

Abstract
Most viruses enter cells via receptor-mediated endocytosis. However, the entry mechanisms used by many of them remain unclear. How viruses are targeted to cellular endocytic machinery is also largely unknown. We have studied the entry mechanisms of influenza viruses by tracking the interaction of single viruses with cellular endocytic structures in real time using fluorescence microscopy. Our results show that influenza can exploit clathrin-mediated and clathrin- and caveolin-independent endocytic pathways in parallel, both pathways leading to viral fusion with similar efficiency. Remarkably, viruses taking the clathrin-mediated pathway enter cells via the de novo formation of clathrin-coated pits (CCPs) at viral binding sites. CCP formation at these sites is much faster than elsewhere on the cell surface, suggesting a virus-induced CCP formation mechanism that may be commonly exploited by many other types of viruses.
Many viruses infect their host cells via endocytosis. While the endocytic pathways used by some viruses have been determined, the entry mechanisms for most remain poorly understood1–9. This is in part due to the presence of multiple endocytic pathways that may be exploited by viruses, including clathrin-mediated endocytosis, caveolin-mediated endocytosis, and clathrin- and caveolin-independent pathways10–14. Another important question in viral entry is how viruses are targeted to endocytic machinery for internalization. This is in fact a critical and largely open question for other endocytic ligands as well11. For clathrin-mediated endocytosis, ligands and bound receptors are targeted to CCPs, then the CCPs mature into coated vesicles (CCVs), resulting in the internalization of ligands and receptors. Two different targeting mechanisms may be possible: 1) ligands and bound receptors may be targeted to pre-existing CCPs on the cell surface, 2) clathrin and cofactors may be recruited to the site of the bound ligand, leading to de novo formation of a CCP at that site. So far, only the first mechanism has been directly observed15,16, by detecting single CCPs in living cells17. A direct observation of the second scenario may be more challenging as it requires the real-time detection of single ligand molecules or particles simultaneously with single CCPs.
Because of its medical importance, influenza has long been used as a model system to understand viral entry mechanisms. Influenza viruses enter cells via receptor-mediated endocytosis and then progress to late endosomes where viral fusion leads to the release of the viral genome1,9,18–23. While it has been established that influenza binds to the terminal sialic acid of glycoproteins and glycolipids on cell surfaces18, little is known about how these viruses are targeted to cellular endocytic structures after binding.
The exact endocytic pathway exploited by influenza is also unclear. Electron microscopy of virus infected cells has revealed influenza viruses inside CCPs and CCVs as well as inside smooth pits and vesicles1, while Semliki Forest viruses were only found to associate with coated structures24. However, without real-time imaging, it is not clear whether these smooth pits lead to influenza entry, nor is it known whether the smooth virus-bearing vesicles result from uncoating of CCVs or from clathrin-independent endocytosis. A recent study shows that dominant negative mutants and drugs that specifically block clathrin-mediated and caveolin-mediated endocytosis do not significantly affect the infectivity of influenza8. This indicates that the virus can infect cells via a clathrin- and caveolin-independent pathway(s) although it is unclear whether this pathway is utilized by influenza under unperturbed cellular conditions. The relative significance of the clathrin-mediated and clathrin- and caveolin-independent pathways in influenza infection is at present unknown. The molecular mechanism of the clathrin- and caveolin-independent pathway also remains largely elusive.
In this work, we have used real-time fluorescence microscopy to simultaneously track individual influenza viruses and endocytic structures in living cells. This has allowed us to characterize the endocytic mechanism exploited by each virus without inhibiting specific endocytic pathways. In addition, the ability to monitor individual viruses in real time provides previously unavailable information about the dynamics of endocytic processes and is particularly well suited to address how viruses are targeted to endocytic machinery. Our single-virus studies show that influenza viruses enter cells via multiple endocytic pathways, with both the clathrin-mediated pathway and a clathrin- and caveolin-independent pathway(s) significantly populated. Both pathways lead to viral fusion with intracellular membrane compartments and thus, presumably, to the release of the viral genome. Simultaneous tracking of single viruses and single CCPs show that viruses exploiting the clathrin-mediated pathway are internalized via the de novo formation of CCPs around the viruses. We have resolved the formation and disassembly dynamics of these CCPs/CCVs in real time. Remarkably, the CCP formation rate is much higher at the sites of bound viruses than elsewhere on the cell surface. The kinetics of CCP formation at these sites suggest that the formation is most likely induced by viral binding.
RESULTS
Imaging endocytic structures and viruses
To visualize endocytic structures in cells, we express clathrin or caveolin fused to fluorescent proteins following previously established procedures7,17,25,26. Briefly, we constructed a plasmid encoding enhanced yellow fluorescent protein (EYFP) attached to clathrin light chain a (LCa). This fusion protein was expressed in BS-C-1 cells. It has been shown previously that the functional integrity of clathrin is not compromised by fluorescent protein labels17,25,27,28. The distribution of EYFP-labeled clathrin (EYFP-clathrin) in cells shows discrete, dynamic fluorescent structures (Fig. 1, Supplementary Video 1). To test whether the EYFP image reveals all CCPs and CCVs, we have immunolabeled these structures with an antibody against the AP2 complex, an abundant protein in CCPs and CCVs. Over 97% of discrete structures in the immunofluorescence image colocalize with those in the EYFP image (Fig. 1c)17,25. In addition, we have imaged fluorescently-labeled transferrin, a known marker for clathrin-mediated endocytosis, and found that over 96% of transferrin spots colocalize with EYFP-clathrin structures before their internalization (Fig. 1d). These spots were subsequently internalized as indicated by microtubule-dependent movement and disappearance of the associated EYFP-clathrin signal (data not shown). These observations suggest that our EYFP fluorescence images reveal nearly all CCPs and CCVs in living cells.
Figure 1.
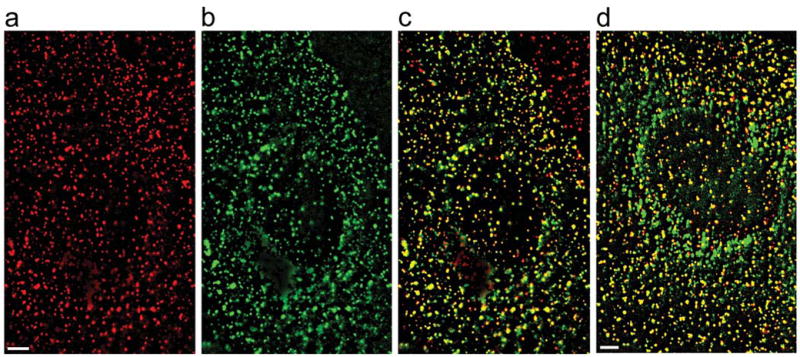
Fluorescence images of clathrin-coated structures in BS-C-1 cells expressing EYFP-clathrin. For a clearer visualization of discrete fluorescent spots, a low-spatial frequency background due to cytoplasmic EYFP-clathrin has been subtracted from the images. Scale bars: 3 μm (a) The immunofluorescence image of clathrin-coated structures in a cell transiently transfected with EYFP-clathrin. (b) The EYFP fluorescence image of clathrin-coated structures in the same cell. (c) Overlay of the immunofluorescence signal (red) with the EYFP signal (green). Pixels with both red and green signal appear yellow. Counting spots in multiple cells shows that over 97% of the clathrin-coated structures that appeared in the immunofluorescence image colocalized with those in the EYFP image. The red-only spots in the upper-right corner of the image belong to a neighboring cell not transfected with EYFP-clathrin. (d) Overlay of the transferrin image (red) and EYFP image (green) of a cell stably expressing EYFP-clathrin. Alexa Fluor 647-labeled transferrin (Molecular Probes) was bound to cells at 4°C for 15 minutes. After removing unbound transferrin, cells were imaged at room temperature. An image of the first 5 s is shown. Over 96% of the transferrin spots colocalize with clathrin-coated structures and appear yellow.
