

Abstract
Background
The neuron-derived orphan receptor-1 (NOR1) belongs to the evolutionary highly conserved and most ancient NR4A subfamily of the nuclear hormone receptor superfamily. Members of this subfamily function as early response genes regulating key cellular processes including proliferation, differentiation, and survival. Although NOR1 has previously been demonstrated to be required for smooth muscle cell (SMC) proliferation in vitro, the role of this nuclear receptor for the proliferative response underlying neointima formation and target genes trans-activated by NOR1 remain to be defined.
Methods and Results
Using a model of guide wire-induced arterial injury, we demonstrate decreased neointima formation in NOR1-/- mice compared to wildtype mice. In vitro, NOR1-deficient SMC exhibit decreased proliferation due to a G1→S phase arrest of the cell cycle and increased apoptosis in response to serum deprivation. NOR1-deficiency alters phosphorylation of the retinoblastoma protein by preventing mitogen-induced cyclin D1 and D2 expression. Conversely, overexpression of NOR1 induces cyclin D1 expression and the transcriptional activity of the cyclin D1 promoter in transient reporter assays. Gel shift and chromatin immunoprecipitation assays identified a putative response element for NR4A receptors in the cyclin D1 promoter, to which NOR1 is recruited in response to mitogenic stimulation. Finally, we provide evidence that these observations are applicable in vivo by demonstrating decreased cyclin D1 expression during neointima formation in NOR1-deficient mice.
Conclusions
These experiments characterize cyclin D1 as a NOR1-regulated target gene in SMC and demonstrate that NOR1 deficiency decreases neointima formation in response to vascular injury.
Keywords: nuclear receptor, smooth muscle cell, cell cycle
In an era marked by the increasing prevalence of obesity, diabetes, and cardiovascular disease, members of the nuclear hormone receptor superfamily have emerged as transcription factors that regulate diverse aspects of metabolism1. In addition to their function to act as molecular sensors of lipid and carbohydrate homeostasis, many nuclear receptors exert pleiotropic effects to control inflammatory and proliferative responses during vascular remodeling. This ability to integrate metabolic and vascular signaling networks has been well described for the ligand-activated peroxisome proliferator-activated receptors (PPAR) and liver X receptors (LXR)2, 3. However, the nuclear receptor superfamily comprises a large number of receptors, for which ligands have not been identified, and these remain classified as orphan nuclear receptors1. For many of these orphan receptors, the physiological functions and their target genes remain unknown, yet the high degree of conservation points to an important role in the control of gene expression.
The NR4A subfamily of orphan nuclear receptors consists of the members Nur77, Nurr1, and NOR14, 5. These receptors bind as monomers to the nerve growth factor-induced clone B (NBFI-B) response element (NBRE), and as homodimers to the Nurr1 response element (NurRE) in the promoter of their target genes4, 5. In contrast to other members of the nuclear receptor superfamily, the ligand-binding pocket of these NR4A receptors is covered by hydrophobic residues, which has resulted in the characterization of NR4A receptors as ligand-independent and constitutively-active transcription factors6. Consistent with this concept, these receptors function as early response genes, which are rapidly induced in response to various extracellular cues4, 5.
In the vascular system, NOR1 is expressed in atherosclerotic lesions and in the neointima following vascular injury7-9. At the cellular level, NOR1 expression is rapidly induced by inflammatory stimuli in macrophages10 and by growth factors in endothelial cells11. Finally, NOR1 is highly expressed in SMC following mitogenic stimulation7, 9 and recent studies7, including our experiments using NOR1-deficient SMC9, pointed to a key role of this receptor in the transcriptional control of SMC proliferation. In the present study, we extend these observations by demonstrating that NOR1-deficient mice are protected from neointima formation due to altered cyclin D1 expression. These results indicate that NOR1 may constitute a potential novel target for the intervention for proliferative vascular diseases.
Methods
Animals
All experiments were performed using littermate NOR1-/- and NOR1+/+ mice as recently described9, 12. The institutional animal care and use committee at the University of Kentucky approved all animal procedures.
Cell Culture
Rat and mouse aortic SMC were cultured as described9. Human coronary artery SMC were commercially obtained (Lonza Inc.). Cells were grown to 60–70% confluence, serum-deprived in 0.1% Fetal Bovine Serum (FBS) for at least 24 hours and subjected to stimulation with FBS at a final concentration of 10%. For all data shown, cells were used between passages 3 and 7 and individual experiments were repeated at least three times with different preparations of cells.
Cell Cycle Distribution and Apoptosis Assays
Following mitogenic stimulation, cells were collected by trypsinization, washed twice in PBS, and re-suspended in staining buffer containing 20 μg/ml RNase A and 100 μg/ml propidium iodide. Cells were stained for 30 minutes at 4 °C, and 1 × 106 cells were analyzed for cell cycle distribution with a FACSCalibur sorting system (Becton Dickinson). Apoptosis was assessed using a fluorescent-based FragEL™ DNA fragmentation kit (Calbiochem, QIA39).
Western Blot Analysis
Western blotting was performed as described9 using the following antibodies: Ser807/811 Phospho-Rb (9308, Cell Signaling), NOR1 (PP-H7833, R & D Systems), Nurr1 (AF2156, R & D Systems), Nur77 (LS-B114, Lifespan Biosciences), cyclin D1 (05-815, Millipore), cyclin D2 (M-20) and GAPDH (FL 335) (Santa Cruz Biotechnology).
Quantitative real-time RT-PCR
RNA was isolated and reverse transcribed as described9. PCR reactions were performed using the iCycler and SYBR Green I system (Bio-Rad, Hercules, CA). Each sample was analyzed in triplicate and normalized to values for transcription factor IIB (TFIIB) mRNA expression. All primer sequences are provided in Supplemental Table SI.
Plasmids, Transient Transfections and Luciferase Assay
The human and murine cyclin D1 promoter constructs were kindly provided by Drs. Richard G. Pestell13 and Nicholas H. Heintz14, respectively. The NBRE sites in the murine and human cyclin D1 promoter located between -2197 to -2190 and -1061 to -1054 were mutated from AAAGGTCA to AAAGAACA and from AAAGGTGA to AAAGAAGA, respectively, using the QuickChange II site–directed mutagenesis kit (Stratagene). The NOR1 expression vector was employed as previously described9. The E2F-Luc reporter construct was commercially obtained (Stratagene). SMC were transfected with 1.5 μg of reporter DNA using LipofectAMINE 2000 (Invitrogen). Following transfection, cells were serum-deprived in 0.1 % FBS for 12 hours and stimulated with 10% FBS for the indicated time points. Luciferase activity was assayed using a Dual Luciferase Reporter Assay (Promega). Transfection efficiency was normalized to renilla luciferase activities generated by cotransfection of 5 ng pRLCMV (Promega).
Electrophoretic Mobility Shift Assay
Electrophoretic mobility shift assays (EMSA) were performed using 5 μg nuclear extract as described15 and the oligonucleotides described in Supplemental Table SI. Supershift experiments were performed by incubating 5 μg of nuclear extract with 2 μg of NOR1 antibody (PP-H7833-00; R & D Systems) for 20 minutes before addition of the radiolabeled probe.
Chromatin Immunoprecipitation (ChIP) Assays
ChIP assays were performed using the EZ-ChIP kit (Millipore) as described9. Chromatin was immunoprecipitated using 5μg antibody directed against NOR1 (PP-H7833-00, R & D Systems). Final DNA extractions were amplified by real-time PCR using primer pairs covering the NBRE cognate sequences in the human or mouse cyclin D1 promoter (Supplemental Table S1).
Endovascular Femoral Artery Guide Wire Injury
Guide wire endothelial denudation injuries were performed on the left femoral artery of littermate NOR1+/+ (n=9) and NOR1-/- (n=8) mice at 8 weeks of age using a 0.25-mm SilverSpeed-10 hydrophilic guide wire (Micro Therapeutics Inc.) as described16. The denudation injury was accomplished upon four passages of the wire. Sham surgery without injury was performed on the right side. Mice were euthanized at the indicated time points and femoral arteries were isolated for tissue analysis.
Tissue Preparation and Morphometry
Following sacrifice, mice were perfused with PBS for 5 minutes, followed by 4% paraformaldehyde for 30 minutes at 100 cm H2O via canulation of the left ventricle. Femoral arteries were embedded in paraffin and cut into 10 μm sections. Serial sections 2 mm proximal from the incision site of the wire insertion were evaluated by staining with an elastic Verhoeff-Van Gieson staining kit (Sigma-Aldrich) to visualize the internal and external elastic lamina. The intimal and medial areas were measured by computerized morphometry using the software Image-Pro Plus 4.0 (MediaCybernetics). Intimal hyperplasia was defined as the formation of a neointimal layer medial to the internal elastic lamina (IEL). Media area was defined as the area encircled by the external elastic lamina (EEL) and the IEL. The intima-to-media (I/M) ratio was calculated as the intimal area divided by the media area.
