative explanations for the event that triggered the beliefs?); and fourth, identify the themes in the content of the thoughts and images that occur across a range of situations.
Outcomes of CT versus ADM
Findings from initial studies that compared CT with ADM, which were conducted in the 1970s and 1980s, suggested that CT is as efficacious as ADM in reducing acute distress, and that its effects are more enduring.23-30 However, in a major placebo-controlled trial of ADM versus CT, Elkin et al. reported that, for patients who began the trial with severe symptoms, ADM was more efficacious than CT, and CT was not demonstrably more efficacious than placebo.31 Moreover, there was little evidence for an enduring effect of CT.32 However, there were suggestions in these data that the outcome of CT varied across research sites as a function of therapist experience, which might account for the apparent discrepancy between these findings and those that have been obtained in other randomized trials of CT and ADM.33 Indeed, a mega-analysis of data pooled across the major CT-ADM comparisons showed that the two types of treatment are comparably effective in severely depressed patients.34
None of the above-cited studies was designed to test these treatments specifically in severely depressed patients. Even in the Elkin et al. study, the sample sizes of severely depressed patients were under 30 per condition. In a more recent CT -ADM placebo-controlled comparison, 240 severely depressed patients were randomized to ADM (n=120), CT (n=60) or a (pill) placebo control (n=60) treatment.35 ADM involved paroxetine (an SSRI), augmented with lithium or desipramine as needed. The providers of both the ADM and the CT were experienced practitioners who received feedback and supervision throughout the period of the study. As shown in FIG. 1, both CT and ADM outperformed placebo at the 8-week assessment point (after which the placebo condition was discontinued). At the end of the 16-week treatment phase of the study, there were no differences in outcome between ADM and CT, with 58% of patients in both treatment groups meeting the criteria for ‘response’. Curiously, there was no indication that the two treatments affected different symptom clusters of depression: patients treated with either ADM or CT showed comparable rates of change of both cognitive and vegetative symptoms of depression.36 This contrasts with results from studies that compared ADM with other psychosocial interventions. For example, one study reported that ADM, when compared with interpersonal psychotherapy, produced a more rapid resolution of vegetative symptoms such as insomnia, despite the fact that both treatments caused comparable levels of overall improvement in depression-symptom severity.37
Figure 1. Cognitive therapy and antidepressant medication have comparable short-term effects.

This graph shows the response of outpatients who had moderate to severe depression to cognitive therapy (CT), antidepressant medication (ADM) or placebo (PLA). Patients who were assigned to either ADM or to CT showed a significantly higher response rate after 8 weeks of treatment than those who were assigned to PLA. After 16 of treatment weeks the percentages of patients who responded to ADM and CT were almost identical.35 Figure modified, with permission, from REF #35.
In the continuation phase of the recent CT versus ADM study, patients who responded to 16 weeks of ADM were randomly assigned to either continue the treatment or change to a (pill) placebo condition.38 Patients who responded to 16 weeks of CT were withdrawn from treatment and allowed no more than three booster sessions (never more than one per month) during the first year of the follow-up period. As shown in FIG. 2, 76% of the ADM responders relapsed following medication withdrawal, compared with only 31% of the patients who had been treated with CT.38 Patients who continued ADM also fared better than patients who were assigned to the placebo treatment, with a relapse rate of 47% (which did not differ significantly from the 31% relapse rate in the CT group). After the continuation phase had ended, the patients who had not relapsed while on ADM were withdrawn from medication. Of these patients, 54% experienced a recurrence (the onset of a new depressive episode), compared with only 17% of the patients who had previously been given CT.
Figure 2.

Less relapse after cognitive therapy compared to antidepressant medication. The second phase of the parent study35 followed patients who had responded to antidepressant medication (ADM) or to cognitive therapy (CT).38 Patients who responded to ADM were randomly assigned to either continue ADM treatment for one year (beige and red lines), or to change to placebo treatment for one year (green line). Patients who responded to CT were allowed three sessions of CT during the 1-year continuation period. In the follow-up period, none of the patients received any treatment. The figure shows that prior treatment with CT protected against relapse of depression at least as well as the continued provision of ADM, and better than ADM treatment that was subsequently discontinued. Note that the patient group that was given ADM in the continuation year contained a number of patients who did not adhere to the medication regimen. The red line indicates the response of the ADM-continuation group including these non-compliant patients, whereas the beige line shows the response of the patients in this group after the non-compliant patients had been removed from the analysis. Figure modified, with permission, from REF #38, Figure 1 within that paper – need to obtain permission from the Archives of General Psychiatry.
These findings, which are consistent with the pattern observed in previous studies, indicate that CT has an enduring effect that is not found with ADM. This suggests that, whereas the acute responses to CT and ADM might be due to changes in similar mechanisms (consistent with the similar rates of change of different symptom clusters resulting from the two types of treatment), CT can be assumed to also produce changes that ADM does not. Attempts to determine whether the two types of treatment affect different symptom domains, which could acount for the difference in their enduring effects, have not thus far succeeded. It might therefore be useful to consider differences in more-subtle underlying mechanisms of change that are not apparent at the symptom level.
Brain mechanisms of cognitive aspects of depression
For two reasons, neuroscience provides an excellent platform for studying the mechanisms that underlie depression and the effects of ADM and CT treatment. First, understanding how alterations in brain activity are associated with the initial response to treatment and subsequent symptom return could contribute to a mechanistic explanation for the treatment patterns that have been observed (and which were described above). Second, initial data are already suggestive, in that changes in brain activity that have been associated with depression have also been observed in individuals who have recovered from depression but are at risk for recurrence.39,40 These data suggest that assessments of brain function could be used to predict the onset and recurrence of depressive episodes.
The following sections thus introduce one account of how changes in activity in brain areas that are thought to be involved in depression might interact, and also describe some implications for an understanding of the short-term similarities and the long-term differences between the effects of CT and ADM. What follows is not meant to be a comprehensive summary of the many investigations that have contributed to our understanding of the nature of brain function in depression. Rather, as the direct aim of CT is to bring about changes in the way that patients process emotion-relevant information, with the assumption that the behavioural and vegetative features of depression will remit as a consequence of the cognitive changes, we emphasize evidence from studies that made use of emotional stimuli or that focused on the brain circuits that have been found, experimentally, to be involved in emotion regulation. In particular, aspects of emotional information processing that are often observed in patients with depression, such as repetitive negative thinking, have been associated with increased limbic activity and decreased activity in the prefrontal cortex. As described below, converging evidence for these associations has been obtained with various neuroimaging methods, predominantly positron-emission tomography (PET) and functional MRI (fMRI).