Similarly, we expressed enhanced green fluorescent protein (EGFP)-tagged caveolin-1 (caveolin-1-EGFP) in BS-C-1 cells7,26,29,30. The distribution of caveolin-1-EGFP in cells shows discrete fluorescent structures (Supplementary Video 2), which were previously suggested to be individual caveolae and caveosomes7,26,29,30. Nearly perfect colocalization has been found between the caveolin structures in the EGFP image and those in the immunofluorescence image obtained using anti-caveolin antibodies (data not shown)7. The cholera toxin subunit β, a known marker for caveolin-mediated endocytosis31, was successfully internalized via labeled caveolae in these caveolin-1-EGFP expressing cells (Supplementary Fig. 1), indicating that the EGFP label did not significantly perturb the internalization of caveolae7.
To visualize individual viruses in living cells32–36, we labeled the influenza virus with a lipophilic dye, DiD. This dye-labeling does not affect viral infectivity36. Tracking single viruses revealed a three-stage active transport process (Fig. 2) similar to what we have observed previously in CHO cells, with stage I being an actin-dependent movement in the cell periphery, stage II being a rapid, unidirectional movement on the microtubule towards the perinuclear region, and stage III being a bi-directional, microtubule-dependent movement36. The actin- or microtubule-dependence of these movements were tested with actin- or microtubule-disrupting drugs in ways similar to our previous study36. Here we use the onset of the microtubule-dependent stage II movement to indicate that internalization of the virus has already occurred. We then determine the endocytic mechanism responsible by examining the colocalization of the virus with CCPs and caveolae prior to this movement.
Figure 2.
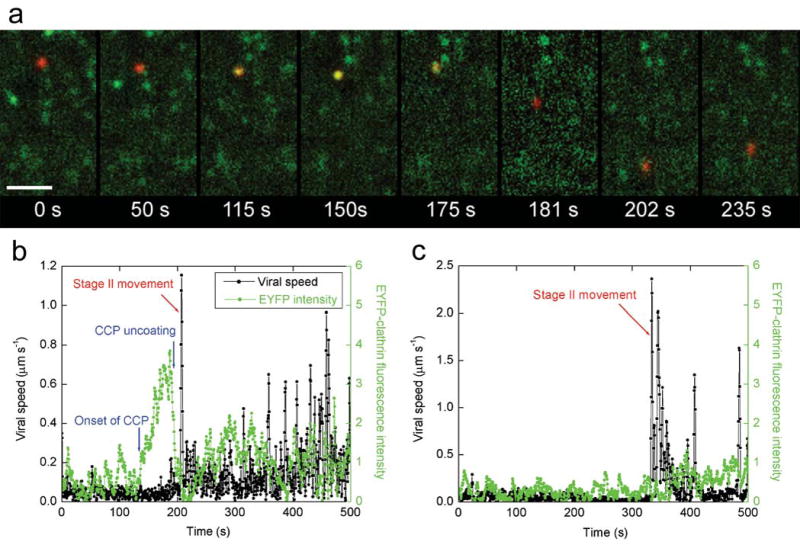
Internalization of influenza viruses via multiple pathways. (a) Snapshots of a virus internalized via a CCP. A live movie of this virus is available as Supplementary Video 3. Scale bar: 10 μm. t = 0 s: the virus (red) binds to the cell. t = 50 s: the virus is undergoing stage I movement. t = 115 s: a CCP labeled with EYFP (green) begins to form at the virus site. t = 150 s, the clathrin coat reaches its peak fluorescence intensity. t = 175 s: the clathrin coat rapidly disassembles. t = 181 seconds: the virus is transported towards the nucleus on a microtubule (stage II movement). t = 202 s: the virus enters stage III transport involving both plus- and minus-ended-directed motilities on microtubules. t = 235 s: the virus continues stage III movement. (b) The time-trajectories of a virus internalized via de novo formation of a CCP. Black symbols are the velocity time-trajectories of the virus. Stage II movement is identified as the rapid unidirectional translocation from the cell periphery to the perinuclear region (red arrows). Green symbols are the integrated fluorescence intensity of EYFP-clathrin associated with the virus. (c) The time-trajectories of a virus internalized without association with a clathrin-coated structure. Symbols are as defined in b. Live movies of the two viruses in b and c are also available (Supplementary Videos 4 and 5).
Multiple influenza entry pathways
To determine whether influenza viruses are internalized via clathrin-mediated endocytosis, we infected BS-C-1 cells expressing EYFP-clathrin with DiD-labeled viruses and imaged individual viruses and clathrin-coated structures simultaneously (Supplementary Video 1). We found that most viruses had been internalized into the cells as indicated by the onset of stage II movement. The average time between viral binding and stage II movement is about 5.6 min, in quantitative agreement with that obtained in cells not expressing EYFP-clathrin36. About 65% of the internalized viruses had associated with a discrete clathrin-coated structure for an extended period of time, followed by a rapid disappearance of the clathrin signal (Fig. 2a,b, Supplementary Videos 3 and 4), suggesting the internalization of the viruses via CCPs and the rapid uncoating of CCVs afterwards. The remaining 35% of the viruses did not show any association with clathrin-coated structures before stage II movement (Fig. 2c, Supplementary Video 5), suggesting their internalization via a clathrin-independent pathway. Similar partition values were obtained for infection started at various times ( ≥ 24 hours) post-transfection with EYFP-clathrin and for infection in cells stably expressing EYFP-clathrin. This partition was also independent of the excitation laser intensity used. The same partition ratio was obtained when we used the initial acidification of virus particles to pH 636, instead of stage II movement, as an indication that they have already been internalized into intracellular endocytic compartments (data not shown).
De novo formation of CCPs around viruses
For those viruses endocytosed via the clathrin-mediated pathway, an interesting question arises as to how these viruses are targeted to CCPs. Our quantitative analysis shows that only 6% of the viruses joined with a pre-existing CCP. The remaining 94% were internalized via the de novo formation of a CCP at the virus-binding sites: these viruses initially bound to sites that showed no discrete EYFP-clathrin signal and then a fluorescent EYFP spot appeared centered on the virus, typically 2–3 minutes after viral binding. The EYFP fluorescence intensity gradually increased and then rapidly disappeared before the virus started stage II movement (Fig. 2a,b, Supplementary Videos 3 and 4). Our quantitative analysis of the relative displacement between the CCP and the virus confirms this visual impression that CCP formation begins directly centered on the virus (Supplementary Fig. 2). We occasionally observed clathrin-structures passing by, temporarily colocalizing with and then separating from viruses. These events are not counted as clathrin-mediated endocytosis.
Dynamics of clathrin-mediated endocytosis
Real-time observation of the entire formation and disassembly process of CCPs/CCVs around the viruses allows us to determine the dynamics of the process (Fig. 3). The formation of CCPs is initiated on average 190 s after the viruses bind to the cell (Fig. 3a). The probability of CCP formation increases with time initially and peaks after a lag of 170 s following viral binding (Fig. 3a). The clathrin signal persists for about 70 s on average (Fig. 3b). This period includes a clathrin accumulation phase with a roughly linearly increasing clathrin intensity that indicates the recruitment of clathrin to form a CCP, which then pinches off from the plasma membrane to form a CCV, followed by rapid uncoating of the CCV that is typically completed in only a few seconds (Fig. 3b). The lifetimes of the virus-bound CCPs are similar to those of the constitutive CCPs not associated with viruses (data not shown).