Immunohistochemistry and TUNEL Staining
Paraffin sections were incubated with a Cy3-conjugated α-smooth muscle actin antibody (C6198; Sigma Aldrich), a macrophage antiserum (AIA31240; Accurate Chemical & Scientific Company), or an antibody against von Willebrand Factor antibody (SIG-3115; Covance Research Products). Sections analyzed for macrophage staining and von Willebrand Factor were subsequently incubated with Alexa 594-conjugated goat-anti rabbit IgG (A11012; Invitrogen). Sections were counterstained with 4′,6′-diamidinophenylindole (DAPI) and visualized using confocal microscopy. Immunohistochemistry for cyclin D1, NOR1, Nurr1, and Nur77 was performed as described9. Sections were incubated with a primary antibody (cyclin D1, 92G2, Cell Signaling; NOR1, IMG-71915 Imgenex; Nurr1, AF2156, R & D Systems; Nur77, LS-B114, Lifespan Biosciences) followed by incubation with a biotinylated goat anti-rabbit IgG (BA-1000, Vector Laboratories Inc.). Apoptotic cells were detected using a fluorescent-based FragEL™ DNA fragmentation kit (Calbiochem, QIA39) and counterstaining with DAPI. Quantification of cyclin D1 expression and of apoptotic cells was performed by counting the number of positively stained nuclei in the neointima and the total number of nuclei within the neointimal area. The ratio in each section provided the fraction of cyclin D1-positive or apoptotic cells.
Statistical Analysis
To compare wildtype and NOR1-deficient mice on a single variable, we used an unpaired Student's t-test. One-way and 2-way ANOVA was used to compare multiple groups as appropriate. P values <0.05 were considered to be statistically significant. Results are expressed as mean ± SEM.
The authors had full access to and take full responsibility for the integrity of the data. All authors have read and agree to the manuscript as written.
Results
NR4A receptors are expressed during Neointima Formation
Previous studies demonstrated that NOR1, Nur77, and Nurr1 are expressed in atherosclerotic lesions8, 17 and that NOR1 mRNA is induced following balloon angioplasty7. To complement these studies, we analyzed their expression profiles during neointima formation in the mouse femoral artery using endothelial denudation injuries. As depicted in Fig. 1A, all three members of the NR4A subfamily were expressed in the neointima of NOR1+/+ mice. In contrast, NOR1 protein was undetectable in mice deficient for the locus confirming the phenotype of the mouse strain. In response to vascular injury of wildtype mice, transcript levels of all three receptors were rapidly induced within three to six hours (Fig. 1B). Although Nur77 and Nurr1 were induced to a comparable extent in wildtype and NOR1-deficient mice, their induction was delayed in NOR1-deficient mice. In vitro, NOR1 mRNA expression was induced by more than 600-fold upon mitogenic stimulation of wildtype SMC, which is consistent with previous studies7, 9 (Fig. 1C). While Nur77 mRNA expression increased by more than 20-fold in wildtype and NOR1-/- SMC, Nurr1 transcript levels were induced only to a modest extent in both genotypes. Finally, complementary results were obtained in Western Blotting experiments analyzing protein levels of all three receptors in response to mitogenic stimulation (Fig. 1D).
Figure 1. NOR1, Nur77, and Nurr1 Expression during Neointima Formation and in Response to mitogenic Stimulation of SMC.
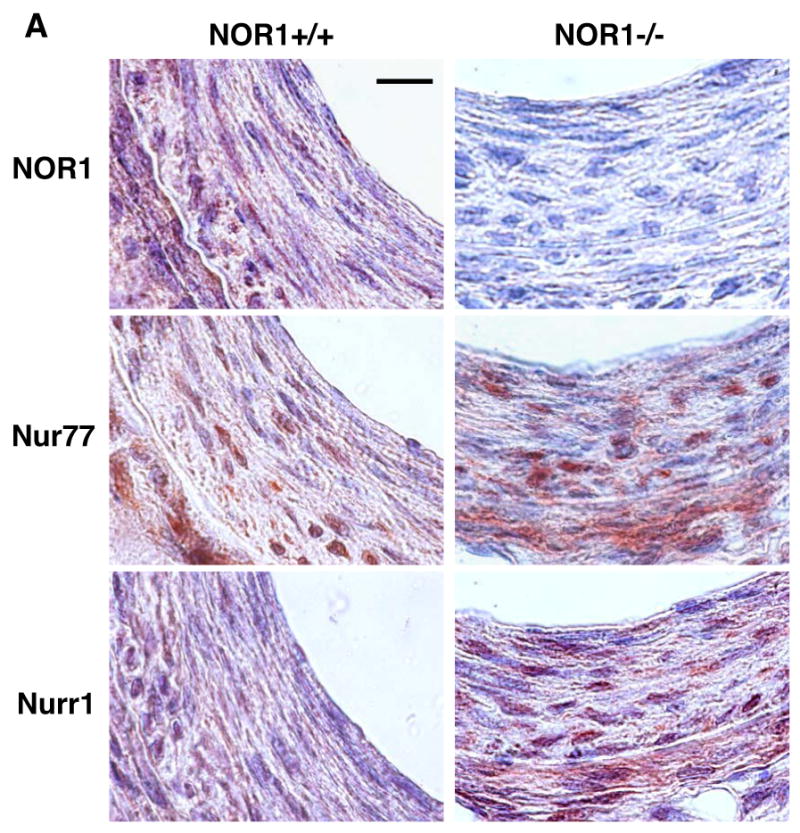


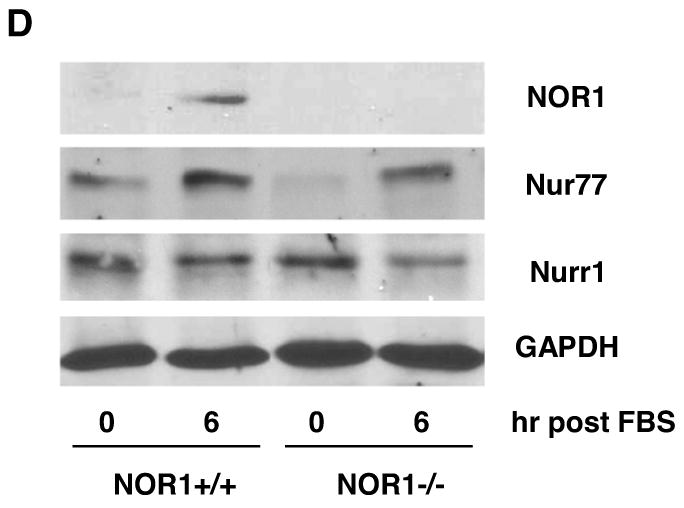
(A-B) Endothelial denudation injuries were performed in littermate NOR1+/+ and NOR1-/- mice. (A) Tissues were harvested after 28 days and NOR1, Nur77, and Nurr1 expression was analyzed by immunohistochemistry (scale bar 10 μM). (B) mRNA was harvested from sham-operated and injured vessels at the indicated time points and analyzed for NOR1, Nur77, and Nurr1 expression using real-time RT-PCR. Data are normalized to TFIIB mRNA expression levels and presented as mean ± SEM fold increase relative to sham-operated vessels (* P < 0.05 vs. time point 0 of the same genotype, 2-way ANOVA). (C-D) SMC were isolated from NOR1+/+ and NOR1-/- mice, serum-deprived for 48h, and stimulated with 10% FBS. (C) mRNA was harvested at the indicated time points and analyzed for NOR1, Nur77, and Nurr1 mRNA expression using real-time RT-PCR. Data are normalized to TFIIB mRNA expression levels and presented as fold increase relative to quiescent cells (* P < 0.05 vs. quiescent, 2-way ANOVA). (D) NOR1, Nur77, and Nurr1 protein expression was analyzed by Western blotting at the indicated time point. Cohybridization for GAPDH was performed to assess equal loading. The autoradiograms are representative of three independently performed experiments.
NOR1 Deficiency attenuates Neointima Formation following Vascular Injury
The profound increase in NOR1 expression during vascular remodeling and the reduction of cell proliferation in NOR1-deficient SMC reported in our previous in vitro assays9, prompted us to examine neointima formation in NOR1-deficient mice. As depicted in Fig. 2A, neointima formation was substantially decreased in NOR1-deficient mice compared to wildtype mice. Quantitative morphometry confirmed a significant 56.7 % reduction of the intima/media (I/M) ratio and a concomitant increase in lumen size in NOR1-/- mice (Table 1). Cells present in the neointima were primarily SMC staining positive for α-SMA (Fig. 2B). Staining for von Willebrand factor identified an intact endothelial layer above the neointima while no endothelial cells were detected within the neointima. Macrophages were largely absent within the neointimal layer and sparsely detected within the adventitial layer.