Increased or sustained amygdala activity
The amygdala, a limbic region that is important for processing the emotional aspects of information and generating emotional reactions,41 projects to cortical and subcortical regions, such as the hippocampus, which subserves the creation and maintenance of emotional associations in memory.42 Through these connections, increases in amygdala activity could foster sustained repetitive negative associations. Alterations in amygdala volume and activity have been observed in samples of depressed patients,43 with the most-consistent evidence suggesting increased and relatively unmodulated amygdala activity.20,44-46 Such abnormalities in neural processes and associated cognitive processes are understood to be central to the expression of depressive symptoms,47 although anxiety disorders have also been linked to amygdala dysfunction.48
Decreased prefrontal control
Given that the prefrontal cortex exerts an inhibitory effect on limbic regions such as the amygdala,49-54 sustained emotional reactivity might result from decreased prefrontal executive control. The existence of inhibitory connections from the orbital prefrontal cortex (OFC) and ventromedial regions, such as the rostral and subgenual cingulate gyri, to the amygdala,55,56 suggests that these regions might exert a ‘damping’ effect on the amygdala, and therefore on emotional reactivity. In particular, alterations in the activity of the rostral and subgenual cingulate regions (Brodmann areas 24 and 25), which have direct connections to the amygdala, have been consistently implicated in depression.53,57,58 However, it is likely that these regions are themselves enervated by ‘upstream’ processes in regions that are directly involved in executive control. The dorsolateral prefrontal cortex (DLPFC), in particular, is implicated in activating brain mechanisms that are necessary for completing tasks,59 yielding decreased activity in other, non-task-relevant regions. The DLPFC might specifically inhibit the amygdala through cortico-cortical connections to the OFC.60 Consequently, depressed individuals might suffer from decreased control of amygdala activity as a result of deficits in early automatic prefrontal mechanisms and in the higher-level areas that have been implicated in executive control, such as the DLPFC.44,50,61,62 An important result of this disinibition is continued increased amygdala activity, potenitally facilitating prolonged emotional reactivity. Inhibitory connections from the amygdala back to the prefrontal cortex could further contribute to these deficits in the regulation of emotion.
In support of the hypothesis of decreased prefrontal control in depression, findings from both resting-state PET imaging and task-related fMRI studies suggest that depressed individuals have decreased prefrontal activity compared with healthy individuals.46,52,63-65 Depressed individuals also perform poorly on tasks that assess executive control and working memory,66,67 particularly those that engage the DLPFC.68,69 Impairment in these functions in depressed individuals is especially evident under conditions that require sustained activation of executive resources. For example, in a study in which the participants' task was to judge whether degraded pictures of digits were zeroes, depressed individuals' performance worsened over time, whereas controls' did not.70-In support of the idea that increased limbic activity interferes with prefrontal regulatory function, processes that engage sustained elaborative emotional processing, such as rumination, also seem to interfere with cognitive tasks in patients with depression.71 Findings from several studies that examined resting-state electroencephalograms (EEGs) suggest that there is decreased left prefrontal activity in depressed individuals, relative both to depressed participants' right frontal EEG activity and to EEG measures taken from the left prefrontal regions of healthy individuals.60 Consistent with the picture that is suggested by findings from other studies, a recent study used fMRI to demonstrate, in a sample of depressed patients, decreased left DLPFC recruitment during executive control and emotional information-processing tasks, as well as decreased functional relationships between DLPFC activity and amygdala activity.46
In the following sections we thus consider two mechanisms that could be implicated in maintenaining depressive functioning: abnormally increased and sustained limbic (especially amygdala) activity, and abnormally low prefrontal control. The amygdala and the prefrontal cortex are but two of the brain regions for which there is evidence of dysfunction in depression,62 and some accounts of brain abnormalities in patients with depression have emphasized the significance of neurochemical systems rather than brain regions.50,52,58,74-76 For example, Mayberg and colleagues have cited evidence in support of their hypothesis that an extensive brain network is impaired in indivuals with the full syndrome of depression.77,78 Their work has included examinations of activity in the DLPFC, the rostral and posterior cingulate gyri and the OFC in association with cognitive processes, including attention and self-referential focus. They have also reported evidence that links the vegetative and circadian aspects of depression to the insula, the subgenual cingulate gyrus and the hypothalamus.79 These regions have been shown not only to interact with the regions that we are concentrating on in this article, but also, as a full network, to yield insights into differential aspects of remission and recovery in CT and ADM treatments.78 This broader conception has also led this group to consider, for example, non-pharmacological and non-psychotherapeutic interventions that might have specific effects on this circuitry.80 However, the purpose of this Perspective is to formulate a hypothesis that, on the one hand, describes the brain functions that are thought to be disrupted during depression and ameliorated by short-term treatments with ADM or CT, and that on the other hand can account for the observation that the effects of CT endure, relative to those of ADM. This approach emphasizes the cognitive and emotion-dysregulation aspects of depression rather than the neurovegetative symptoms, and consequently focuses on the brain regions that are typically associated with those features.
Effects of treatment on brain circuitry
Both CT and ADM probably affect limbic and prefrontal circuitry, although their proximal mechanisms of action might differ. A primary goal of CT is to replace automatic emotional reactivity with more-controlled processing.81 CT might thus increase inhibitory executive control, helping to interrupt or dampen automatic limbic reactions. In fact, functions of the PFC that are impaired in depression, such as task-related direction of attention,82 willful regulation of emotional responses53,54.83,84, and reappraisal,85 are the focus of therapeutic activity in CT. Many of these cognitive processes are also associated with decreased limbic, especially amygdala, activity.53,54,83,84 Possibly as a consequence, probable correlates of sustained limbic reactivity, such as dysfunctional negative cognitions, are decreased following CT,86-89 although these causal links have yet to be firmly established.
The few findings that have emerged from neuroimaging studies of changes in emotional reactivity during CT are consistent with this formulation. One of the authors of this Perspective (G.J.S.) observed, using fMRI, increased amygdala activity in depressed patients (relative to controls) before CT in a task that required them to rate the personal relevance of emotional information, and decreased DLPFC activity (relative to controls) in a task that required them to arrange digits in numerical order.46,90 As shown in FIG. 3, in a small sample of participants that was assessed following 14 weeks of CT, these abnormalities normalized: amygdala activity in response to emotionally relevant information decreased, and prefrontal activity on cognitive tasks increased, nearly to the level that was observed in healthy controls.91 This result is conceptually similar to results that have been obtained in fMRI studies of cognitive-behavioural therapy for specific phobia, in which patients who were treated successfully with cognitive-behavioural therapy showed normalization in the activity of structures that have been implicated in emotional reactivity.92,93 In further support of the idea that CT is associated with increased functional use of the DLPFC is the finding that training in prefrontal executive tasks is associated with increased DLPFC activity during cognitive tasks and with decreased amygdala reactivity in response to emotional stimuli after therapy.94 Yet Mayberg et al. have described, using resting-state PET studies, that recovery in CT is associated with decreased, rather than increased, resting DLPFC metabolism.95 The apparent contrast between this finding and those that suggest that CT results in greater frontal activity during frontal tasks could be resolved if one considers that recovery might involve the lowering of tonic resting-state activity, to allow for greater reactivity when executive control is recruited; however, this hypothesis has yet to be tested in the context of neuroimaging investigations.
Figure 3. Changes in BOLD signal in response to cognitive and emotional tasks associated with cognitive therapy.

Nine depressed participants and 24 controls completed tasks that involved rating the personal relevance of negative words and arranging digits in numerical order before and after 12 weeks of cognitive therapy (CT). As shown in the figure, CT was associated with normalization of amygdala activity in response to emotional words as well as normalization of dorso-lateral prefrontal activity during a cognitive task that involved putting digits in numerical order in working memory.91
ADM might target limbic regions directly, rather than relying on inhibition through the prefrontal cortex. SSRIs increase the availability of serotonin at the synapse, which could lead to inhibition of the amygdala as well as of other ventral limbic regions, as has been observed in resting-state neuroimaging studies.96-103 Although activity in dorsal prefrontal regions also increased in these studies, such effects could have resulted from decreased limbo-cortical inhibition. Findings from fMRI studies suggest that symptom decreases produced by SSRIs lead to a functional decrease in limbic reactivity to emotional faces.20,104 Increases in DLPFC and perigenual cingulate activity as the result of treatment with SSRIs and SNRIs have also been observed,104,105 again consistent with the possibility that SSRIs and SNRIs act on inhibitory mechanisms. However, direct comparisons of these results with those that have been obtained with CT have not been made.