Figure 3.
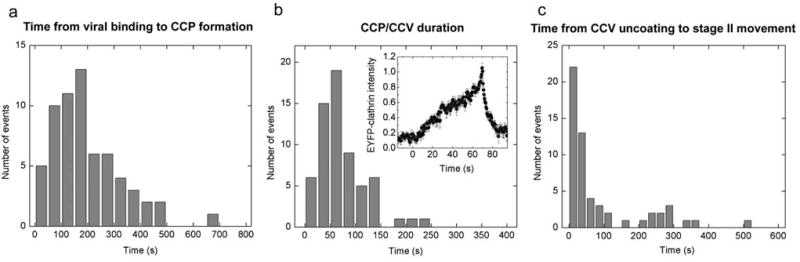
Dynamics of the CCPs and CCVs formed de novo at the virus-binding sites. (a) A histogram of the time between viral binding and onset of CCP formation. (b) A histogram of the time between the onset of CCP formation and CCV uncoating. Inset: an averaged curve of 60 time-trajectories of the EYFP-clathrin intensity associated with viruses shows the life cycle of CCPs. Each EYFP-clathrin trajectory was normalized to the peak height and scaled in time so that the onset of the CCP formation occurs at t = 0 sec and the peak clathrin intensity occurs at t = 69 sec, which is the average duration of the clathrin-accumulation phase. (c) A histogram of the time between the first CCV uncoating event and the onset of stage II viral movement.
The above results show that the viruses were not typically targeted to pre-existing CCPs, but were predominantly internalized via CCPs that formed de novo at the site of the bound viruses. However, this does not necessarily imply that the formation of these pits is induced by viral binding. As CCPs are constitutively forming on the cell surface, the possibility exists that a CCP would by chance form at the virus site. To address this possibility, we analyzed the formation rate of CCPs at random sites on the cell surface. Remarkably, we found that the formation rate at random sites was 20 times lower than that at the sites of bound viruses.
Is it then possible that the viruses are captured by “hot spots” on the cell surface that have a higher CCP formation rate17,25? To look for these hot spots, we chose sites of pre-existing CCPs and analyzed the waiting time for another CCP to form at the same or nearby sites after the initial CCP had disappeared. The CCP formation rate obtained at these sites is, however, similar to the rate obtained at random sites on the cell surface. Only at less than 5% of the pre-existing-CCP sites, did we observe a rapidly ‘blinking” fluorescent spot that may indicate repetitive CCP formation17,25. In addition, the “dark” times at these “blinking” sites are only about 10–20 s, much shorter than the average time it takes for a CCP to appear at a virus binding site both before and after the viral binding event. These results indicate that the de novo CCP formation at the virus-binding sites are not due to static “hot spots” on the cell surface.
The possibility remains that the observed high CCP formation rate is due to preferential association of viruses with mobile “hot spots” that have a higher CCP formation rate. If the viruses were indeed internalized by binding to these hypothetical “hot spots” and did not influence the CCP formation rate there, then the probability that a CCP appears at these sites should be independent of time relative to viral binding and the distribution of the relative time (T1) between viral binding and CCP formation should follow an exponential decay without any lag. This is inconsistent with our observed T1 distribution, which shows a significant lag (Fig. 3a). Could the lag be caused by the relative movement between viruses and “hot spots” on the cell surface before their association? If so, the average time it takes a CCP to appear at the “hot spots” should be less than <T1> (190 s). Considering that the average duration of a CCP is 70 s, the probability of finding a CCP at the “hot spots” should be greater than 27% at any time. Thus if viruses move to the “hot spots” to get internalized, the fraction of viruses that joined with a pre-existing CCP should be greater than 27%, again inconsistent with our observed value (6%). These results suggest that the CCPs that appear de novo at the sites of the bound viruses are most likely induced or influenced by viral binding.
After clathrin uncoating, the viruses show heterogeneous transport dynamics. The majority of viruses started their microtubule-dependent movement less than 40 s after uncoating; whereas the rest remained in the cell periphery for several minutes before stage II movement (Fig. 3c and Supplementary Fig. S3). For the latter fraction of viruses, the relatively long lag between clathrin uncoating and stage II movement leaves open the possibility that the viruses were recycled back to the cell surface and were then re-endocytosed. Indeed, we occasionally observed a second round of CCP formation during that period. If the hypothetical re-endocytosis events occur via a clathrin-independent mechanism, it could lead to a moderate effect on the partition ratio of viruses between the clathrin-dependent and -independent pathways, as we detail in Supplementary Fig. 3.
Effect of neuraminidase on influenza entry
Influenza viruses bind to sialic acids on cell surfaces via hemagglutinin in a highly multivalent manner. Such multivalent binding could restrict viral mobility on the cell surface. Influenza neuraminidase cleaves hemagglutinin-sialic acid bonds37. It is thus possible that neuraminidase activity facilitates viral movement on the cell surface and targeting to endocytic sites and, therefore, plays a significant role in cellular entry of influenza. However, our observation of de novo CCP formation at viral binding sites suggests that neuraminidase activity may not be critical for the internalization of influenza. To test this, we investigated the cellular entry of influenza in the presence of neuraminidase inhibitors RWJ-270201 and Oseltamivir38,39. While these inhibitors effectively block influenza infection at the concentration of a few nM (data not shown)38,39, we fail to observe their effect on viral endocytosis, trafficking and fusion even at 1μM concentration. The relative time between viral binding and the stage II movement is very similar to that in the absence of drugs (Fig. 4), suggesting that neuraminidase is not essential for the cellular entry of influenza and these recently developed influenza drugs function to block other stages of the infection37.
Figure 4.

The effect of neuraminidase (NA) inhibitors on the endocytosis of influenza viruses. Grey columns show a histogram of the time between viral binding and the onset of stage II movement in the absence of neuraminidase inhibitors. Red columns show the histogram in the presence of 1 μM RWJ270201. The histogram in the presence of 1 μM oseltamivir is similar (data not shown).
Clathrin- and caveolin-independent pathway
Next, we address the internalization mechanism of those viruses that entered cells without colocalization with CCPs. To this end, we infected BS-C-1 cells expressing caveolin-1-EGFP with DiD-labeled viruses and imaged individual viruses together with caveolae and caveosomes (Supplementary Video 2). We found that most viruses had been internalized into the cells as indicated by the onset of stage II movement and the average time between viral binding and stage II movement was similar to that in cells not expressing caveolin-1-EGFP. Among the internalized viruses, less than 5% showed co-localization with caveolae/caveosomes before internalization. In cells treated with filipin (5 μg/ml), an inhibitor of caveolin-mediated endocytosis40, we observed a similar partition ratio of viruses between the clathrin-dependent and -independent pathways to that in untreated cells, while the same filipin treatment blocks the internalization of cholera toxin subunit β [M.L., M.J.R., X.Z. unpublished]. The above observations indicate that the clathrin-independent endocytic pathway exploited by influenza is also caveolin- and lipid raft-independent14.
To initiate infection, influenza viruses release their genome into the cytoplasm by fusing with endosomes1,9,18–23. We tested whether the clathrin-mediated pathway and clathrin- and caveolin-independent pathway both lead to viral fusion. To this end, we labeled the viruses with a high surface density of DiD such that its fluorescence is partially quenched. Viral fusion events can be identified by a dramatic increase in the DiD fluorescence intensity due to fusion-induced dequenching (Fig. 5, Supplementary Videos 6 and 7)36. Among the viruses that fused, 69% had shown association with clathrin-coated structures before stage II movement, whereas 31% had not. This partition is almost identical to that obtained for all viruses showing microtubule-dependent movement.