Figure 2. Neointima Formation is decreased in NOR1-deficient Mice.
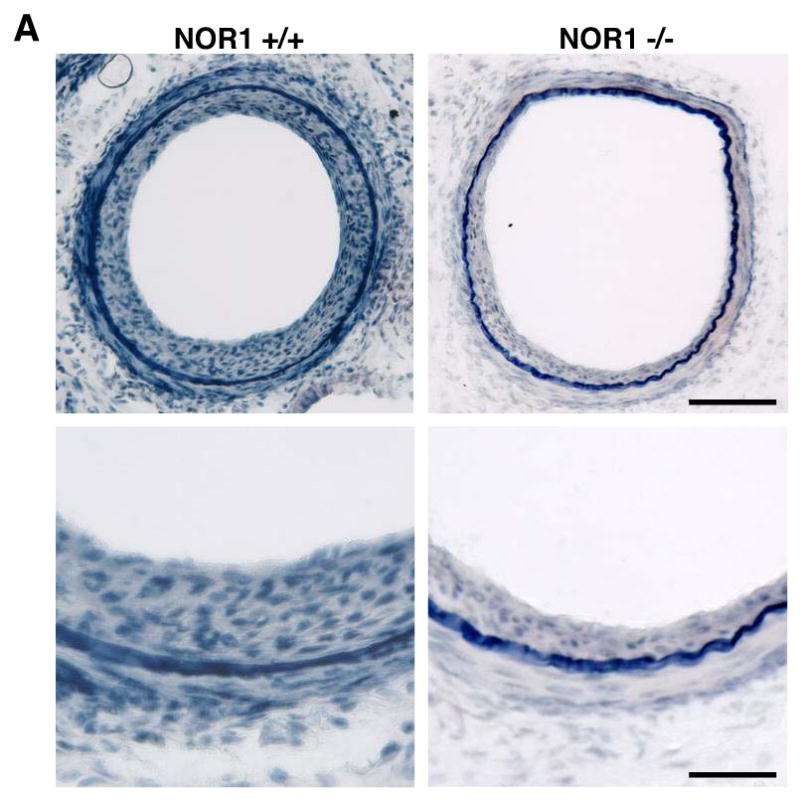
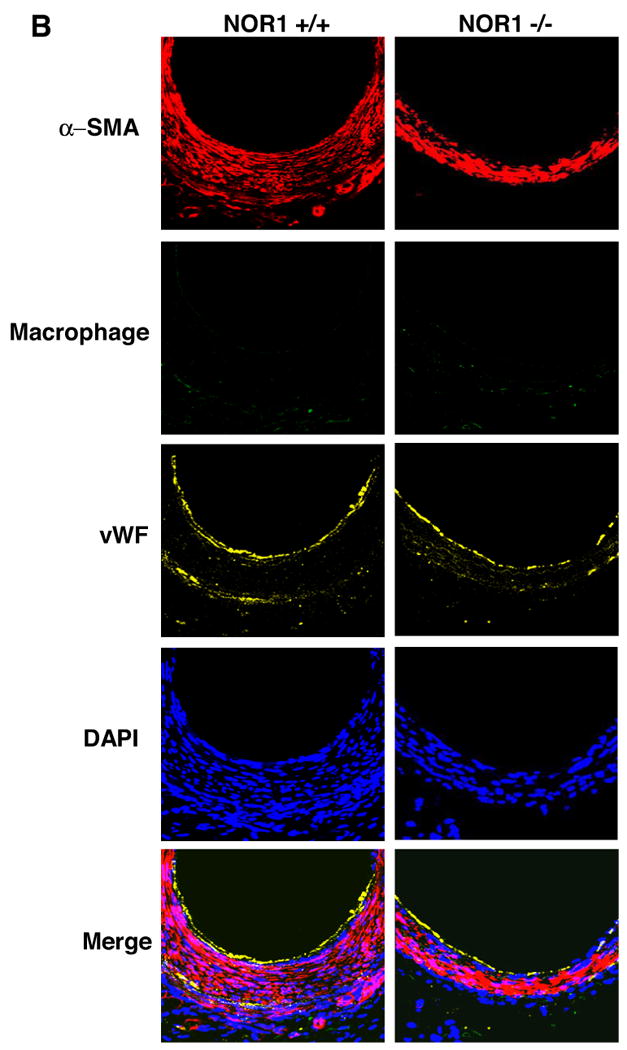
Endothelial denudation injury was performed in NOR1+/+ (n=9) and NOR1-/- (n=8) mice. (A) Tissues were harvested after 28 days and elastic Verhoeff Van Gieson stain was performed to visualize the internal and external elastic lamina. Upper panel: magnification ×200, scale bar 50 μM; lower panel: magnification ×400, scale bar 25 μM (B) Representative immunofluorescence image of injured femoral arteries from NOR1+/+ and NOR1-/- mice showing α-SMA-positive SMC (red), macrophages (green), and endothelial cells stained for von Willebrand Factor (yellow). Sections were counterstained with 4′,6′-diamidinophenylindole to visualize nuclei (blue) and merged to show the composite staining.
Table 1.
Morphological Analysis of Mouse Femoral Arteries
| NOR1+/+ sham | NOR1+/+ injured | NOR1-/- sham | NOR1-/- injured | |
|---|---|---|---|---|
|
Vessel size (μM2) |
55875 ± 3854 | 59845 ± 3638 | 52284 ± 4174 | 62784 ± 4445 |
|
Luminal area (μM2) |
48826 ± 2687 | 21145 ± 3147 * | 46478 ± 2154 | 39785 ± 3196 # |
|
Intimal area (μM2) |
0 ± 0 | 30049 ± 3998 * | 0 ± 0 | 11589 ± 3689 *# |
|
Medial area (μM2) |
6587 ± 548 | 10926 ± 645 * | 5748 ± 987 | 9739 ± 895 * |
| Intima/Media Ratio | 0 ± 0 | 2.75 ± 0.6 * | 0 ± 0 | 1.19 ± 0.41 *# |
NOR1+/+ wildtype (n=9) and NOR1-/-mice (n=8) were analyzed for vessel size (total area encircled by external elastic lamina), luminal area (area encircled by the endoluminal perimeter), intimal area (area encircled by internal elastic lamina – luminal area), and medial area (area encircled by external elastic lamina – area encircled by internal elastic lamina) 28 days after vascular injury. The ratio of intima to media was calculated as the intimal area/medial area. Data is expressed as mean ± SEM
P < 0.05, injured vs. sham by two-way ANOVA.
P< 0.05, NOR1+/+ vs. NOR1-/- in the injured group by two-way ANOVA.
NOR1 is required for Cell Cycle Progression and Survival of SMC
To determine the mechanisms underlying decreased neointima formation and altered SMC proliferation in NOR1-/- mice9, we analyzed cell cycle distribution of wildtype and NOR1-/- SMC. Upon mitogenic stimulation with 10 % FBS, NOR1 wildtype SMC progressed into S phase while NOR1-/- SMC remained arrested in the G0/G1 phase (29.95 ± 0.57 % vs. 4.18 ± 1.23 % of cells in S phase, respectively, P<0.01 (unpaired Student's t-test), Fig. 3A and B). Analysis of cell cycle distribution further revealed that serum-deprivation increased apoptosis of NOR1-/- SMC compared to wildtype mice (9.3 ± 0.19 % vs. 1.66 ± 1.28 %, P<0.05 (unpaired Student's t-test), Fig. 3A and B), an observation that was confirmed by an increased number of NOR1-deficient SMC revealing DNA fragmentation (Fig. 3C). In concert, these data point to an increased propensity for apoptosis in NOR1-/- SMC and corroborate previous observations using NOR1 antisense to demonstrate that this receptor is required for S phase entry of the cell cycle 7.
Figure 3. NOR1 Expression is required for Cell Cycle Progression and Survival of SMC.