FIGURE 4 shows the patterns of response to CT and ADM that we propose on the basis of the evidence and theory that are summarized in this Perspective. As shown in the figure, acutely depressed individuals could be characterized by decreased prefrontal function, possibly arising in part from increased amygdala reactivity. We propose that CT operates by bolstering prefrontal function whereas ADM operates more directly on the amygdala. These treatments might thus result in end states that are, in one important respect, similar; normalized amygdala and PFC activity might result from either ADM or CT. Further investigation of the differences in short-term responses will be crucial, then, for understanding why CT produces a sustained effect whereas ADM does not. If the amygdala is ‘the point of entry’ for environmental stressors, cessation of ADM treatment could leave a person at risk of having strong maladaptive reactions to new environmental insults. By contrast, if CT works by building skills that require the active operation of the PFC, the effects of CT on PFC activity and function might be relatively enduring.
Figure 4. Hypothetical time-course of changes to amygdala and prefrontal function associated with ADM and CT.
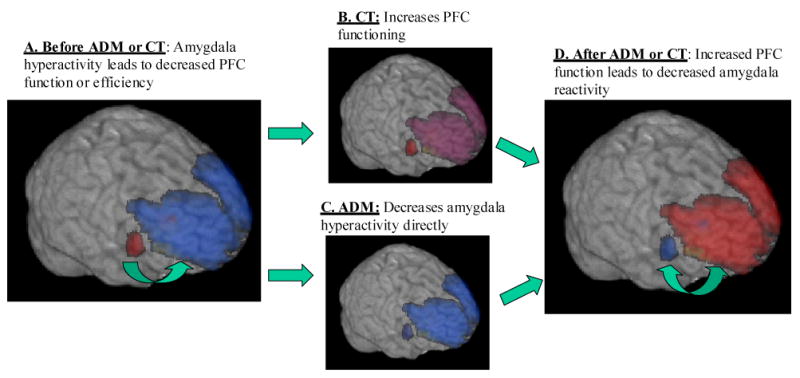
A. During acute depression, amygdala activity is increased (red) and prefrontal activity is decreased (blue) compared to healthy individuals; B. CT effectively exercises the PFC, yielding increased inhibitory function; C. ADM targets amygdala function more directly, decreasing its activity; D. After ADM or CT, amygdala function is decreased and prefrontal function is increased. The double-headed arrow between the amygdala and PFC represents the bidirectional homeostatic influences which are believed to operate in healthy individuals.
Consistent with this conceptualization, some of the mechanisms of action would be expected to differ between CT and ADM. Findings from randomized comparisons of CT and ADM suggest that they indeed engage different psychological mechanisms. For example, a change in negative expectations predicts subsequent change in depression in people treated with CT but not in people treated with ADM,89 and a change in information-processing biases, such as the kinds of causal attributions that people make for negative life events, predicts absence of relapse following treatment termination and seems to mediate the enduring effect of CT.106 Although there are as yet no published fMRI studies that compare patients treated with CT and patients treated with ADM, PET studies have shown post-treatment differences in resting-state measures of brain function between these groups.78,95,98,107-108 As discussed previously, the response to CT has been associated with decreased resting-state activity in the prefrontal cortx, and with increased resting-state activity in the hippocampus and the dorsal cingulate.96 This pattern was in some important respects the opposite of that which was exhibited by a matched group of patients who responded to the ADM paroxetine, in whom prefrontal increases and hippocampal and subgenual cingulate decreases were observed.95 Thus, whereas CT might allow a resetting of tonic prefrontal activity, to yield greater capacity for ‘top-down’ emotion regulation when it is needed (such as when skills taught in CT are engaged), ADMs might increase subcortical cingulate metabolism tonically, creating a ‘bottom-up’ effect whereby relevant limbic regions are tonically inhibited during ADM administration.
This interpretation, although speculative, is consistent with fMRI data obtained from patients who had responded to ADMs, in whom decreased amygdala responses to neutral as well as emotional faces was observed.20 In another fMRI study, depressed patients who were treated successfully with SSRIs displayed increased corticolimbic connectivity at rest, but not in response to emotional stimuli.109 This finding is consistent with the idea that ADM decreases limbic activity, which in turn leads to prefrontal disinhibition.
Prediction of change
A neuroimaging-informed understanding of the brain mechanisms that are engaged by CT and ADM treatment could lead to the development of algorithms that predict which treatment is more likely to benefit a given patient.76 Moreover, given the evidence of enduring effects for CT, there is reason to think that changes in the brain mechanisms that are mobilized by CT should be especially likely to endure. Measures of alterations in these mechanisms might thus allow one to differentiate between patients who would benefit most from CT or from ADM, and might predict resistance to relapse and recurrence following the termination of treatment.76 Ideally a model such as the one we have proposed could be used to explain the heterogeneity of depressive symptom,s and thereby serve as a basis for guiding treatment selection. Whereas the ultimate goal of treatment would be to produce the kind of synchronized modulation of the final, common limbic-cortical pathways that is considered to be crucial for illness remission,76 the targeted mechanisms would depend on individual vulnerability factors. To the extent that CT and ADM both address abnormal limbic function, high levels of amygdala reactivity should predict recovery in either intervention. In fact, fMRI data suggest that high levels of amygdala reactivity to emotional stimuli predict recovery from depression with CT90 as well as with other interventions, including ADM.110 Amygdala reactivity has similarly been shown to predict recovery after both CT and ADM in patients with anxiety pathology.111
CT's focus on helping depressed individuals to increase their emotion-regulation skills could similarly explain why patients with the lowest levels of activity in regulatory structures, such as the rostral and subgenual anterior cingulate gyri, would benefit most from CT.90,112 By contrast, because ADM targets limbic activity directly, patients with decreased rostral and subgenual cingulate activity would not be expected to recover from depression on ADM treatment. Rather, increased rostral cingulate activity, both in resting- state studies79,113-115 and in an fMRI study of reactivity, has predicted recovery during ADM treatment.105,113-116 Taken together, these findings suggest that amygdala activity might represent a general predictor of recovery. Decreased rostral and subgenual cingulate activity might predict a good response to CT, whereas increased rostral and subgenual cingulate might predict a better response to ADM. An algorithm of this kind might therefore be used for selecting treatments for individual patients.
Conclusions and future directions
The prevalence of depression is high, and the burdens that it places on society are enormous. Research in the past few decades has led to the discovery, refinement and testing of new, effective treatments for depression. Nonetheless, our best treatments currently produce symptom remission in fewer than two-thirds of the patients who receive them, and sustained recovery is achieved in approximately one-third of treated patients. Neuroscience research has the potential to enhance our ability to match patients to the treatments that would most benefit them, and to provide clues as to how to refine the treatments so that they can be delivered more effectively and efficiently.