Figure 5.
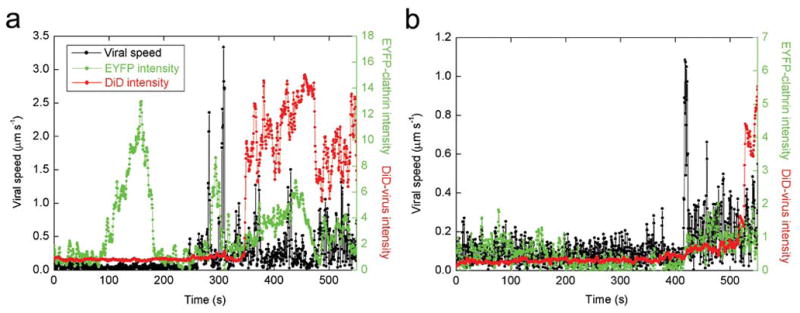
Time-trajectories of viruses that successfully fused after endocytosis. (a) An example of a virus that was internalized via a CCP. (b) An example that was internalized without association with a CCP. Live movies of these two viruses are available (Supplementary Videos 6 and 7). Black symbols are the velocity time-trajectories of the viruses. Red symbols are the integrated DiD fluorescence intensities of the viruses. Viral fusion can be identified as a dramatic increase of the DiD signal. Green symbols are the integrated fluorescence intensities of EYFP-clathrin associated with the viruses.
Next, we compare the trafficking kinetics of viruses along the clathrin-dependent and -independent pathways. The distributions of time between viral binding and stage II movement are similar for the two pathways (Supplementary Fig. 4a). This is not surprising if the two endocytic pathways are competing for viral entry. The time distributions between stage II movement and viral fusion are also similar for the two pathways (Supplementary Fig. 4b). This may in part be caused by the convergence of clathrin-mediated and clathrin-independent pathways in post-endocytic trafficking41. We have indeed observed merging events of virus-containing vesicles internalized via the two pathways prior to viral fusion. These results are consistent with a previous finding that influenza viruses enter endosomes at similar times whether or not dominant negative mutants were present to block clathrin-mediated endosytosis42. The distances between the fusion sites and the nuclear envelope are also similar for viruses entering via the two pathways (Supplementary Fig. 4c).
The above results indicate that the clathrin-mediated and -independent pathways are equally efficient for viral fusion once the viruses are internalized. Considering previous results that influenza viruses fuse with endosomes to release their genome and initiate infection1,9,18–23 and that influenza infects cells in the presence of drugs and mutants that block clathrin- and caveolin-mediated endocytosis8, our data suggest that the two pathways are likely equally efficient for infection once the viruses are internalized. Our results further show that clathrin- and caveolin-independent endocytosis is not an alternative means exploited by influenza only when clathrin-dependent endocytosis is blocked, but a parallel pathway that influenza takes under normal cellular conditions.
DISCUSSION
While receptor-mediated endocytosis is known to be one of the major mechanisms that viruses exploit to enter cells, the molecular mechanisms underlying the complex processes of viral endocytosis are still poorly understood. In this work, we have visualized single influenza viruses and endocytic structures in living cells and tracked the interaction between them in real time using fluorescence microscopy. This has allowed us to determine the endocytic mechanisms exploited by influenza without inhibiting specific endocytic pathways and to directly address how influenza viruses are targeted to endocytic machinery for internalization.
Our single-virus trajectories show that influenza viruses enter cells via multiple endocytic pathways. In any given infection experiment, roughly two thirds of the viruses enter cells via clathrin-mediated endocytosis and the remaining via a clathrin- and caveolin-independent endocytic pathway(s). Both pathways lead to viral fusion with intracellular membrane compartments with a similar efficiency.
By tracking the interaction between viruses and CCPs in real time, we show that the viruses exploiting the clathrin-mediated pathway are predominantly (94%) internalized via de novo formation of CCPs around the viruses. While recruitment to pre-existing CCPs has been observed previously for G-protein coupled receptors15,16, the de novo formation of CCPs at the sites of bound ligands has not been directly observed before. We have resolved the dynamics of CCP formation around the viruses: clathrin begins to appear at the viral binding sites a few minutes after binding; this is followed by a roughly linear increase in the clathrin intensity for about one minute, during which time a CCP is formed and matures into a CCV. Immediately after the clathrin intensity reaches its maximum, the clathrin coat rapidly disassembles, typically in only a few seconds. Remarkably, we found that the formation rate of CCPs was 20 times higher at the sites of bound viruses than elsewhere. The formation kinetics of CCPs at these sites suggest that the formation is most likely induced by viral binding.
Ligand-induced clathrin-redistribution to the plasma membrane has been previously reported for epidermal growth factor (EGF) and nerve growth factor (NGF)43. There, the binding of EGF to its receptor causes phosphorylation of the clathrin heavy chain and a global redistribution of clathrin to the cell periphery to form CCPs44. The majority of newly induced CCPs do not actually colocalize with the NGF receptors45,46. This is distinct from our observations of de novo formation of CCPs at the virus binding sites without a global redistribution of clathrin upon viral binding.
An interesting question arises: since influenza viruses bind to sialic acids of general glycolipids and glycoproteins on the cell surface instead of specific receptors with known internalization motifs, how is the signal of viral binding transmitted across the plasma membrane to initiate the recruitment of clathrin and associated factors and induce the formation of a CCP at the binding site? One possible mechanism stems from the highly multivalent binding of influenza to the cell via multiple hemagglutinin-sialic acid bonds. This highly multivalent binding of the nearly spherical virus to the cell surface may induce local curvature on the plasma membrane, thereby promoting formation of a CCP at the binding site. It is particularly interesting to note the most conserved domain (BAR domain) of amphiphysin, a component of CCPs, forms a crescent-shaped dimer structure that preferentially binds to highly-curved negative-charged membranes47. While the wildtype amphiphysin can efficiently recruit clathrin to liposomes and polymerize clathrin into an invaginated lattice on a lipid monolayer, an amphiphysin with mutations in the BAR domain lacks such ability47. We speculate that amphiphysin or other proteins with a similar BAR domain preferentially bind to the curved plasma membrane at the influenza virus binding sites and promote CCP formation at those sites.
METHODS
Viruses and fluorescent labeling
Influenza virus X-31 was purchased from Charles River Laboratories. To label with lipophilic dyes, the viruses were incubated with DiD (Molecular Probes) for 2 hours at 22°C. Unbound dye was removed by buffer exchange into 50 mM HEPES buffer (pH 7.4, 145 mM NaCl) using gel filtration columns. Immediately before experiments, viral aggregates were removed with 0.2 μm pore size filters.
Cell culture
BS-C-1 cells were maintained in a 5% CO2 environment in Minimum Essential Eagle Medium (MEM, Invitrogen) with 10% (v/v) fetal bovine serum (FBS) and passaged every 2–3 days. For fluorescence imaging, BS-C-1 cells were cultured in MEM with 10% (v/v) FBS in Petri dishes with glass coverslips on the bottom. Prior to fluorescence experiments, cells were washed in serum-free, phenol red-free medium fortified with 100 mM pH 8.0 Hepes buffer.
Plasmids
We obtained the GFP-clathrin-LCa construct as a gift from J. H. Keen17. From this construct, the clathrin LCa cDNA was PCR amplified to introduce an Xho I restriction site at the 5′ end and an Age I restriction site at the 3′ end. Similarly, we amplified the EYFP cDNA, introducing an Xho I restriction site at the 3′ end. The EYFP PCR product was cloned into the pcDNA3.1/V5-His-TOPO vector (Invitrogen). This construct was then digested with Age I and Xho I (New England Biolabs), and the clathrin-LCa PCR product was ligated using the these sites into the vector containing EYFP, yielding a cDNA coding for a chimeric protein with EYFP fused to the N-terminus of clathrin-LCa. The caveolin-1-EGFP construct was a gift from A. Helenius7.