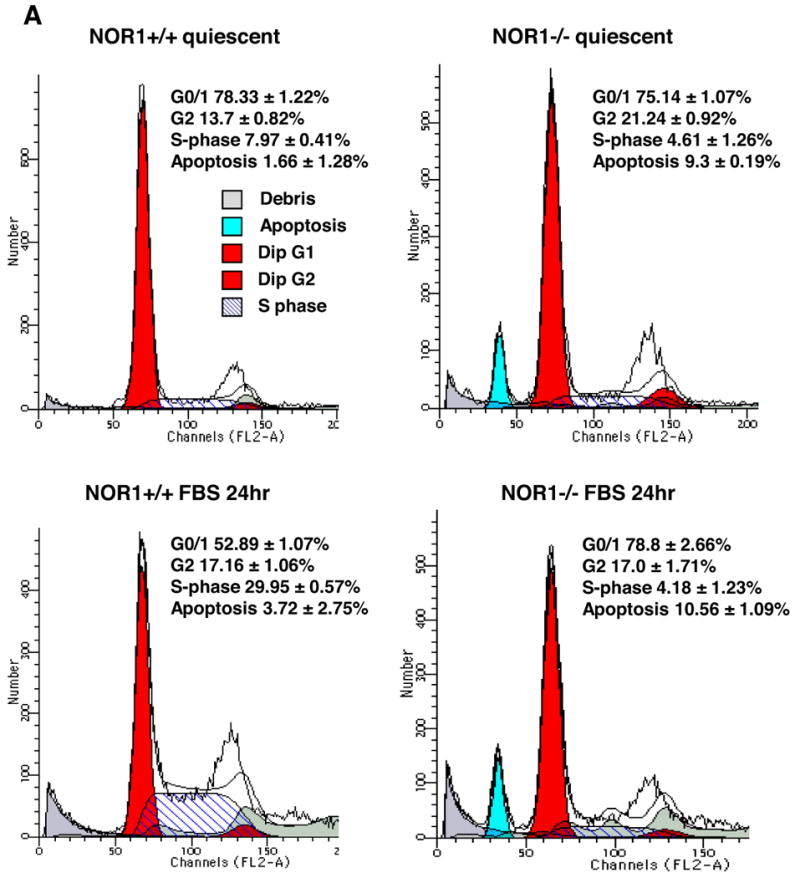
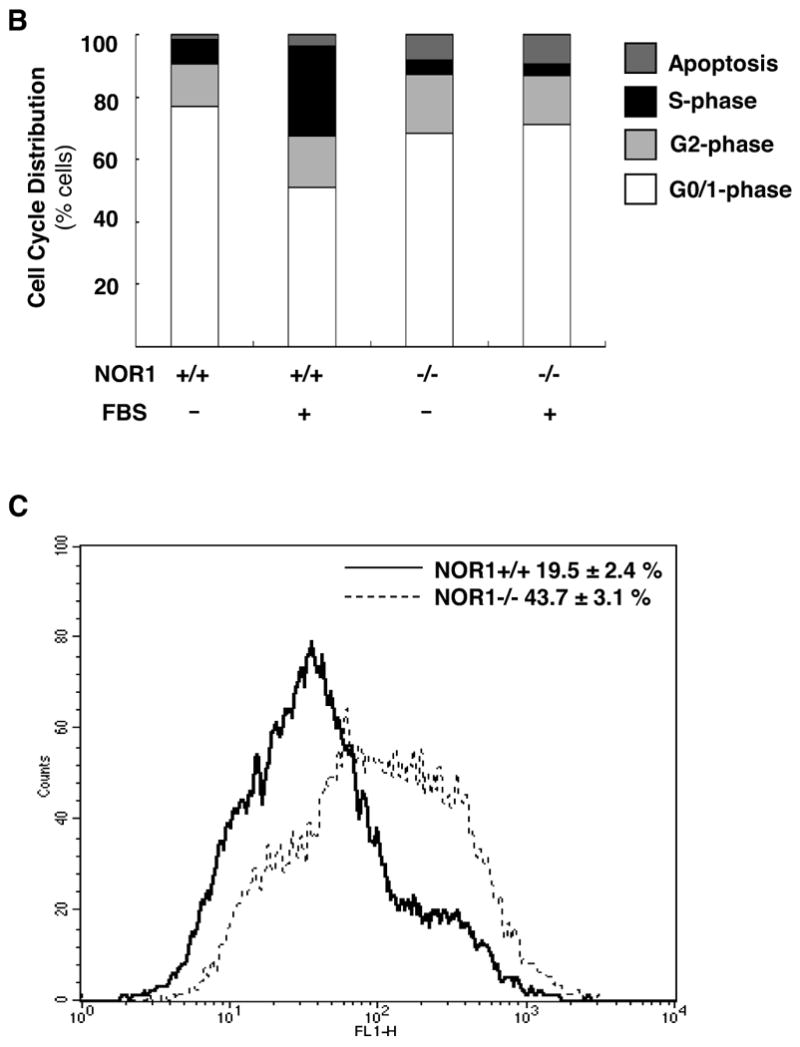
Quiescent aortic SMC isolated from NOR1+/+ and NOR1-/- were stimulated to enter the S phase by treatment with 10 % FBS for 24 h. (A) Representative cell cycle distribution assessed by DNA staining with propidium iodide and subsequent flow cytometry. (B) Depicted are DNA histograms for cell cycle distribution of quiescent and mitogen-stimulated NOR1+/+ and NOR-/-SMC. (C) NOR1+/+ and NOR1-/- SMC were serum deprived for 48h and assayed by FACS for fluorescent-based DNA fragmentation. A representative histogram is depicted and the percentage of apoptotic cells is indicated as mean ± SEM in the upper right corner.
NOR1 Deficiency increases Apoptosis in Neointimal SMC
To determine whether reduced neointima formation in NOR1-/- mice may be in part due to increased apoptosis, we next quantified SMC apoptosis in neointimal lesions. As shown in a representative Fig. 4A and quantified in Fig. 4B, less than 10 % of the total number of neointimal SMC in wildtype mice demonstrated the presence of DNA fragmentation. In contrast, NOR1-deficiency increased the number of apoptotic SMC in the neointima by approximately 3-fold compared to wildtype mice (P < 0.05). These results confirm the above-described in vitro observations and indicate that increased SMC apoptosis may in part be responsible for the decreased neointima formation in NOR1-deficient mice.
Figure 4. NOR1 Deficiency increases neointimal SMC apoptosis.
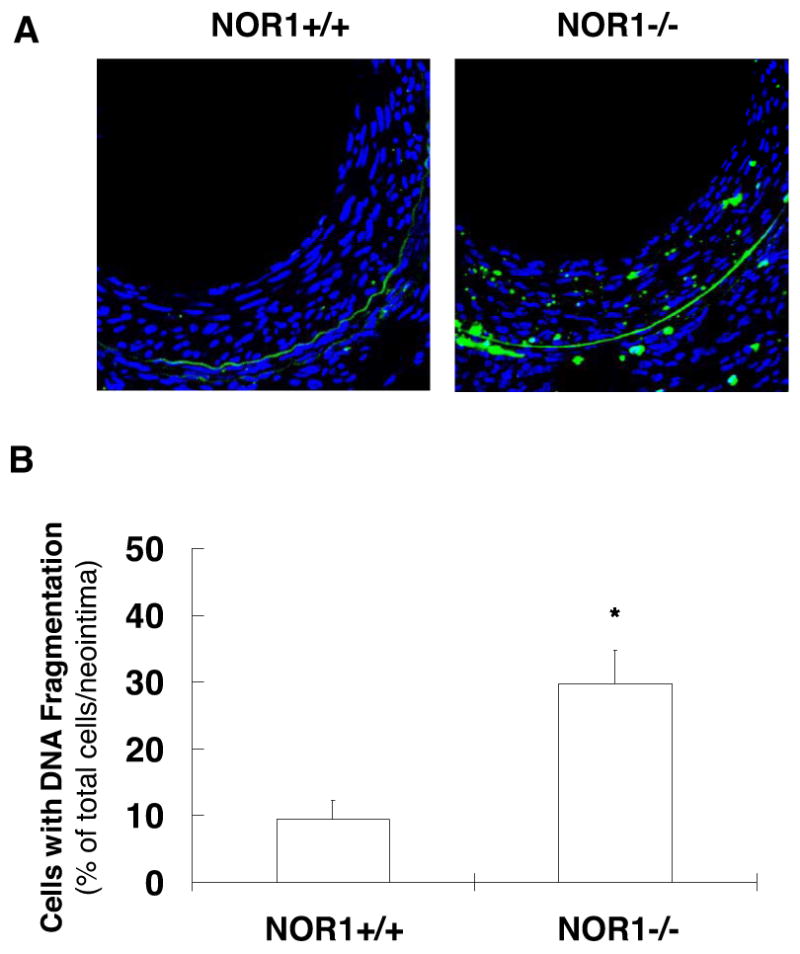
(A) Arterial tissues from NOR1+/+ and NOR1-/- mice were harvested 28 days after injury and analyzed for DNA fragmentation (green). Sections were counterstained with DAPI to visualize nuclei (blue) and merged to show the composite staining. (B) Apoptotic cells with DNA fragmentation were quantified in the neointima and expressed as percent of DAPI-stained nuclei (* P < 0.05 vs. wildtype, unpaired Student's t-test).