Future investigations should include, in addition to neuroimaging studies aimed at identifying potential predictors, assessments of other important depression-relevant phenomena, so that imaging, symptom, cognitive, genetic and patient-history variables can be linked to the outcomes of psychotherapeutic or medication treatments. A study by Nemeroff and colleagues provides an excellent example of the prediction of differential outcomes of ADM and Cognitive Behavioural Analysis System of Psychotherapy (CBASP,117 a variant of CT), using an important early-history variable.118 Adult patients who reported early-childhood trauma before randomization to ADM or CBASP improved more with CBASP than with ADM, suggesting a correspondence between the targets of CBASP and the pathological processes that were set in motion by the early-childhood trauma.118 Genetic studies have also shown the potential to reveal links between polymorphisms and the cognitive processes that are targeted in psychotherapies such as CT,119 suggesting the possibility that in the future genotyping could be used as a prognostic tool.
The formulation of depression that we have discussed, which focuses on uncontrolled limbic reactivity in conjunction with decreased prefrontal control, could inform the development and refinement of treatments for depression, as well as the differential prediction of response to generally effective treatments. Detecting such moderating effects for treatment response is in its infancy. The formulation suggests that patients with increased limbic reactivity in the absence of decreased regulatory control would be prime candidates for ADM, whereas those with prominent difficulties in emotion regulation, especially if it is associated with decreased prefrontal function, might benefit more from CT. As noted by Kazdin, differential moderation always implies differences in the mechanisms that are engaged by the treatments.120 Direct comparisons in which patients who are heterogeneous with respect to these underlying neural characteristics are randomized to the respective modalities are needed if models such as the one we have proposed are to be subjected to rigorous tests.
The evidence shows that that CT is as efficacious as ADM, and that its effects are more enduring. Thus, even if CT and ADM work through the same mechanisms in the same temporal order to reduce depressive symptoms, any enduring effects of CT must be produced by mechanisms that are not mobilized in the same way by ADM. The model that is proposed in this article suggests that CT helps patients learn to recruit prefrontal regulatory brain mechanisms – a skill that these patients could continue to use long after treatment ends. Exploring the neural bases of these effects should enhance our understanding of the nature of depression and allow us to develop more powerful and targeted preventive interventions.
Most of the published research that compared the changes in brain function that are associated with CT and ADM used resting-state assessments. Challenge paradigms, in which emotional stimuli are presented to depressed individuals during neuroimaging assessment after the end of treatment, could elucidate some of the mechanisms of recovery that are associated with online emotion regulation. Such paradigms, alone or in combination with resting-state studies, could also indicate which neurocognitive features can be used to predict which patients are most likely to benefit from continued treatment.
Another virtually unexplored area is the examination of the time-course of recovery from depression after treatment. That is, although CT and ADM might initially work through different mechanisms, the initial increases in PFC activity that are associated with CT could lead, after a prolonged period of recovery, to stable decreases in the activity or reactivity of the amygdala. Similarly, decreased amygdala activity associated with ADM treatment could disinhibit prefrontal resources and lead, over time, to increased reliance on prefrontal function, so that in the presence of an affective stimulus normal emotion regulation can occur. Thus, examination of brain function months or years into recovery could reveal similar effects of CT and ADM. Research examining these hypotheses would require the imaging of depressed individuals not only before and immediately following treatment, but also long after recovery.
Careful attention to the temporal relations between cognition and emotion and their underlying neural substrates may be needed to tease apart the mechanisms that underlie different interventions, especially when these interventions produce comparable short-term outcomes, as seems to be the case for CT and ADM. Different treatments might produce similar outcomes through the same or different mechanisms, whereas different outcomes always reflect differential mechanisms or between-treatment differences in the potency of the effects on common mechanisms.121 Given that a measure of an underlying process might reflect a cause of change in one modality and a consequence of change in another, it is important to assess purported mechanisms repeatedly across the course of treatment and to examine temporal patterns of covariation with subsequent outcomes as a function of differential treatment.89
Clinical experience suggests that patients treated with CT first learn to apply the CT strategies each time they experience their habitual tendency to process information in a negatively biased manner. That is, they acquire and then implement ‘compensatory skills’. Repeated application of these skills seems to result in the alteration of the patient's general beliefs about themselves, as inferred from an increasing tendency, over time, for patients' prepotent reactions to be free of the negative biases that are characteristic of depressed people and of people who are prone to depression. This process is referred to as the ‘accommodation’ of beliefs to new evidence.122 It is tempting to speculate that this progression in clinical course reflects, and is reflected by, a progression of changes over time in distinct neural mechanisms, with initial gains being mediated by increased inhibitory cortical control exercised by conscious processes, and the maintenance of these gains resulting from a reduction in the frequency and intensity of maladaptive negative reactions to life events, produced by a tempered reactivity in relevant limbic regions.
Contributor Information
Robert J. DeRubeis, University of Pennsylvania
Greg J. Siegle, University of Pittsburgh
Steven D. Hollon, Vanderbilt University
References
- 1.Kessler RC, et al. The epidemiology of major depressive disorder: Results from the National Comorbidity Survey Replication (NCS-R) JAMA. 2003;289:3095–3105. doi: 10.1001/jama.289.23.3095. [DOI] [PubMed] [Google Scholar]