Transfection of cells
Cells were transfected with EYFP-clathrin-LCa or caveolin-1-EGFP using FuGENE 6 transfection reagent (Roche). For a 60 mm petri dish to be transfected, 2 μl of FuGENE 6 reagent was combined with MEM and 1 μg of DNA for a final volume of 100 μl. The FuGENE/DNA mixture was incubated at room temperature for 20 min, added to the cell culture, and then incubated in 5% CO2 environment.
Cells stably expressing EYFP-clathrin
The stable cell line was created using the RetroMax retroviral expression system (Imgenex). The EYFP-LCa gene was cloned into the Hind III/Cla I restriction sites of the pCLNCX retrovirus expression vector (Imgenex) that confers G418 resistance. 293T cells, growing in Dulbecco’s Modified Eagle’s Medium (DMEM) containing 10% (v/v) FBS, were then transfected using this vector and the pCL-10A1 retrovirus packaging vector (Imgenex). 293T supernatant was collected after 2 days and filtered through 0.45 micron pore size filters. The target BS-C-1 cells were then infected with an equal volume mix of the 293T supernatant with cell medium (MEM containing 10% (v/v) FBS). 24 hours after infection, the cells were split into various dilutions (1:20 to 1:100) and selection was begun by adding medium containing 1.15 mg ml−1 G418. New medium containing G418 was then introduced every three days. Visible antibiotic resistant colonies were formed approximately 14 days after infection. These colonies were then picked and seeded onto new plates and propagated.
Immunofluorescence
Cells transfected with EYFP-clathrin-LCa were fixed in 2% (v/v) formaldehyde for 40 min. After washing with PBS, cells were permeablized in PBS containing 10% (v/v) FBS, 3% (w/v) bovine serum albumin (BSA) and 0.5% (v/v) triton-X 100, and then incubated at 4°C overnight with the monoclonal antibody against α-adaptin (Affinity Bioreagents), a subunit of the AP2 complex25. The cells were then washed extensively with PBS containing 0.2% (w/v) BSA and 0.1% (v/v) triton-X 100 and incubated at room temperature for 30 min with FITC-conjugated goat anti-mouse IgG (Novus Biologicals). The FITC-conjugated antibodies were excited with 488nm Argon-ion laser (Melles Griot) and the fluorescence emission was collected using a 535/50 nm filter. At the laser power used, the emission from EYFP in this detection wavelength range is negligible.
Fluorescence imaging
To obtain a simultaneous image of DiD-labeled viruses and EYFP-tagged clathrin, DiD was excited with a 633 nm helium-neon laser (Melles-Griot) while EYFP was excited with a 532 nm diode-pumped Nd:YAG laser (Crystalaser). A custom designed polychroic beam splitter (Chroma) that reflects at wavelengths 520–550 nm and 615–655 nm was used to direct the laser lines onto the sample. The fluorescent emission from DiD and EYFP was collected by an oil-immersion objective with numerical aperture 1.45 (Olympus), spectrally separated by 610 nm long-pass dichroic mirrors (Chroma) and imaged onto two separate areas of the CCD camera (Roper Scientific, CoolSnap HQ). A 665 long pass filter was used for DiD emission and a 585/70 nm band pass filter was used for EYFP emission. Image series were recorded at 2 frames per second. For co-imaging of DiD-labeled viruses and EGFP-tagged caveolin, a 488 nm argon-ion laser (Melles Griot) was used to excite EGFP. A custom designed polychroic beam splitter (Chroma) that reflects at 475–495 nm and 615–640 nm was used to direct the laser lines onto the sample. Differential interference contrast optics were used to obtain cell images before and after the fluorescence imaging. Experiments were conducted at 37°C unless otherwise mentioned.
Image analysis
Each frame of the DiD fluorescence image series was processed by convolving with a Gaussian spatial filter to remove background and noise. Virus peaks were detected by recursively integrating over bright regions connected to each local maximum. The location of each virus was computed as the centroid of the bright region. Physical trajectories of viruses were reconstructed by pairing peaks in each frame to previously established trajectories according to proximity and similarity in intensity. The EYFP intensity associated with the virus was determined by subtracting a low-spatial frequency background signal in the EYFP channel and then integrating the EYFP signal weighted by the DiD signal of the virus. Only those viruses that start in the peripheral region of the cell surface and move roughly within the focal plane are analysed. Viruses that move by a substantial amount orthogonal to the focal plane (out of focus) are not considered. As BS-C-1 cells are comparatively flat, most virus trajectories remain close to the focal plane.
Supplementary Material
Videos were taken under the following imaging condition. For Videos 1–5, the excitation laser for the EYFP-clathrin or Caveolin-1-EGFP was turned on for 0.5 s every 1.5 s while that for the DiD-labeled viruses was always on. The camera integration time for each frame is 0.5 s. All camera pixels were read out individually. For Videos 6 and 7, the excitation laser for the EYFP-clathrin was turned on for 0.5 s every 1.5 s while that for the DiD-viruses was turned on for 0.5 s every 1s. The pixels were binned in a 2×2 fashion. Videos 6 and 7 thus do not allow an accurate determination of whether CCPs form at or off the virus-binding sites, but are only meant to show that both clathrin-dependent and -independent endocytosis can lead to viral fusion.
All videos are processed by subtracting the low-spatial frequency background signal generated by cytoplasmic EYFP-clathrin or Caveolin-1-EGFP. The videos are compressed significantly and compression compromises video quality.
As the viruses were added to the cells in situ the binding of viruses to cells is highly asynchronous. Thus some viruses appear to move only slowly while the circled viruses show the specified behavior of three-stage movement. These slow-moving viruses in the field of view are either in stage I or in stage III during the exhibited time window.
Supplementary Video 1. A dual-color movie of EYFP-tagged clathrin structures (green) and DiD-labeled viruses (red) in a live cell.
Supplementary Video 2. A dual-color movie of EGFP-tagged caveolin structures (green) and DiD-labeled viruses (red) in a live cell.
Supplementary Videos 3 and 4. The internalization of influenza viruses via CCPs. Both movies show the de novo formation of a CCP (green) around the virus (red, in white circles), the gradual increase of clathrin intensity, and the rapid disappearance of the clathrin signal immediately before the virus exhibit a rapid, unidirectional movement towards the perinuclear region (stage II movement).
Supplementary Video 5. The internalization of an influenza virus without association with a CCP. The virus (red, in a white circle) did not associate with a CCP before its stage-II movement inside the cell.
Supplementary Video 6 The internalization and fusion of an influenza virus (red, in a white circle) after association with a CCP (green). Fusion is indicated by a dramatic increase of the DiD signal (red).
Supplementary Video 7. The internalization and fusion of an influenza virus (red, in a white circle) without association with a CCP.