NOR1 Deficiency alters Phosphorylation of the Retinoblastoma Protein and E2F Activity
Cell cycle transition through the G1 checkpoint is regulated by the retinoblastoma tumor suppressor protein (Rb) and requires its phosphorylation18. Therefore, we next examined whether Rb phosphorylation is altered in NOR1-deficienct SMC. As depicted in Fig. 5A, quiescent NOR1+/+ SMC exhibited low levels of Rb phosphorylated at Ser807/811, which increased following mitogenic stimulation. In marked contrast, deficiency of NOR1 resulted in significantly decreased Rb phosphorylation. Hypophosphorylated Rb sequesters the E2F transcription factor restricting the expression of E2F-regulated target genes, whose protein products are required for DNA replication18. To further determine whether NOR1-regulated Rb phosphorylation affects downstream E2F activity, we performed trans-activation assays using a reporter construct driven by multiple E2F consensus sites. As demonstrated in Fig. 5B, overexpression of NOR1 resulted in a profound 5.03 ± 0.95-fold induction of E2F activity compared to cells transfected with the empty control vector. Taken together, these studies demonstrate that NOR1 is required for mitogen-induced Rb phosphorylation and sufficient to increase the transcriptional activity of E2F.
Figure 5. NOR1 Deficiency alters Rb phosphorylation and E2F activity.
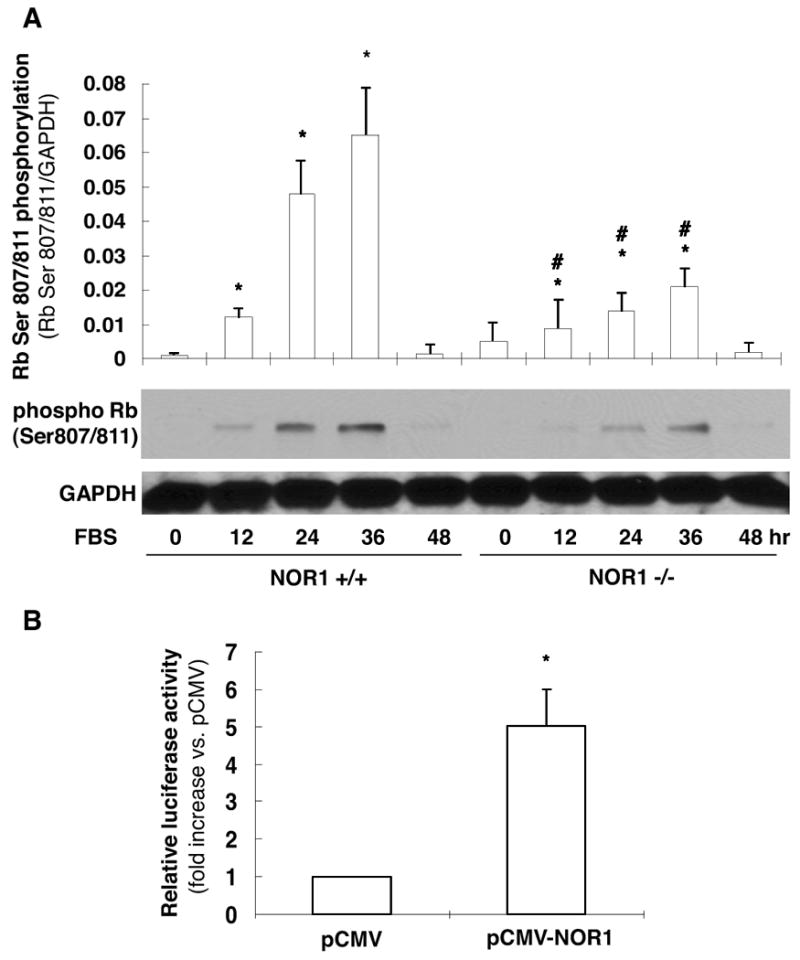
(A) Aortic SMC isolated from NOR1+/+ or NOR1-/- mice were serum-deprived and stimulated with 10% FBS. Cells were harvested at the indicated time points and analyzed for Rb (ser807/811) phosphorylation and GAPDH to assess equal loading. The upper panel represents the mean ± SEM of Rb phosphorylation/GAPDH protein levels quantified by densitometry (n=3, * P < 0.05 vs. timepoint 0, # P < 0.05 vs. wildtype, 2-way ANOVA). (B) Rat SMC were cotransfected with either a NOR1 expression vector or empty control vector and a luciferase reporter construct driven by multiple E2F binding sites. Following transfection, cells were harvested after 24 hours and luciferase activities were analyzed as described in the Methods. Data are presented as mean ± SEM from three independently performed experiments (*P < 0.01 vs. control vector, unpaired Student's t-test).
NOR1 regulates Cyclin D1 and D2 Expression in Response to mitogenic Stimulation
The phosphorylation of Rb is regulated by cyclin/cyclin-dependent kinase (CDK) complexes and cyclin D1 expression is limiting for cell proliferation and survival13, 19. In addition, recent studies identified cyclin D2 as a target gene for Nur77 in monocytes10, and we have previously demonstrated that NOR1 expression is required for cyclin D1 and D2 expression in response to platelet-derived growth factor 9. Consistent with these prior studies, cyclin D1 and D2 protein expression was completely abolished in NOR1-/- cells stimulated with FBS compared to wildtype SMC (Fig. 6A). Conversely, overexpression of NOR1 in quiescent human coronary artery SMC using an eukaryotic expression vector increased cyclin D1 protein expression and downstream Rb phosphorylation in the absence of mitogenic stimulation (Fig. 6B). The mechanisms, by which NOR1 induces cyclin D1 and D2 expression occur at the transcriptional level since mitogen-induced mRNA expression of cyclin D1 (Fig. 6C) and D2 (Fig. 6D) was completely repressed in NOR1-deficient SMC. In concert, these data demonstrate that NOR1 is necessary and sufficient to induce cyclin D1 protein expression providing a potential mechanism, by which NOR1 regulates Rb phosphorylation, cell cycle progression, and survival.
Figure 6. NOR1 regulates mitogen-induced Cyclin D Expression.
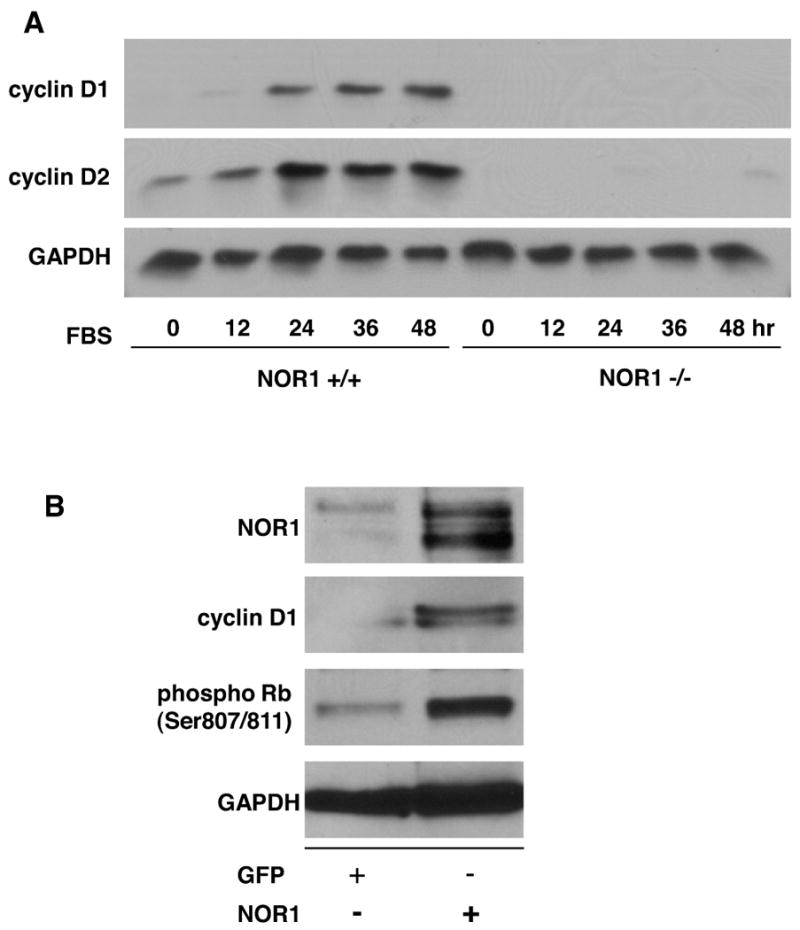

(A) Serum-deprived SMC isolated from NOR1+/+ or NOR1-/- mice were stimulated with 10% FBS. Cyclin D1 and D2 protein expression was analyzed at the indicated time points. (B) Quiescent human coronary artery SMC cells were transfected with either a NOR1 expression vector or a GFP expression vector. Forty-eight hours post-transfection, Rb phosphorylation, NOR1, cyclin D1, and GAPDH protein expression was analyzed. The autoradiograms shown are representative of three independently performed experiments. (C) Cyclin D1 and (D) cyclin D2 mRNA expression was analyzed by real-time RT-PCR in NOR1+/+ and NOR1-/- SMC before (white bars) and after stimulation with 10% FBS for 24 hours (black bars). Data are normalized to TFIIB mRNA expression levels and presented as mean ± SEM relative to quiescent SMC (*P < 0.05 compared to quiescence, 2-way ANOVA).