- 2.Murray CJL, Lopez AD. Global mortality, disability, and the contribution of risk factors: Global Burden of Disease Study. Lancet. 1997;349:1436–1442. doi: 10.1016/S0140-6736(96)07495-8. [DOI] [PubMed] [Google Scholar]
- 3.Greenberg PE, Burnham HG, Lowe SW, Corey-Lisle PK. The economic burden of depression in the United States: how did it change from 1990 to 2000. J Clin Psychiatry. 2003;64:1465–1475. doi: 10.4088/jcp.v64n1211. [DOI] [PubMed] [Google Scholar]
- 4.Moussavi S, et al. Depression, chronic diseases, and decrements in health: rsults from the World Health Surveys. Lancet. 2007;370:851–8. doi: 10.1016/S0140-6736(07)61415-9. [DOI] [PubMed] [Google Scholar]
- 5.DeRubeis RJ, Young PR, Dahlsgaard KK. Affective Disorders. In: Bellack AS, Hersen M, editors. Comprehensive clinical psychology. Vol. 6. Pergamon; Oxford: pp. 339–366. [Google Scholar]
- 6.American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders. 4th ed, Text Rev. American Psychiatric Association Press; Washington DC: 2000. [Google Scholar]
- 7.Frank E, et al. Conceptualization and rationale for consensus definitions of terms in major depressive disorder: Remission, recovery, relapse, and recurrence. Arch Gen Psychiatry. 1991;48:851–855. doi: 10.1001/archpsyc.1991.01810330075011. [DOI] [PubMed] [Google Scholar]
- 8.de Mello MF, de Jesus Mari J, Bacaltchuk J, Verdeli H, Neugebauer R. A systematic review of research findings on the efficacy of interpersonal therapy for depressive disorders. Eur Arch Psychiatry Clin Neurosci. 2005;255:75–82. doi: 10.1007/s00406-004-0542-x. [DOI] [PubMed] [Google Scholar]
- 9.UK ECT Review Group. Efficacy and safety of electroconvulsive therapy in depressive disorders: A systematic review and meta-analysis. Lancet. 2003;361:799–808. doi: 10.1016/S0140-6736(03)12705-5. [DOI] [PubMed] [Google Scholar]
- 10.Nahas Z, et al. Two-year outcome of vagus nerve stimulation (VNS) for treatment of major depressive episodes. J Clin Psychiatry. 2005;66:1097–104. doi: 10.4088/jcp.v66n0902. [DOI] [PubMed] [Google Scholar]
- 11.Dimidjian S, et al. Behavioral activation, cognitive therapy, and anti-depressant medication in the acute treatment of major depression. J Consult Clin Psychol. 2006;74:658–670. doi: 10.1037/0022-006X.74.4.658. [DOI] [PubMed] [Google Scholar]
- 12.Hammond DC. Neurofeedback with anxiety and affective disorders. Child Adolesc Psychiatr Clin N Am. 2005;14:105–123. doi: 10.1016/j.chc.2004.07.008. [DOI] [PubMed] [Google Scholar]
- 13.Baehr E, Rosenfeld JP, Baehr R. The clinical use of an alpha asymmetry protocol in the neurofeedback treatment of depression: Two case studies. J Neurotherapy. 1997;2:10–23. [Google Scholar]
- 14.Mayberg HS, et al. Deep brain stimulation for treatment-resistant depression. Neuron. 2005;45:651–660. doi: 10.1016/j.neuron.2005.02.014. [DOI] [PubMed] [Google Scholar]
- 15.Depression Guideline Panel. Depression in Primary Care, Vol 2: Treatment of Major Depression (Clinical Practice Guideline No 5; AHCPR Publ No 93-0551) U.S. Department of Health and Human Services, Public Health Service, Agency for Health Care Policy and Research; Rockville, MD: 1993. [Google Scholar]
- 16.Agency for Health Care Policy and Research. Treatment of depression – newer pharmacotherapies: Summary, evidence report/technology assessment number 7. Rockville, MD: US Department of Health and Human Services; 1999. http://www.ahcpr.gov/clinic/deprsumm.htm. [Google Scholar]
- 17.Thase ME, Rush AJ. When at first you don't succeed…sequential strategies for antidepressant nonresponders. J Clin Psychiatry. 1997;58(Suppl 13):23–29. [PubMed] [Google Scholar]
- 18.Hollon SD, Thase ME, Markowitz JC. Treatment and prevention of depression. Psychol Sci Public Interest. 2002;3:39–77. doi: 10.1111/1529-1006.00008. [DOI] [PubMed] [Google Scholar]
- 19.American Psychiatric Association. Practice guideline for the treatment of patients with major depressive disorder (revision) Am J Psychiatry. 2000;157(suppl 4):1–45. [PubMed] [Google Scholar]
- 20.Sheline YI, et al. Increased amygdala response to masked emotional faces in depressed subjects resolves with antidepressant treatment: an fMRI study. Biol Psychiatry. 2001;50:651–8. doi: 10.1016/s0006-3223(01)01263-x. [DOI] [PubMed] [Google Scholar]
- 21.Warner-Schmidt JL, Duman RS. Hippocampal neurogenesis: opposing effects of stress and antidepressant treatment. Hippocampus. 2006;16:239–49. doi: 10.1002/hipo.20156. [DOI] [PubMed] [Google Scholar]
- 22.Beck AT, Rush AJ, Shaw BF, Emery G. Cognitive therapy of depression. Guilford; New York: 1979. [Google Scholar]
- 23.Rush AJ, Beck AT, Kovacs M, Hollon SD. Comparative efficacy of cognitive therapy and pharmacotherapy in the treatment of depressed outpatients. Cognit Ther Res. 1977;1:17–38. [Google Scholar]
- 24.Blackburn IM, Bishop S, Glen AIM, Whalley LJ, Christie JE. The efficacy of cognitive therapy in depression: A treatment trial using cognitive therapy and pharmacotherapy, each alone and in combination. Br J Psychiatry. 1981;139:181–189. doi: 10.1192/bjp.139.3.181. [DOI] [PubMed] [Google Scholar]
- 25.Murphy GE, Simons AD, Wetzel RD, Lustman PJ. Cognitive therapy and pharmacotherapy, singly and together, in the treatment of depression. Arch Gen Psychiatry. 1984;41:33–41. doi: 10.1001/archpsyc.1984.01790120037006. [DOI] [PubMed] [Google Scholar]
- 26.Hollon SD, et al. Cognitive therapy, pharmacotherapy and combined cognitive-pharmacotherapy in the treatment of depression. Arch Gen Psychiatry. 1992;49:774–781. doi: 10.1001/archpsyc.1992.01820100018004. [DOI] [PubMed] [Google Scholar]
- 27.Kovacs M, Rush AT, Beck AT, Hollon SD. Depressed outpatients treated with cognitive therapy or pharmacotherapy: A one – year follow – up. Arch Gen Psychiatry. 1981;38:33–39. doi: 10.1001/archpsyc.1981.01780260035003. [DOI] [PubMed] [Google Scholar]
- 28.Blackburn IM, Eunson KM, Bishop S. A two year naturalistic follow up of depressed patients treated with cognitive therapy, pharmacotherapy and a combination of both. J Affective Disorders. 1986;10:67–75. doi: 10.1016/0165-0327(86)90050-9. [DOI] [PubMed] [Google Scholar]
- 29.Simons AD, Murphy GE, Levine JL, Wetzel RD. Cognitive therapy and pharmacotherapy for depression: Sustained improvement over one year. Arch Gen Psychiatry. 1986;43:43–48. doi: 10.1001/archpsyc.1986.01800010045006. [DOI] [PubMed] [Google Scholar]
- 30.Evans MD, et al. Differential relapse following cognitive therapy, pharmacotherapy, and combined cognitive-pharmacotherapy for depression. Arch Gen Psychiatry. 1992;49:802–808. doi: 10.1001/archpsyc.1992.01820100046009. [DOI] [PubMed] [Google Scholar]