Supplementary Figure 1. Internalization of cholera toxin via caveolae. Alexa Fluor 647-conjugated cholera toxin subunit β (Molecular Probes) was incubated with cells expressing Caveolin-1-EGFP at 4°C for 15 min. After removing excess cholera toxin, the temperature was raised to 37°C to initiate endocyosis. Cholera toxin was found to localize into discrete spots (red images), all of which colocalize with caveolin-1-EGFP structures (green images). Three examples of these cholera toxin-containing structures are indicated by arrows. For each example, three consecutive images, each 10 s apart in time, are shown to illustrate the movement of these structures. Treating cells with nocodazole (60 μM) completely inhibited this movement, indicating that these EGFP-labeled caveolin structures containing cholera toxin were internalized and moving in a microtubule-dependent manner. Such structures were not observed in cells treated with filipin (5 μg/ml), an inhibitor of caveolin-dependent endocytosis, suggesting that the internalization of cholera toxin here is via caveolae. During the observation window (50–100 s), 54% of the cholera toxin-containing structures showed microtubule-dependent movement in Caveolin-1-EGFP expressing cells; and this fraction is similar in untransfected cells (52%). In both transfected and untransfected cells, we started to observe such mobile structures within 5 minutes of the initiation of endocytosis. These results indicate that the expression of Caveolin-1-EGFP did not significantly perturb caveolin-mediated endocytosis. Scale bars: 3 μm.
Supplementary Figure 2. Relative displacements of CCPs from their associated viruses. Black and red symbols are the displacements in the x and y directions in units of camera pixels (95 nm/pixel), respectively. We used least-squares fitting to determine the location of any EYFP-labeled clathrin structures near the virus prior to its stage II movement. In each frame, we considered a 4 μm2 area centered on the virus peak. The EYFP signal in this region was fit to a Gaussian function I (x, y) = I0+Ae−[(x−x0)2 +(y−y0)2]/2σ2, where I0 is the average background fluorescence level, A is the amplitude of the peak, x0 and y0 are the center coordinates, and σ is the width. I0, A, x0, and y0 were treated as free fitting parameters, while σ was fixed to be the experimentally measured width of a diffraction-limited spot. The error bars are determined from the variance matrix obtained by the least-squares fitting. Visual inspection of the movies shows that we are able track the peak as soon as a clathrin structure discernable from the background appears in the 4 μm2 area. In 96% of the trajectories, the clathrin peak position (x0, y0) starts centered on the virus to within 1 pixel, as indicated by examples shown in a–e. Among these, only 2% of viruses seem to land on a pre-existing CCP. The rest show the appearance of a clathrin spot after viral binding. The EYFP-clathrin intensity gradually increased and then rapidly disappeared before the virus started stage II movement. After the clathrin-structure has disappeared, tracking is no longer possible, as indicated by the large error bars appearing at the end of the plots. In the remaining 4% of the trajectories, the initial clathrin peak position is separated from that of the virus (i.e. x0 ≠ 0 and y0 ≠ 0) and the separation in general decreases with time until the virus joins the clathrin structure, as shown in f. This quantitative analysis confirms the results of our visual inspection and indicates de novo formation of CCPs at the site of bound viruses.
Supplementary Figure 3. An integrated histogram of the relative time between the last CCV uncoating event prior to stage II viral movement and the onset of stage II movement. Circles indicate the number of events with the relative time shorter than value indicated on the horizontal axis. The solid curve is a fit to a double exponential decay with time constants 20 s and 270 s. These data clearly show a biphasic behaviour: while the majority of viruses started their stage II movement relatively rapidly after uncoating with a time constant of 20 s, the rest remained in the cell periphery for several minutes before stage II movement. The former population of viruses most likely stays inside the cell during the brief period between clathrin uncoating and stage II movement, as both recycling and endocytosis typical take a few minutes or longer. However, the relatively long lag between clathrin uncoating and stage II movement for the latter population leaves open the possibility that these viruses may have entered the cell via a CCP, then recycled back to the cell surface and re-entered via a clathrin-independent mechanism. We thus reevaluate the partition ratio of viruses between the clathrin-dependent and -independent pathways by classifying endocytosis based on the internalization event immediately preceding stage II movement. This is reasonable as viral fusion was not observed without microtubule-dependent movement. In the extreme case that all of the viruses under the second phase were recycled and re-entered the cell without using a CCP, the fraction of viruses that enter via the clathrin-independent pathway would be increased from the observed 35% to 50%. The true fraction is most likely between these values.
Supplementary Figure 4. Histograms of the time between viral binding and stage II movement (a), the time between stage II movement and viral fusion (b), and the distance between the fusion site and the nuclear envelope (c) for viruses that enter via clathrin-dependent and -independent endocytosis.
Acknowledgments
We thank J. H. Keen and A. Helenius for their generous gifts of GFP-clathrin-LCa and Caveolin-1-EGFP plasmids, respectively. This work is supported in part by a Searle Scholarship, a Beckman Young Investigator award, Office of Naval research and National Science Foundation (to X.Z.). M.J.R. is a National Science Foundation pre-doctoral fellow.
References
- 1.Matlin KS, Reggio H, Helenius A, Simons K. Infectious entry pathway of influenza-virus in a canine kidney-cell line. J Cell Biol. 1981;91:601–613. doi: 10.1083/jcb.91.3.601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Doxsey SJ, Brodsky FM, Blank GS, Helenius A. Inhibition of endocytosis by anti-clathrin antibody. Cell. 1987;50:453–463. doi: 10.1016/0092-8674(87)90499-5. [DOI] [PubMed] [Google Scholar]
- 3.Anderson HA, Chen YZ, Norkin LC. Bound simian virus 40 translocates to caveolin-enriched membrane domains, and its entry is inhibited by drugs that selectively disrupt caveolae. Mol Biol Cell. 1996;7:1825–1834. doi: 10.1091/mbc.7.11.1825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Carbone R, et al. Eps15 and eps15R are essential components of the endocytic pathway. Cancer Res. 1997;57:5498–5504. [PubMed] [Google Scholar]
- 5.Stang E, Kartenbeck J, Parton RG. Major histocompatibility complex class I molecules mediate association of SV40 with caveolae. Mol Biol Cell. 1997;8:47–57. doi: 10.1091/mbc.8.1.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.DeTulle L, Kirchhausen T. The clathrin endocytic pathway in viral infection. EMBO J. 1998;17:4585–4593. doi: 10.1093/emboj/17.16.4585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pelkmans L, Kartenbeck J, Helenius A. Caveolar endocytosis of simian virus 40 reveals a new two-step vesicular-transport pathway to the ER. Nat Cell Biol. 2001;3:473–483. doi: 10.1038/35074539. [DOI] [PubMed] [Google Scholar]
- 8.Sieczkarski SB, Whittaker GR. influenza virus can enter and infect cells in the absence of clathrin-mediated endocytosis. J Virol. 2002;76:10455–10464. doi: 10.1128/JVI.76.20.10455-10464.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pelkmans L, Helenius A. Insider information: what viruses tell us about endocytosis. Curr Opin Cell Biol. 2003;15:414–422. doi: 10.1016/s0955-0674(03)00081-4. [DOI] [PubMed] [Google Scholar]
- 10.Nichols BJ, Lippincott-Schwartz J. Endocytosis without clathrin coats. Trends Cell Biol. 2001;11:406–412. doi: 10.1016/s0962-8924(01)02107-9. [DOI] [PubMed] [Google Scholar]
- 11.Conner SD, Schmid SL. Regulated portals of entry into cells. Nature. 2003;422:37–44. doi: 10.1038/nature01451. [DOI] [PubMed] [Google Scholar]
- 12.Brodsky FM, Chen CY, Knuehl C, Towler MC, Wakeham DE. Biological basket weaving: Formation and function of clathrin-coated vesicles. Annu Rev Cell Dev Biol. 2001;17:517–568. doi: 10.1146/annurev.cellbio.17.1.517. [DOI] [PubMed] [Google Scholar]