NOR1 activates the Cyclin D1 Promoter
To further investigate whether NOR1 induces cyclin D1 promoter activity, we performed trans-activation assays using a NOR1 expression vector cotransfected with reporter constructs driven by a murine 3.6 kb or human 1.7 kb cyclin D1 promoter fragment (Fig. 7A). Compared to cells transfected with the control vector, overexpression of NOR1 resulted in a significant induction of basal and mitogen-induced activity of the cyclin D1 promoter. To characterize the transcriptional mechanisms governing this regulation, we screened both cyclin D1 promoter sequences for putative NBRE consensus sites. Using this approach, we identified a canonical consensus NBRE site within the murine cyclin D1 promoter (-2197 to -2190, AAAGGTCA) and a putative NBRE site with one base substitution in the human cyclin D1 promoter (-1061 to -1054, AAAGGTGA). To establish the functional relevance of these NBRE sites for the regulation of the cyclin D1 promoter by NOR1, site-directed mutations were next introduced into these NBRE sequences. In marked contrast to the regulation of the wildtype promoters, both murine and human cyclin D1 promoter constructs bearing mutations of the predicted NBRE sites exhibited low basal activity and were not inducible by FBS.
Figure 7. NOR1 activates the Cyclin D1 Promoter.

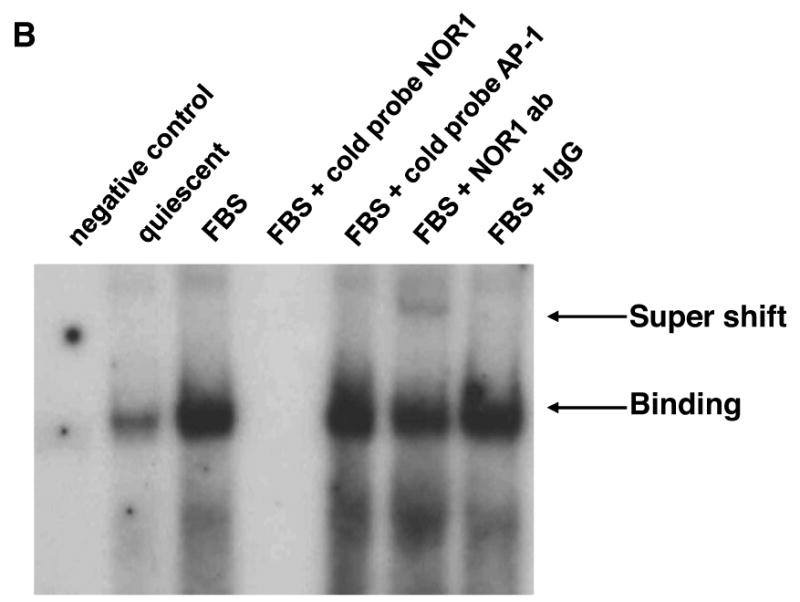

(A) Rat SMC were cotransfected with either a NOR1 or GFP expression vector and a luciferase reporter construct driven by the wildtype or NBRE-mutated murine or human cyclin D1 promoter. Following transfection, cells were starved for 12 hours, stimulated with 10% FBS for 48 hours, and analyzed for luciferase activities. Data are presented as mean ± SEM from three independently performed experiments (*P < 0.05 vs. quiescence, #P < 0.05 vs. GFP, 2-way ANOVA). (B) Serum-deprived SMC isolated from NOR1+/+ mice were stimulated with 10% FBS for 12 hours. DNA binding activities in nuclear extracts recognizing a radiolabeled murine cyclin D1 promoter region containing the NBRE site between -2197 to -2190 were detected by EMSA. Specificity was confirmed by competition with cold cyclin D1 or AP-1 oligonucleotides and by supershift using a NOR1 antibody. The autoradiogram shown is representative of three independently performed experiments. (C) Quiescent wildtype SMC were stimulated with 10% FBS and harvested at the indicated time points cells for ChIP assays. Following chromatin immunoprecipitation using a NOR1 antibody or control IgG, quantitative real-time PCR analysis was performed with primer pairs that cover the NBRE site between -2197 and -2190 in the murine cyclin D1 promoter. (D) Quiescent human coronary artery SMC were pretreated with vehicle (DMSO) or the MEK1 inhibitor PD98059 (10 μM) for 30 minutes prior to stimulation with 10% FBS for 24h. Following chromatin immunoprecipitation using a NOR1 antibody or control IgG, quantitative real-time PCR analysis was performed with primer pairs that cover the NBRE site between -1061 and -1054 in the human cyclin D1 promoter. Data in (C) and (D) are normalized to total extract (input) and presented as fold ± SEM increase relative to quiescent SMC (*P<0.001 compared to quiescent SMC, 1-way ANOVA (C) and 2-way ANOVA (D)).
EMSA and ChIP assays were next performed to confirm that NOR1 directly binds to these NBRE sites. As depicted in Fig. 7B, EMSA experiments revealed a specific complex at the murine NBRE site that was induced by FBS and completely abolished by competition with an unlabeled NOR1 oligonucleotide but not by an AP-1 oligonucleotide. Diminished binding and supershift with an antibody raised against NOR1 further verified specificity of this complex. ChIP assays corroborated occupancy of the endogenous NBRE sites in the murine and human cyclin D1 promoter by NOR1 using murine wildtype SMC and human coronary artery SMC. As shown in Fig. 7C, quantitative analysis by real-time PCR revealed that mitogenic stimulation results in a profound increase of NOR1 binding to the NBRE site located between -2197 and -2190 in the murine cyclin D1 promoter. Similarly, FBS induced NOR1 binding to the NBRE site located between -1061 and -1054 in the human cyclin D1 promoter (Fig. 7D). This binding of NOR1 to the human cyclin D1 promoter was prevented by PD98059, an inhibitor of ERK-MAPK signaling, consistent with this signaling cascade being a key pathway in the control of SMC proliferation. In concert, these experiments demonstrate that NOR1 binds to an NBRE motif in the murine and human cyclin D1 promoter resulting in the activation of the promoter and cyclin D1 transcription.
Cyclin D1 Expression during Neointima Formation is attenuated in NOR1-deficient Mice
To finally investigate whether cyclin D1 expression during neointimal SMC proliferation is decreased in NOR1-deficient mice in vivo, we performed cyclin D1 immunohistochemistry of guide wire-injured femoral arteries isolated from NOR1+/+ and NOR1-/- mice. As depicted in Fig. 8A, neointimal SMC of wildtype mice revealed typical nuclear localization of cyclin D1 expression, which was decreased in the neointima of NOR1-/- mice. Quantification of neointimal cyclin D1 expression as means of cyclin D1-positive nuclei divided by the total number of nuclei confirmed a significant decrease of cyclin D1 expression in the neointima of NOR1-/- mice compared to wildtype mice (Fig. 8B, 11.5 ± 3.49 % vs. 40.7 ± 5.27 %, respectively, P<0.001).
Figure 8. NOR1 Deficiency attenuates Cyclin D1 Expression during Neointima Formation.