- 31.Elkin I, et al. Initial severity and differential treatment outcome in the National Institute of Mental Health Treatment of Depression Collaborative Research Program. J Consult Clin Psychol. 1995;63:841–847. doi: 10.1037//0022-006x.63.5.841. [DOI] [PubMed] [Google Scholar]
- 32.Shea MT, et al. Course of depressive symptoms over follow-up: Findings from the National Institute of Mental Health Treatment of Depression Collaborative Research Program. Arch Gen Psychiatry. 1992;49:782–787. doi: 10.1001/archpsyc.1992.01820100026006. [DOI] [PubMed] [Google Scholar]
- 33.Jacobson NS, Hollon SD. Prospects for future comparisons between drugs and psychotherapy: Lessons from the CBT-versus-pharmacotherapy exchange. J Consult Clin Psychol. 1996;64:104–108. doi: 10.1037//0022-006x.64.1.104. [DOI] [PubMed] [Google Scholar]
- 34.DeRubeis RJ, Gelfand LA, Tang TZ, Simons AD. Medications versus cognitive behavioral therapy for severely depressed outpatients: Mega-analysis of four randomized comparisons. Am J Psychiatry. 1999;156:1007–1013. doi: 10.1176/ajp.156.7.1007. [DOI] [PubMed] [Google Scholar]
- 35.DeRubeis RJ, et al. Cognitive therapy vs. medications in the treatment of moderate to severe depression. Arch Gen Psychiatry. 2005;62:409–416. doi: 10.1001/archpsyc.62.4.409. [DOI] [PubMed] [Google Scholar]
- 36.Bhar SS, et al. Sequence of improvement in depressive symptoms across cognitive therapy and pharmacotherapy. Journal of Affective Disorders. 2008;110:161–168. doi: 10.1016/j.jad.2007.12.227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.DiMascio A, et al. Differential symptom reduction by drugs and psychotherapy in acute depression. Arch Gen Psychiatry. 1979;36:1450–1456. doi: 10.1001/archpsyc.1979.01780130068008. [DOI] [PubMed] [Google Scholar]
- 38.Hollon SD, et al. Prevention of relapse following cognitive therapy vs medications in moderate to severe depression. Arch Gen Psychiatry. 2005;62:417–422. doi: 10.1001/archpsyc.62.4.417. [DOI] [PubMed] [Google Scholar]
- 39.Ramel W, Goldin PR, Eyler LT, Brown GG, Gotlib IH, McQuaid JR. Amygdala reactivity and mood-congruent memory in individuals at risk for depressive relapse. Biol Psychiatry. 2007;61:231–9. doi: 10.1016/j.biopsych.2006.05.004. [DOI] [PubMed] [Google Scholar]
- 40.Liotti M, Mayberg HS, McGinnis S, Brannan SL, Jerabek P. Unmasking disease-specific cerebral blood flow abnormalities: mood challenge in patients with remitted unipolar depression. American Journal of Psychiatry. 2002;159:1830–40. doi: 10.1176/appi.ajp.159.11.1830. [DOI] [PubMed] [Google Scholar]
- 41.LeDoux J. The Emotional Brain. Simon Schuster; New York: 1996. [Google Scholar]
- 42.Sheline YI, Sanghavi M, Mintun MA, Gado MH. Depression duration but not age predicts hippocampal volume loss in medically healthy women with recurrent major depression. Journal of Neuroscience. 1999;19:5034–5043. doi: 10.1523/JNEUROSCI.19-12-05034.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Abercrombie H, et al. Metabolic rate in the right amygdala predicts negative affect in depressed patients. Neuroreport. 1998;9:3301–3307. doi: 10.1097/00001756-199810050-00028. [DOI] [PubMed] [Google Scholar]
- 44.Siegle GJ, Steinhauer SR, Thase ME, Stenger VA, Carter CS. Can't shake that feeling: fMRI assessment of sustained amygdala activity in response to emotional information in depressed individuals. Biol Psychiatry. 2002;51:693–707. doi: 10.1016/s0006-3223(02)01314-8. [DOI] [PubMed] [Google Scholar]
- 45.Drevets WC. Prefrontal cortical amygdalar metabolism in major depression. Annals of the New York Academy of Sciences. 1999;877:614–637. doi: 10.1111/j.1749-6632.1999.tb09292.x. [DOI] [PubMed] [Google Scholar]
- 46.Siegle GJ, Thompson W, Carter CS, Steinhauer SR, Thase ME. Increased amygdala and decreased dorsolateral prefrontal BOLD responses in unipolar depression: Related and independent features. Biol Psychiatry. 2007;61 doi: 10.1016/j.biopsych.2006.05.048. [DOI] [PubMed] [Google Scholar]
- 47.Dougherty D, Rauch S. Neuroimaging and neurobiological models of depression. Harvard Review of Psychiatry. 1997;5:138–159. doi: 10.3109/10673229709000299. [DOI] [PubMed] [Google Scholar]
- 48.Evans KC, Wright CI, Wedig MM, Gold AL, Pollack MH, Rauch SL. A functional MRI study of amygdala responses to angry schematic faces in social anxiety disorder. Depress Anxiety. 2007;25:496–505. doi: 10.1002/da.20347. [DOI] [PubMed] [Google Scholar]
- 49.Metcalfe J, Mischel W. A hot/cool-system analysis of delay of gratification: dynamics of willpower. Psychological Review. 1999;106:3–19. doi: 10.1037/0033-295x.106.1.3. [DOI] [PubMed] [Google Scholar]
- 50.Davidson RJ. Affective style psychopathology and resilience: Brain mechanisms and plasticity. American Psychologist. 2000;55:1196–1214. doi: 10.1037//0003-066x.55.11.1196. [DOI] [PubMed] [Google Scholar]
- 51.Drevets WC, Raichle M. Reciprocal suppression of regional cerebral blood flow during emotional versus higher cognitive processes: Implications for interactions between emotion and cognition. Cognition and Emotion. 1998;12:353–385. [Google Scholar]
- 52.Mayberg HS, et al. Reciprocal limbic cortical function and negative mood Converging PET findings in depression and normal sadness. American Journal of Psychiatry. 1999;156:675–682. doi: 10.1176/ajp.156.5.675. [DOI] [PubMed] [Google Scholar]
- 53.Ochsner KN, Bunge SA, Gross JJ, Gabrieli JDE. Rethinking feelings: An fMRI study of the cognitive regulation of emotion. Journal of Cognitive Neuroscience. 2002;14:1215–1229. doi: 10.1162/089892902760807212. [DOI] [PubMed] [Google Scholar]
- 54.Ochsner K, et al. For better or for worse: neural systems supporting the cognitive down- and up-regulation of negative emotion. Neuroimage. 2004;23:483–99. doi: 10.1016/j.neuroimage.2004.06.030. [DOI] [PubMed] [Google Scholar]
- 55.Ray JP, Price JL. The organization of projections from the mediodorsal nucleus of the thalamus to orbital and medial prefrontal cortex in macaque monkeys. Journal of Comparative Neurology. 1993;337:1–31. doi: 10.1002/cne.903370102. [DOI] [PubMed] [Google Scholar]
- 56.Ghashghaei HT, Barbas H. Pathways for emotion: interactions of prefrontal and anterior temporal pathways in the amygdala of the rhesus monkey. Neuroscience. 2002;115:1261–79. doi: 10.1016/s0306-4522(02)00446-3. [DOI] [PubMed] [Google Scholar]
- 57.Mayberg HS. Limbic Cortical Dysregulation A proposed model of depression. Journal of Neuropsychiatry and Clinical Neurosciences. 1997;9:471–481. doi: 10.1176/jnp.9.3.471. [DOI] [PubMed] [Google Scholar]
- 58.Drevets WC. Neuroimaging studies of mood disorders. Biological Psychiatry. 2000;48:813–29. doi: 10.1016/s0006-3223(00)01020-9. [DOI] [PubMed] [Google Scholar]