- 13.Kirchhausen T. Clathrin. Annu Rev Biochem. 2000;69:699–727. doi: 10.1146/annurev.biochem.69.1.699. [DOI] [PubMed] [Google Scholar]
- 14.Nabi IR, Le PU. Caveolae/raft-dependent endocytosis. J Cell Biol. 2003;161:673–677. doi: 10.1083/jcb.200302028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Scott MGH, Benmerah A, Muntaner O, Marullo S. Recruitment of activated G protein-coupled receptors to pre-existing clathrin coated pits in living cells. J Biol Chem. 2002;277:3552–3559. doi: 10.1074/jbc.M106586200. [DOI] [PubMed] [Google Scholar]
- 16.Santini F, Gaidarov I, Keen JH. G protein-coupled receptor/arrestin3 modulation of the endocytic machinery. J Cell Biol. 2002;156:556–676. doi: 10.1083/jcb.200110132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gaidarov I, Francesca S, Warren RA, Keen JH. Spatial control of coated-pits dynamics in living cells. Nat Cell Biol. 1999;1:1–7. doi: 10.1038/8971. [DOI] [PubMed] [Google Scholar]
- 18.Skehel JJ, Wiley DC. Receptor binding and membrane fusion in viral entry: the influenza hemagglutinin. Annu Rev Biochem. 2000;69:531–569. doi: 10.1146/annurev.biochem.69.1.531. [DOI] [PubMed] [Google Scholar]
- 19.White J, Helenius A, Gething MJ. Heaemagglutinin of influenza virus expressed from a cloned gene promote membrane fusion. Nature. 1982;300:658–659. doi: 10.1038/300658a0. [DOI] [PubMed] [Google Scholar]
- 20.Yishimura A, Ohnishi S. Uncoating of influenza viruses in endosomes. J Virol. 1984;51:497–504. doi: 10.1128/jvi.51.2.497-504.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Martin K, Helenius A. Transport of incoming influenza virus nucleocapsids into the nucleus. J Virol. 1990;65:232–244. doi: 10.1128/jvi.65.1.232-244.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lamb RA, Krug RM. Orthomyxoviridae: the viruses and their replication. In: Knipe DM, Howley PM, editors. Fields Virology. Lippincott Williams and Wilkins; Philadelphia: 2001. pp. 1487–1531. [Google Scholar]
- 23.Klasse PJ, Bron R, Marsh M. Mechanisms of enveloped virus entry into animal cells. Adv Drug Del Rev. 1998;34:65–91. doi: 10.1016/S0169-409X(98)00002-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Helenius A, Kartenbeck J, Simon K, Fries E. On the entry of Semliki Forest viruses into BHK-21 cells. J Cell Biol. 1980;84:404–420. doi: 10.1083/jcb.84.2.404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Marrifield CJ, Feldman ME, Wan L, Almers W. Imaging actin and dynamin recruitment during invagination of single clathrin-coated pits. Nat Cell Biol. 2002;4:691–698. doi: 10.1038/ncb837. [DOI] [PubMed] [Google Scholar]
- 26.Volonte D, Galbiati F, Lisanti MP. Visualization of cavelin-1, a caveolar marker protein, in living cells using green fluorescent protein (GFP) chimeras. FEBS Lett. 1999;445:431–439. doi: 10.1016/s0014-5793(99)00164-7. [DOI] [PubMed] [Google Scholar]
- 27.Wu X, et al. Clathrin exchange during clathrin-mediated endocytosis. J Cell Biol. 2001;155:291–300. doi: 10.1083/jcb.200104085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rappoport JZ, Simon SM. Clathrin-mediated endocytosis during cell migration. J Cell Sci. 2003;116:847–855. doi: 10.1242/jcs.00289. [DOI] [PubMed] [Google Scholar]
- 29.Pelkmans L, Punterner D, Helenius A. Local actin polymerization and dynamin recruitment in SV-40-induced internalization of caveolae. Science. 2002;296:535–539. doi: 10.1126/science.1069784. [DOI] [PubMed] [Google Scholar]
- 30.Thomsen P, Roepstorff K, Stahlhut M, Deurs BV. Caveolar are highly immobile plasma membrane microdomains, which are not involved in constitutive endocytic trafficking. Mol Biol Cell. 2002;13:238–250. doi: 10.1091/mbc.01-06-0317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schnitzer JE, Oh P, Pinney E, Allard J. Filipin-sensitive caveolae-mediated transport in endothelium-reduced transcytosis, scavenger endocytosis, and capillary-permeability of select macromolecules. J Cell Biol. 1994;127:1217–1232. doi: 10.1083/jcb.127.5.1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Georgi A, Mottola-hartshorn C, Warner W, Fields B, Chen LB. Detection of individual fluorescently labelled reovirions in living cells. Proc Natl Acad Sci USA. 1990;87:6579–6583. doi: 10.1073/pnas.87.17.6579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Suomalainen M, et al. Microtubule-dependent plus- and minus end-directed motilities are competing processes for nuclear targeting of adenovirus. J Cell Biol. 1999;144:657–672. doi: 10.1083/jcb.144.4.657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Seisenberger G, et al. Real-time single-molecule imaging of the infection pathway of an Adeno-associated virus. Science. 2001;294:1929–1932. doi: 10.1126/science.1064103. [DOI] [PubMed] [Google Scholar]
- 35.McDonald D, et al. Visualization of the intracellular behavior of HIV in living cells. J Cell Biol. 2002;159:441–452. doi: 10.1083/jcb.200203150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lakadamyali M, Rust MJ, Babcock HP, Zhuang X. Visualizing infection of individual influenza viruses. Proc Natl Acad Sci, USA. 2003;100:9280–9285. doi: 10.1073/pnas.0832269100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wagner R, Matrosovich M, Klenk H-D. Functional balance between haemagglutinin and neuraminidase in influenza virus infection. Rev Med Viol. 2002;12:159–166. doi: 10.1002/rmv.352. [DOI] [PubMed] [Google Scholar]
- 38.Babu YS, et al. BCX-1812 (RWJ-270201): Discovery of a novel, highly potent, orally active, and selective influenza neuraminidase inhibitor through structure-based drug design. J Med Chem. 2000;43:3482–3486. doi: 10.1021/jm0002679. [DOI] [PubMed] [Google Scholar]
- 39.Sidwell RW, et al. In vivo influenza virus-inhibitory effects of the cyclopentane neuraminidase inhibitor RWJ-270201. Antimicrob Agents Chemo. 2001;45:749–757. doi: 10.1128/AAC.45.3.749-757.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rothberg KG, Ying YS, Kamen BA, Anderson RGW. Cholesterol controls the clustering of the glycophospholipid-anchored membrane-receptor for 5-methyltetrahydrofolate. J Cell Biol. 1990;111:2931–2938. doi: 10.1083/jcb.111.6.2931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Naslavsky N, Weigert R, Donaldson JG. Convergence of non-clathrin-and clathrin-derived endosomes involves Arf6 inactivation and changes in phosphoinositides. Mol Biol Cell. 2003;14:417–431. doi: 10.1091/mbc.02-04-0053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sieczkarski SB, Whittaker GR. Differential requirements of Rab5 and Rab7 for endocytosis of influenza and other enveloped viruses. Traffic. 2003;4:333–343. doi: 10.1034/j.1600-0854.2003.00090.x. [DOI] [PubMed] [Google Scholar]
- 43.Connolly JL, Green SA, Greene LA. Pit formation and rapid changes in surface morphology of sympathetic neurons in response to nerve growth factors. J Cell Biol. 1981;90:176–180. doi: 10.1083/jcb.90.1.176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wilde A, et al. EGF receptor signaling stimulates SRC kinase phosphorylation of clathrin, influencing clathrin redistribution and EGF uptake. Cell. 1999;96:677–687. doi: 10.1016/s0092-8674(00)80578-4. [DOI] [PubMed] [Google Scholar]
- 45.Grimes ML, et al. Endocytosis of activated TrkA: Evidence that nerve growth factor induces formation of signaling endosomes. J Neuro. 1996;16:7950–7964. doi: 10.1523/JNEUROSCI.16-24-07950.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Beattie EC, Howe CL, Wilde A, Brodsky FM, Mobley WC. NGF signals through TrkA to increase clathrin at the plasma membrane and enhance clathrin mediated membrane trafficking. J Neuro. 2000;20:7325–7333. doi: 10.1523/JNEUROSCI.20-19-07325.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Peter BJ, et al. BAR domains as sensors of membrane curvature: the amphiphysin BAR structure. Science. 2004;303:495–499. doi: 10.1126/science.1092586. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Videos were taken under the following imaging condition. For Videos 1–5, the excitation laser for the EYFP-clathrin or Caveolin-1-EGFP was turned on for 0.5 s every 1.5 s while that for the DiD-labeled viruses was always on. The camera integration time for each frame is 0.5 s. All camera pixels were read out individually. For Videos 6 and 7, the excitation laser for the EYFP-clathrin was turned on for 0.5 s every 1.5 s while that for the DiD-viruses was turned on for 0.5 s every 1s. The pixels were binned in a 2×2 fashion. Videos 6 and 7 thus do not allow an accurate determination of whether CCPs form at or off the virus-binding sites, but are only meant to show that both clathrin-dependent and -independent endocytosis can lead to viral fusion.