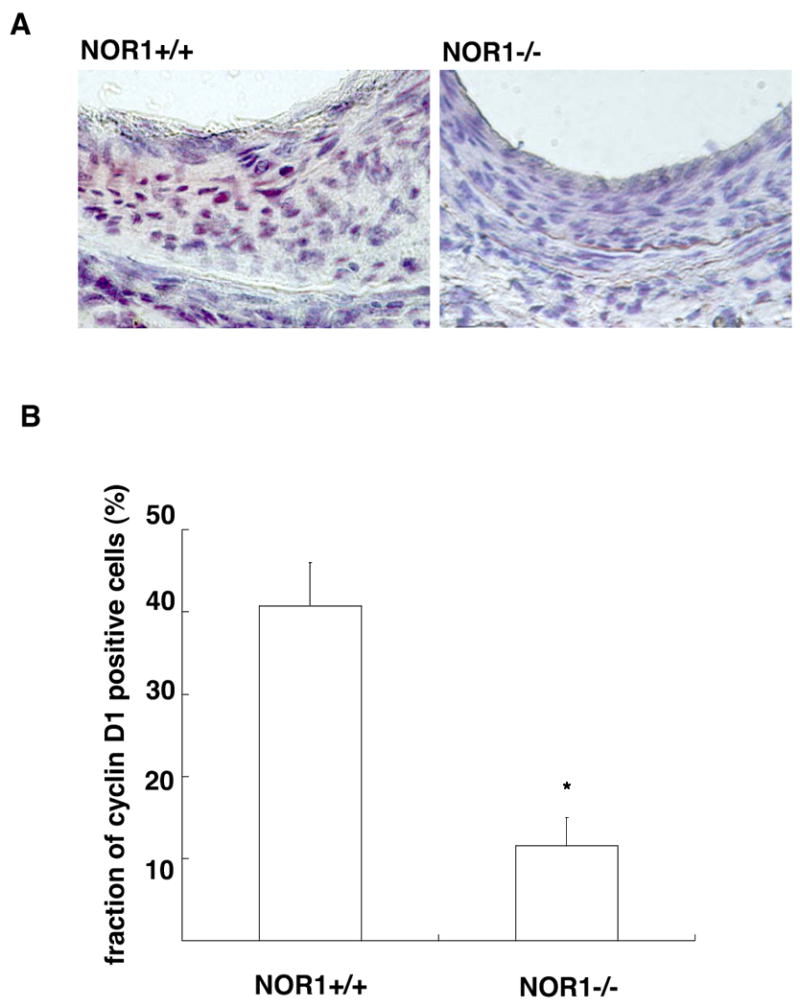
Femoral artery sections were isolated 28 days after injury of NOR1+/+ and NOR1-/- mice. (A) Cyclin D1 immunohistochemistry was performed on tissues obtained from NOR1+/+ and NOR1-/- mice. (B) Cyclin D1 positive cells were quantified by analyzing the fraction of cyclin D1-stained nuclei in the neointima relative to the total number of nuclei in the neointimal area (n=8-9/group). Values are expressed as mean ± SEM (*P<0.001 compared to NOR1+/+, unpaired Student's t-test).
Discussion
Previous studies by Martínez-González et al. using NOR1 silencing7 and our experiments in NOR1-deficient SMC9 provided evidence that this receptor promotes SMC proliferation in vitro. Although these studies point to an important role for NOR1 in the regulation of SMC proliferation, the contribution of NOR1 to the proliferative response underlying neointima formation and the molecular mechanisms responsible for the mitogenic activity of NOR1 remain unknown. In the present study, we extend these earlier observations and demonstrate that NOR1-deficiency attenuates neointima formation suggesting an important role of NOR1 signaling for proliferative vascular remodeling.
Consistent with the mitogenic activity of NOR1 in vitro7, 9, our data establish that NOR1 is required for cell cycle progression and cell survival. The main gatekeeper of G1→S phase progression is the Rb tumor suppressor protein18, and we observed altered Rb phosphorylation in NOR1-deficient SMC. Conversely, overexpression of NOR1 in SMC induced Rb phosphorylation and downstream E2F-dependent promoter trans-activation. Among the cyclin/CDK complexes that phosphorylate Rb, mammalian D-type cyclins drive cells into S phase and prevent apoptosis13, 19, and mitogen-induced cyclin D1 and D2 expression was completely abolished as a consequence of NOR1-deficiency. Furthermore, we demonstrate that cyclin D1 expression is decreased in neointimal SMC of NOR1-/- mice suggesting that the regulation of cyclin D1 by NOR1 is also applicable in vivo. Interestingly, Pei et al. previously demonstrated that NOR1 overexpression in macrophages induces cyclin D2 mRNA10. In addition, we have recently observed that cyclin D1 and D2 protein levels were decreased in NOR1-deficient cells9. These studies combined with our present data indicate that cyclin D1 may constitute a key target gene responsible for the mitogenic activity of NOR1.
In comparison to the well-studied early response genes encoding proteins of the AP-1 complex, little is known about the endogenous binding activity of NOR1 to consensus sites in target promoters. A single recent study identified that NOR1 binds to an atypical NBRE with one base substitution in the uncoupling protein-1 (UCP1) promoter20. In the present study, we identified a canonical NBRE site in the murine cyclin D1 promoter at -2197 to -2190 and a putative NBRE site with one base substitution in the human cyclin D1 promoter located at -1061 to -1054. Using gel shift and ChIP assays, we confirmed that NOR1 is recruited to these NBRE sites in response to mitogenic signals. The functional relevance of NOR1 binding to these NBRE sites was provided by the observation that overexpression of NOR1 induces cyclin D1 promoter activity while mutation of the NBRE sites resulted in a complete loss of both FBS- and NOR1-induced cyclin D1 promoter activity. Although it is possible, if not likely, that NOR1 regulates additional genes involved in the regulation of cell proliferation, these studies characterize NOR1-regulated cyclin D1 expression as a novel transcriptional cascade promoting cell cycle progression.
Considering that all three members of the NR4A receptor subfamily bind to an NBRE site, functional redundancy between Nur77 and NOR1 has been suggested21. However, two lines of evidence suggest that a possible redundancy is likely context and tissue dependent. First, Nur77-/- mice exhibit no overt phenotype22 while NOR1-/- mice develop an obvious phenotype with inner ear defects and bidirectional circling behavior12. Considering these differences between Nur77-/- and NOR1-/- mice, one might hypothesize that Nur77 function can be replaced by NOR1 but not vice versa. Consistent with this notion would be the superinduction of NOR1 in Nur77-/- mice21 and the lack of increased Nur77 expression in NOR-/- SMC in our studies. Furthermore, since the maximal induction of Nur77 and Nurr1 mRNA during neointima formation was delayed in NOR1-deficient mice, it is possible that NOR1 is required for maximal expression of Nur77 and Nurr1, particularly since a positive autoregulation of NR4A receptors has recently been confirmed23. Second, in the context of neointima formation it appears that Nur77 and NOR1 exert distinct effects on SMC proliferation. Arkenbout et al. recently reported that overexpression of Nur77 protects against neointima formation17. This observation along with our data suggests that Nur77 and NOR1 exhibit opposite roles in the control of SMC proliferation. An obvious question for future investigation is how to reconcile the distinct biological effects of NOR1 and Nur77. Although the transcriptional target gene mediating the inhibition of SMC proliferation by Nur77 remains unknown, one possibility is that its biological effect may occur as a result of its heterodimeric partner RXR. Nurr77, unlike NOR1, can heterodimerize with RXR and mediate efficient trans-activation in response to RXR-specific agonists from canonical DR5 sites24. Ligand-induced RXR activation has been well established to inhibit mitogen-induced SMC proliferation25 and interaction between Nur77 and RXR may at least need to be considered as an explanation for the distinct effect of Nur77 and NOR1 on SMC proliferation.
In conclusion, our results demonstrate that NOR1 induces cyclin D1 promoter activity allowing subsequent gene transcription. NOR1-induced cyclin D1 expression results in the phosphorylation of Rb and subsequent G1→S phase transition, an effect that will ultimately act mitogenic on SMC. Finally, NOR1 deficiency limits the proliferative response underlying neointima formation by compromising cyclin D1 expression. These findings identify NOR1 as a critical component of a transcriptional cascade regulating SMC proliferation and suggest that NOR1 might play a pivotal role for the development of proliferative vascular diseases, including atherosclerosis and in-stent restenosis.
Supplementary Material
Acknowledgments/Funding Sources
These studies were supported by the NIH (RO1 HL084611 to D.B. and RO1 CA111411 to O. M. C.).
Footnotes
Author Disclosures:
Takashi Nomiyama: No disclosures
Yue Zhao: No disclosures
Florence Gizard: No disclosures
Hannes M. Findeisen: No disclosures
Elizabeth B. Heywood: No disclosures
Karrie L. Jones: No disclosures
Orla M. Conneely: No disclosures
Dennis Bruemmer: No disclosures
Journal Subject Codes: Basic Science Research/Vascular biology/[162] Smooth muscle proliferation and differentiation.
Clinical Perspective: SMC proliferation contributes to the initiation of atherosclerosis and the failure of interventional approaches used to treat occlusive atherosclerotic diseases, including in-stent restenosis following percutaneous coronary intervention, vein-graft atherosclerosis, and transplant vasculopathy. Therefore, understanding the molecular mechanisms governing SMC proliferation is of particular interest for the development of novel therapeutic approaches to prevent atherosclerosis and its complications. In previous studies, NR4A receptors have been characterized as key regulators of macrophage inflammation and SMC proliferation. Based on this evidence, we hypothesized that deficiency of NOR1 might prevent pathologic vascular remodeling. In the present study, we confirm this concept and demonstrate that NOR1-deficiency prevents neointima formation in response to injury due to altered cyclin D1 expression. These experiments provide first evidence that modulation of NOR1 expression might provide a rational approach to prevent proliferative vascular disease.
Disclaimer: The manuscript and its contents are confidential, intended for journal review purposes only, and not to be further disclosed.