- 59.Carter CS, et al. Parsing executive processes: strategic vs evaluative functions of the anterior cingulate cortex. Proceedings of the National Academy of Sciences of the United States of America. 2000;97:1944–1948. doi: 10.1073/pnas.97.4.1944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Davidson RJ. Affective neuroscience and psychophysiology: Toward a synthesis. Psychophysiology. 2003;40:655–665. doi: 10.1111/1469-8986.00067. [DOI] [PubMed] [Google Scholar]
- 61.Phillips ML, Ladouceur CD, Drevets WC. A neural model of voluntary and automatic emotion regulation: implications for understanding the pathophysiology and neurodevelopment of bipolar disorder. Mol Psychiatry. 2008 June 24; doi: 10.1038/mp.2008.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Beck AT. The evolution of the cognitive model of depression and its neurobiological correlates. Am J of Psychiatry. 2008;165:969–977. doi: 10.1176/appi.ajp.2008.08050721. [DOI] [PubMed] [Google Scholar]
- 63.Davidson RJ. Assymetric brain function affective style and psychopathology: The role of early experience and plasticity. Dev Psychopathol. 1994;6:741–758. [Google Scholar]
- 64.Baxter L, et al. Reduction of prefrontal glucose metabolism common to three types of depression. Arch Gen Psychiatry. 1989;46:243–250. doi: 10.1001/archpsyc.1989.01810030049007. [DOI] [PubMed] [Google Scholar]
- 65.Bench CJ, Friston KJ, Brown RG, Frackowiak RS, Dolan RJ. Regional cerebral blood flow in depression measured by positron emission tomography the relationship with clinical dimensions. Psychol Med. 1993;23:579–590. doi: 10.1017/s0033291700025368. [DOI] [PubMed] [Google Scholar]
- 66.Ottowitz WE, Dougherty DD, Savage CR. The neural network basis for abnormalities of attention and executive function in major depressive disorder: Implications for application of the medical disease model to psychiatric disorders. Harv Rev Psychiatry. 2002;10:86–99. doi: 10.1080/10673220216210. [DOI] [PubMed] [Google Scholar]
- 67.Liotti M, Mayberg HS. The role of functional neuroimaging in the neuropsychology of depression. J Clin Exp Neuropsychol. 2001;23:121–36. doi: 10.1076/jcen.23.1.121.1223. [DOI] [PubMed] [Google Scholar]
- 68.Cohen JD, et al. Temporal dynamics of brain activation during a working memory task. Nature. 1997;386:604–608. doi: 10.1038/386604a0. [DOI] [PubMed] [Google Scholar]
- 69.Harvey PO, et al. Executive functions and updating of the contents of working memory in unipolar depression. J Psychiatr Res. 2004;38:567–76. doi: 10.1016/j.jpsychires.2004.03.003. [DOI] [PubMed] [Google Scholar]
- 70.Egeland J, et al. Attention profile in schizophrenia compared with depression: differential effects of processing speed, selective attention and vigilance. Acta Psychiatr Scand. 2003;108:276–84. doi: 10.1034/j.1600-0447.2003.00146.x. [DOI] [PubMed] [Google Scholar]
- 71.Watkins E, Brown RG. Rumination and executive function in depression: an experimental study. J Neurol Neurosurg Psychiatry. 2002;72:400–2. doi: 10.1136/jnnp.72.3.400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Phillips ML, Drevets WC, Rauch SL, Lane R. Neurobiology of emotion perception II: Implications for major psychiatric disorders. Biol Psychiatry. 2003;54:515–28. doi: 10.1016/s0006-3223(03)00171-9. [DOI] [PubMed] [Google Scholar]
- 73.Charney DS. Monoamine dysfunction and the pathophysiology and treatment of depression. J Clin Psychiatry. 1998;59(Suppl 14):11–4. [PubMed] [Google Scholar]
- 74.Mayberg HS. Modulating dysfunctional limbic-cortical circuits in depression: towards development of brain-based algorithms for diagnosis and optimised treatment. Br Med Bull. 2003;65:193–207. doi: 10.1093/bmb/65.1.193. [DOI] [PubMed] [Google Scholar]
- 75.Seminowicz DA, Mayberg HS, McIntosh AR, Goldapple K, Kennedy S, Segal Z, Rafi-Tari S. Limbic-frontal circuitry in major depression: a path modeling metanalysis. Neuroimage. 2004;22:409–18. doi: 10.1016/j.neuroimage.2004.01.015. [DOI] [PubMed] [Google Scholar]
- 76.Mayberg HS. Positron emission tomography imaging in depression: a neural systems perspective. Neuroimaging Clin N Am. 2003;13:805–815. doi: 10.1016/s1052-5149(03)00104-7. [DOI] [PubMed] [Google Scholar]
- 77.Ingram RE, Hollon SD. In: Information Processing Approaches to Clinical Psychology. Ingram RE, editor. Academic Press; New York: 1986. pp. 261–284. [Google Scholar]
- 78.Ohira H, Nomura M, Ichikawa N, Isowa T, Iidaka T, Sato A, Fukuyama S, Nakajima T, Yamada J. Association of neural and physiological responses during voluntary emotion suppression. Neuroimage. 2006;29:721–33. doi: 10.1016/j.neuroimage.2005.08.047. [DOI] [PubMed] [Google Scholar]
- 79.O'Reardon JP, Brotman MA, DeRubeis RJ, Wang J, Detre JA, Egeth MJ, Cornew LA, Fara MJ. Prefrontal-amygdala interactions and mood regulation: A perfusion fMRI study. Brain Cogn. 2003;51:185. [Google Scholar]
- 80.Ray RD, Ochsner KN, Cooper JC, Robertson ER, Gabrieli JD, Gross JJ. Individual differences in trait rumination and the neural systems supporting cognitive reappraisal. Cogn Affect Behav Neurosci. 2005;5:156–68. doi: 10.3758/cabn.5.2.156. [DOI] [PubMed] [Google Scholar]
- 81.Rush AJ, Beck AT, Kovacs M, Weissenburger J, Hollon SD. Comparison of the effects of cognitive therapy and pharmacotherapy on hopelessness and self-concept. Am J Psychiatry. 1982;139:862–6. doi: 10.1176/ajp.139.7.862. [DOI] [PubMed] [Google Scholar]
- 82.Blackburn IM, Bishop S. Changes in cognition with pharmacotherapy and cognitive therapy. Br J Psychiatry. 1983;143:609–17. doi: 10.1192/bjp.143.6.609. [DOI] [PubMed] [Google Scholar]
- 83.Garamoni GL, Reynolds CF, 3rd, Thase ME, Frank E, Fasiczka AL. Shifts in affective balance during cognitive therapy of major depression. J Consul Clin Psychol. 1992;60:260–6. doi: 10.1037//0022-006x.60.2.260. [DOI] [PubMed] [Google Scholar]
- 84.DeRubeis RJ, Evans MD, Hollon SD, Garvey MJ, Grove WM, Tuason VB. How does cognitive therapy work? Cognitive change and symptom change in cognitive therapy and pharmacotherapy for depression. J Consult Clin Psychol. 1990;58:862–9. doi: 10.1037//0022-006x.58.6.862. [DOI] [PubMed] [Google Scholar]
- 85.Siegle GJ, Carter CS, Thase ME. Use of fMRI to predict recovery from unipolar depression with cognitive behavior therapy. Am J Psychiatry. 2006;163:735–738. doi: 10.1176/ajp.2006.163.4.735. [DOI] [PubMed] [Google Scholar]
- 86.Siegle GJ, Thompson W, Horner MS, Carter CS, Thase ME. Neural mechanisms of executive control in unipolar depression: Relationships to emotional reactivity and recovery in Cognitive Behavior Therapy. In: Weissman A, editor. Executive function: From basic research to clinical practice; Symp. Meet. Assoc. Behav. Cogn.Ther; Philadelphia, Pennsylvania: 2007. [Google Scholar]
- 87.Straube T, Glauer M, Dilger S, Mentzel HJ, Miltner WH. Effects of cognitive-behavioral therapy on brain activation in specific phobia. Neuroimage. 2006;29:125–35. doi: 10.1016/j.neuroimage.2005.07.007. [DOI] [PubMed] [Google Scholar]