All videos are processed by subtracting the low-spatial frequency background signal generated by cytoplasmic EYFP-clathrin or Caveolin-1-EGFP. The videos are compressed significantly and compression compromises video quality.
As the viruses were added to the cells in situ the binding of viruses to cells is highly asynchronous. Thus some viruses appear to move only slowly while the circled viruses show the specified behavior of three-stage movement. These slow-moving viruses in the field of view are either in stage I or in stage III during the exhibited time window.
Supplementary Video 1. A dual-color movie of EYFP-tagged clathrin structures (green) and DiD-labeled viruses (red) in a live cell.
Supplementary Video 2. A dual-color movie of EGFP-tagged caveolin structures (green) and DiD-labeled viruses (red) in a live cell.
Supplementary Videos 3 and 4. The internalization of influenza viruses via CCPs. Both movies show the de novo formation of a CCP (green) around the virus (red, in white circles), the gradual increase of clathrin intensity, and the rapid disappearance of the clathrin signal immediately before the virus exhibit a rapid, unidirectional movement towards the perinuclear region (stage II movement).
Supplementary Video 5. The internalization of an influenza virus without association with a CCP. The virus (red, in a white circle) did not associate with a CCP before its stage-II movement inside the cell.
Supplementary Video 6 The internalization and fusion of an influenza virus (red, in a white circle) after association with a CCP (green). Fusion is indicated by a dramatic increase of the DiD signal (red).
Supplementary Video 7. The internalization and fusion of an influenza virus (red, in a white circle) without association with a CCP.
Supplementary Figure 1. Internalization of cholera toxin via caveolae. Alexa Fluor 647-conjugated cholera toxin subunit β (Molecular Probes) was incubated with cells expressing Caveolin-1-EGFP at 4°C for 15 min. After removing excess cholera toxin, the temperature was raised to 37°C to initiate endocyosis. Cholera toxin was found to localize into discrete spots (red images), all of which colocalize with caveolin-1-EGFP structures (green images). Three examples of these cholera toxin-containing structures are indicated by arrows. For each example, three consecutive images, each 10 s apart in time, are shown to illustrate the movement of these structures. Treating cells with nocodazole (60 μM) completely inhibited this movement, indicating that these EGFP-labeled caveolin structures containing cholera toxin were internalized and moving in a microtubule-dependent manner. Such structures were not observed in cells treated with filipin (5 μg/ml), an inhibitor of caveolin-dependent endocytosis, suggesting that the internalization of cholera toxin here is via caveolae. During the observation window (50–100 s), 54% of the cholera toxin-containing structures showed microtubule-dependent movement in Caveolin-1-EGFP expressing cells; and this fraction is similar in untransfected cells (52%). In both transfected and untransfected cells, we started to observe such mobile structures within 5 minutes of the initiation of endocytosis. These results indicate that the expression of Caveolin-1-EGFP did not significantly perturb caveolin-mediated endocytosis. Scale bars: 3 μm.
Supplementary Figure 2. Relative displacements of CCPs from their associated viruses. Black and red symbols are the displacements in the x and y directions in units of camera pixels (95 nm/pixel), respectively. We used least-squares fitting to determine the location of any EYFP-labeled clathrin structures near the virus prior to its stage II movement. In each frame, we considered a 4 μm2 area centered on the virus peak. The EYFP signal in this region was fit to a Gaussian function I (x, y) = I0+Ae−[(x−x0)2 +(y−y0)2]/2σ2, where I0 is the average background fluorescence level, A is the amplitude of the peak, x0 and y0 are the center coordinates, and σ is the width. I0, A, x0, and y0 were treated as free fitting parameters, while σ was fixed to be the experimentally measured width of a diffraction-limited spot. The error bars are determined from the variance matrix obtained by the least-squares fitting. Visual inspection of the movies shows that we are able track the peak as soon as a clathrin structure discernable from the background appears in the 4 μm2 area. In 96% of the trajectories, the clathrin peak position (x0, y0) starts centered on the virus to within 1 pixel, as indicated by examples shown in a–e. Among these, only 2% of viruses seem to land on a pre-existing CCP. The rest show the appearance of a clathrin spot after viral binding. The EYFP-clathrin intensity gradually increased and then rapidly disappeared before the virus started stage II movement. After the clathrin-structure has disappeared, tracking is no longer possible, as indicated by the large error bars appearing at the end of the plots. In the remaining 4% of the trajectories, the initial clathrin peak position is separated from that of the virus (i.e. x0 ≠ 0 and y0 ≠ 0) and the separation in general decreases with time until the virus joins the clathrin structure, as shown in f. This quantitative analysis confirms the results of our visual inspection and indicates de novo formation of CCPs at the site of bound viruses.
Supplementary Figure 3. An integrated histogram of the relative time between the last CCV uncoating event prior to stage II viral movement and the onset of stage II movement. Circles indicate the number of events with the relative time shorter than value indicated on the horizontal axis. The solid curve is a fit to a double exponential decay with time constants 20 s and 270 s. These data clearly show a biphasic behaviour: while the majority of viruses started their stage II movement relatively rapidly after uncoating with a time constant of 20 s, the rest remained in the cell periphery for several minutes before stage II movement. The former population of viruses most likely stays inside the cell during the brief period between clathrin uncoating and stage II movement, as both recycling and endocytosis typical take a few minutes or longer. However, the relatively long lag between clathrin uncoating and stage II movement for the latter population leaves open the possibility that these viruses may have entered the cell via a CCP, then recycled back to the cell surface and re-entered via a clathrin-independent mechanism. We thus reevaluate the partition ratio of viruses between the clathrin-dependent and -independent pathways by classifying endocytosis based on the internalization event immediately preceding stage II movement. This is reasonable as viral fusion was not observed without microtubule-dependent movement. In the extreme case that all of the viruses under the second phase were recycled and re-entered the cell without using a CCP, the fraction of viruses that enter via the clathrin-independent pathway would be increased from the observed 35% to 50%. The true fraction is most likely between these values.
Supplementary Figure 4. Histograms of the time between viral binding and stage II movement (a), the time between stage II movement and viral fusion (b), and the distance between the fusion site and the nuclear envelope (c) for viruses that enter via clathrin-dependent and -independent endocytosis.