References
- 1.Chawla A, Repa JJ, Evans RM, Mangelsdorf DJ. Nuclear Receptors and Lipid Physiology: Opening the X-Files. Science. 2001;294:1866–1870. doi: 10.1126/science.294.5548.1866. [DOI] [PubMed] [Google Scholar]
- 2.Gervois P, Fruchart JC, Staels B. Drug Insight: mechanisms of action and therapeutic applications for agonists of peroxisome proliferator-activated receptors. Nat Clin Pract Endocrinol Metab. 2007;3:145–156. doi: 10.1038/ncpendmet0397. [DOI] [PubMed] [Google Scholar]
- 3.Zelcer N, Tontonoz P. Liver X receptors as integrators of metabolic and inflammatory signaling. J Clin Invest. 2006;116:607–614. doi: 10.1172/JCI27883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Maxwell MA, Muscat GE. The NR4A subgroup: immediate early response genes with pleiotropic physiological roles. Nucl Recept Signal. 2006;4:e002. doi: 10.1621/nrs.04002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pols TW, Bonta PI, de Vries CJ. NR4A nuclear orphan receptors: protective in vascular disease? Curr Opin Lipidol. 2007;18:515–520. doi: 10.1097/MOL.0b013e3282ef77d1. [DOI] [PubMed] [Google Scholar]
- 6.Wang Z, Benoit G, Liu J, Prasad S, Aarnisalo P, Liu X, Xu H, Walker NPC, Perlmann T. Structure and function of Nurr1 identifies a class of ligand-independent nuclear receptors. Nature. 2003;423:555–560. doi: 10.1038/nature01645. [DOI] [PubMed] [Google Scholar]
- 7.Martinez-Gonzalez J, Rius J, Castello A, Cases-Langhoff C, Badimon L. Neuron-Derived Orphan Receptor-1 (NOR-1) Modulates Vascular Smooth Muscle Cell Proliferation. Circ Res. 2003;92:96–103. doi: 10.1161/01.es.0000050921.53008.47. [DOI] [PubMed] [Google Scholar]
- 8.Bonta PI, van Tiel CM, Vos M, van Thienen JV, Ferreira V, Arkenbout EK, Seppen J, Spek CA, van der Poll T, Pannekoek H, de Vries CJM. Nuclear Receptors Nur77, Nurr1, and NOR-1 Expressed in Atherosclerotic Lesion Macrophages Reduce Lipid Loading and Inflammatory Responses. Arterioscler Thromb Vasc Biol. 2006;26:2288–94. doi: 10.1161/01.ATV.0000238346.84458.5d. [DOI] [PubMed] [Google Scholar]
- 9.Nomiyama T, Nakamachi T, Gizard F, Heywood EB, Jones KL, Ohkura N, Kawamori R, Conneely OM, Bruemmer D. The NR4A orphan nuclear receptor NOR1 is induced by platelet-derived growth factor and mediates vascular smooth muscle cell proliferation. J Biol Chem. 2006;281:33467–33476. doi: 10.1074/jbc.M603436200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pei L, Castrillo A, Chen M, Hoffmann A, Tontonoz P. Induction of NR4A Orphan Nuclear Receptor Expression in Macrophages in Response to Inflammatory Stimuli. J Biol Chem. 2005;280:29256–29262. doi: 10.1074/jbc.M502606200. [DOI] [PubMed] [Google Scholar]
- 11.Rius J, Martinez-Gonzalez J, Crespo J, Badimon L. NOR-1 is involved in VEGF-induced endothelial cell growth. Atherosclerosis. 2006;184:276–282. doi: 10.1016/j.atherosclerosis.2005.04.008. [DOI] [PubMed] [Google Scholar]
- 12.Ponnio T, Burton Q, Pereira FA, Wu DK, Conneely OM. The nuclear receptor Nor-1 is essential for proliferation of the semicircular canals of the mouse inner ear. Mol Cell Biol. 2002;22:935–945. doi: 10.1128/MCB.22.3.935-945.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Albanese C, D'Amico M, Reutens AT, Fu M, Watanabe G, Lee RJ, Kitsis RN, Henglein B, Avantaggiati M, Somasundaram K, Thimmapaya B, Pestell RG. Activation of the cyclin D1 Gene by the E1A-associated Protein p300 through AP-1 Inhibits Cellular Apoptosis. J Biol Chem. 1999;274:34186–34195. doi: 10.1074/jbc.274.48.34186. [DOI] [PubMed] [Google Scholar]
- 14.Ranjan P, Anathy V, Burch PM, Weirather K, Lambeth JD, Heintz NH. Redox-dependent expression of cyclin D1 and cell proliferation by Nox1 in mouse lung epithelial cells. Antioxid Redox Signal. 2006;8:1447–1459. doi: 10.1089/ars.2006.8.1447. [DOI] [PubMed] [Google Scholar]
- 15.Ogawa D, Stone JF, Takata Y, Blaschke F, Chu VH, Towler DA, Law RE, Hsueh WA, Bruemmer D. Liver X Receptor Agonists Inhibit Cytokine-Induced Osteopontin Expression in Macrophages Through Interference With Activator Protein-1 Signaling Pathways. Circ Res. 2005;96:e59–67. doi: 10.1161/01.RES.0000163630.86796.17. [DOI] [PubMed] [Google Scholar]
- 16.Ogawa D, Nomiyama T, Nakamachi T, Heywood EB, Stone JF, Berger JP, Law RE, Bruemmer D. Activation of peroxisome proliferator-activated receptor gamma suppresses telomerase activity in vascular smooth muscle cells. Circ Res. 2006;98:e50–59. doi: 10.1161/01.RES.0000218271.93076.c3. [DOI] [PubMed] [Google Scholar]
- 17.Arkenbout EK, de Waard V, van Bragt M, van Achterberg TAE, Grimbergen JM, Pichon B, Pannekoek H, de Vries CJM. Protective Function of Transcription Factor TR3 Orphan Receptor in Atherogenesis: Decreased Lesion Formation in Carotid Artery Ligation Model in TR3 Transgenic Mice. Circulation. 2002;106:1530–1535. doi: 10.1161/01.cir.0000028811.03056.bf. [DOI] [PubMed] [Google Scholar]
- 18.Sherr CJ. G1 phase progression: cycling on cue. Cell. 1994;79:551–555. doi: 10.1016/0092-8674(94)90540-1. [DOI] [PubMed] [Google Scholar]
- 19.Baldin V, Lukas J, Marcote MJ, Pagano M, Draetta G. Cyclin D1 is a nuclear protein required for cell cycle progression in G1. Genes Dev. 1993;7:812–821. doi: 10.1101/gad.7.5.812. [DOI] [PubMed] [Google Scholar]
- 20.Kumar N, Liu D, Wang H, Robidoux J, Collins S. Orphan Nuclear Receptor NOR-1 Enhances cAMP-Dependent Uncoupling Protein-1 Gene Transcription. Mol Endocrinol. 2008;22:1057–64. doi: 10.1210/me.2007-0464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cheng LE, Chan FK, Cado D, Winoto A. Functional redundancy of the Nur77 and Nor-1 orphan steroid receptors in T-cell apoptosis. EMBO J. 1997;16:1865–1875. doi: 10.1093/emboj/16.8.1865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lee SL, Wesselschmidt RL, Linette GP, Kanagawa O, Russell JH, Milbrandt J. Unimpaired thymic and peripheral T cell death in mice lacking the nuclear receptor NGFI-B (Nur77) Science. 1995;269:532–535. doi: 10.1126/science.7624775. [DOI] [PubMed] [Google Scholar]
- 23.Zhan Y, Du X, Chen H, Liu J, Zhao B, Huang D, Li G, Xu Q, Zhang M, Weimer BC, Chen D, Cheng Z, Zhang L, Li Q, Li S, Zheng Z, Song S, Huang Y, Ye Z, Su W, Lin SC, Shen Y, Wu Q. Cytosporone B is an agonist for nuclear orphan receptor Nur77. Nat Chem Biol. 2008;4:548–556. doi: 10.1038/nchembio.106. [DOI] [PubMed] [Google Scholar]
- 24.Perlmann T, Jansson L. A novel pathway for vitamin A signaling mediated by RXR heterodimerization with NGFI-B and NURR1. Genes Dev. 1995;9:769–782. doi: 10.1101/gad.9.7.769. [DOI] [PubMed] [Google Scholar]
- 25.Wakino S, Kintscher U, Kim S, Jackson S, Yin F, Nagpal S, Chandraratna RA, Hsueh WA, Law RE. Retinoids inhibit proliferation of human coronary smooth muscle cells by modulating cell cycle regulators. Arterioscler Thromb Vasc Biol. 2001;21:746–751. doi: 10.1161/01.atv.21.5.746. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.