- 88.Paquette V, Levesque J, Mensour B, Leroux JM, Beaudoin G, Bourgouin P, Beauregard M. “Change the mind and you change the brain”: Effects of cognitive-behavioral therapy on the neural correlates of spider phobia. Neuroimage. 2003;18:401–9. doi: 10.1016/s1053-8119(02)00030-7. [DOI] [PubMed] [Google Scholar]
- 89.Siegle GJ, Ghinassi F, Thase ME. Neurobehavioral therapies in the 21st century: Summary of an emerging field and an extended example of Cognitive Control Training for depression. Cogn Ther Res. 2007;31:235–262. [Google Scholar]
- 90.Goldapple K, Segal Z, Garson C, Lau M, Bieling P, Kennedy S, Mayberg H. Modulation of cortical-limbic pathways in major depression: treatment-specific effects of cognitive behavior therapy. Arch Gen Psychiatry. 2004;61:34–41. doi: 10.1001/archpsyc.61.1.34. [DOI] [PubMed] [Google Scholar]
- 91.Saxena S, et al. Differential cerebral metabolic changes with paroxetine treatment of obsessive-compulsive disorder vs major depression. Arch Gen Psychiatry. 2002;59:250–61. doi: 10.1001/archpsyc.59.3.250. [DOI] [PubMed] [Google Scholar]
- 92.Brody AL, et al. Brain metabolic changes in major depressive disorder from pre- to post-treatment with paroxetine. Psychiatry Res. 1999;91:127–39. doi: 10.1016/s0925-4927(99)00034-7. [DOI] [PubMed] [Google Scholar]
- 93.Brody AL, et al. Brain metabolic changes associated with symptom factor improvement in major depressive disorder. Biol Psychiatry. 2001;50:171–8. doi: 10.1016/s0006-3223(01)01117-9. [DOI] [PubMed] [Google Scholar]
- 94.Brody AL, et al. Regional brain metabolic changes in patients with major depression treated with either paroxetine or interpersonal therapy: preliminary findings. Arch Gen Psychiatry. 2001;58:631–40. doi: 10.1001/archpsyc.58.7.631. [DOI] [PubMed] [Google Scholar]
- 95.Kennedy SH, et al. Changes in regional brain glucose metabolism measured with positron emission tomography after paroxetine treatment of major depression. Am J Psychiatry. 2001;158:899–905. doi: 10.1176/appi.ajp.158.6.899. [DOI] [PubMed] [Google Scholar]
- 96.Mayberg HS, et al. Regional metabolic effects of fluoxetine in major depression: serial changes and relationship to clinical response. Biol Psychiatry. 2000;48:830–43. doi: 10.1016/s0006-3223(00)01036-2. [DOI] [PubMed] [Google Scholar]
- 97.Buchsbaum MS, et al. Effect of sertraline on regional metabolic rate in patients with affective disorder. Biol Psychiatry. 1997;41:15–22. doi: 10.1016/s0006-3223(96)00097-2. [DOI] [PubMed] [Google Scholar]
- 98.Nobler MS, Olvet KR, Sackeim HA. Effects of medications on cerebral blood flow in late-life depression. Curr Psychiatry Rep. 2002;4:51–8. doi: 10.1007/s11920-002-0013-x. [DOI] [PubMed] [Google Scholar]
- 99.Fu CH, et al. Attenuation of the neural response to sad faces in major depression by antidepressant treatment: a prospective, event-related functional magnetic resonance imaging study. Arch Gen Psychiatry. 2004;61:877–89. doi: 10.1001/archpsyc.61.9.877. [DOI] [PubMed] [Google Scholar]
- 100.Davidson RJ, Irwin W, Anderle MJ, Kalin NH. The neural substrates of affective processing in depressed patients treated with venlafaxine. Am J Psychiatry. 2003;160:64–75. doi: 10.1176/appi.ajp.160.1.64. [DOI] [PubMed] [Google Scholar]
- 101.Hollon SD, Evans MD, DeRubeis RJ. In: Contemporary Psychological Approaches to Depression: Theory Research, and Treatment. Ingram R, editor. Plenum; New York: 1990. pp. 117–136. [Google Scholar]
- 102.Brody AL, et al. FDG-PET predictors of response to behavioral therapy and pharmacotherapy in obsessive compulsive disorder. Psychiatry Res. 1998;84:1–6. doi: 10.1016/s0925-4927(98)00041-9. [DOI] [PubMed] [Google Scholar]
- 103.Urry HL, et al. Amygdala and ventromedial prefrontal cortex are inversely coupled during regulation of negative affect and predict the diurnal pattern of cortisol secretion among older adults. J Neurosci. 2006;26:4415–25. doi: 10.1523/JNEUROSCI.3215-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Anand A, et al. Antidepressant effect on connectivity of the mood-regulating circuit: an FMRI study. Neuropsychopharmacology. 2005;30:1334–44. doi: 10.1038/sj.npp.1300725. [DOI] [PubMed] [Google Scholar]
- 105.Canli T, et al. Amygdala reactivity to emotional faces predicts improvement in major depression. Neuroreport. 2005;16:1267–70. doi: 10.1097/01.wnr.0000174407.09515.cc. [DOI] [PubMed] [Google Scholar]
- 106.McClure EB, et al. fMRI predictors of treatment outcome in pediatric anxiety disorders. Psychopharmacology (Berl) 2007;191:97–105. doi: 10.1007/s00213-006-0542-9. [DOI] [PubMed] [Google Scholar]
- 107.Fu CH, et al. Neural Responses to Sad Facial Expressions in Major Depression Following Cognitive Behavioral Therapy. Biol Psychiatry. 2008 June 10; doi: 10.1016/j.biopsych.2008.04.033. [DOI] [PubMed] [Google Scholar]
- 108.Mayberg HS, et al. Cingulate function in depression: A potential predictor of treatment response. Neuroreport. 1997;8:1057–1061. doi: 10.1097/00001756-199703030-00048. [DOI] [PubMed] [Google Scholar]
- 109.Pizzagalli D, et al. Anterior cingulate activity as a predictor of degree of treatment response in major depression: evidence from brain electrical tomography analysis. Am J Psychiatry. 2001;158:405–415. doi: 10.1176/appi.ajp.158.3.405. [DOI] [PubMed] [Google Scholar]
- 110.Chen CH, et al. Brain Imaging Correlates of Depressive Symptom Severity and Predictors of Symptom Improvement After Antidepressant Treatment. Biol Psychiatry. 2007;62 doi: 10.1016/j.biopsych.2006.09.018. [DOI] [PubMed] [Google Scholar]
- 111.McCullough JP. Treatment for Chronic Depression: Cognitive Behavioral Analysis System of Psychotherapy (CBASP) Guilford; New York: 2003. [DOI] [PubMed] [Google Scholar]
- 112.Nemeroff CB, et al. Differential responses to psychotherapy versus pharmacotherapy in patients with chronic forms of major depression and childhood trauma. Proc Natl Acad Sci USA. 2003;100:14293–14296. doi: 10.1073/pnas.2336126100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Hayden EP, et al. Early-emerging cognitive vulnerability to depression and the serotonin transporter promoter region polymorphism. J Affect Dis. 2008;107:227–230. doi: 10.1016/j.jad.2007.07.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Kazdin AE. Developing a research agenda for child adolescent psychotherapy. Arch Gen Psychiatry. 2000;57:829–840. doi: 10.1001/archpsyc.57.9.829. [DOI] [PubMed] [Google Scholar]
- 115.Hollon SD, DeRubeis RJ, Evans MD. Causal mediation of change in treatment for depression: Discriminating between nonspecificity and noncausality. Psychol Bull. 1987;102:139–149. [PubMed] [Google Scholar]
- 116.Barber JP, DeRubeis RJ. On second thought: Where the action is in cognitive therapy for depression. Cognit Ther Res. 1989;13:441–457. [Google Scholar]