

Abstract
Epidemiological, clinical and experimental studies have established a positive correlation between green tea consumption and cardiovascular health. Catechins, the major polyphenolic compounds in green tea, exert vascular protective effects through multiple mechanisms, including antioxidative, anti-hypertensive, anti-inflammatory, anti-proliferative, anti-thrombogenic, and lipid lowering effects. (1) Tea catechins present antioxidant activity by scavenging free radicals, chelating redox active transition-metal ions, inhibiting redox active transcription factors, inhibiting pro-oxidant enzymes and inducing antioxidant enzymes. (2) Tea catechins inhibit the key enzymes involved in lipid biosynthesis and reduce intestinal lipid absorption, thereby improving blood lipid profile. (3) Catechins regulate vascular tone by activating endothelial nitric oxide. (4) Catechins prevent vascular inflammation that plays a critical role in the progression of atherosclerotic lesions. The anti-inflammatory activities of catechins may be due to their suppression of leukocyte adhesion to endothelium and subsequent transmigration through inhibition of transcriptional factor NF-kB-mediated production of cytokines and adhesion molecules both in endothelial cells and inflammatory cells. (5) Catechins inhibit proliferation of vascular smooth muscle cells by interfering with vascular cell growth factors involved in atherogenesis. (6) Catechins suppress platelet adhesion, thereby inhibiting thrombogenesis. Taken together, catechins may be novel plant-derived small molecules for the prevention and treatment of cardiovascular diseases. This review highlights current developments in green tea extracts and vascular health, focusing specifically on the role of tea catechins in the prevention of various vascular diseases and the underlying mechanisms for these actions. In addition, the possible structure-activity relationship of catechins is discussed.
Keywords: Green tea catechins, dyslipidemia, oxidative stress, inflammation, endothelial cells, platelets, proliferation, cardiovascular disease
INTRODUCTION
Tea, brewed from the dried leaves of the plant Camellia sinensis, is one of the most widely consumed beverages in the world. Tea can be categorized into three types, depending on the level of oxidation, i.e. green tea (non-oxidized), oolong tea (partially oxidized) and black tea (oxidized). The manufacturing process of tea is designed to either prevent or allow tea polyphenols to be oxidized by naturally occurring polyphenol oxidase in the tea leaves. Green tea is manufactured by inactivating polyphenol oxidase in the fresh leaves by either firing or by steaming, which prevents the enzymatic oxidation of catechins, the most abundant flavonoids in green tea extracts. The major catechins of green tea are (−)-epicatechin (EC), (−)-epicatechin-3-gallate (ECG), (−)-epigallocatechin (EGC) and (−)-epigallocatechin-3-gallate (EGCG) [1–4]. As shown in Fig. 1, catechins are polyphenolic compounds with diphenyl propane skeleton. The chemical structure consists of a polyphenolic ring (A) condensed with six-membered oxygen containing heterocylic ring (C) that carries another polyphenolic ring (B) at the 2 position. Catechins are characterized by multiple of hydroxyl groups on the A and B rings. EC is an epimer containing two hydroxyl groups at 3′ and 4′ position of B ring and a hydroxyl group at 3 position of the C ring (Fig. 2). The only structural difference between EGC and EC is that EGC possesses an additional hydroxyl group at 5′ position of the B ring. ECG and EGCG are ester derivatives of EC and EGC, respectively, through esterification at 3 hydroxyl position of the C ring with a gallate moiety [3,5,6]. In black tea manufacturing, freshly harvested leaves are naturally semi-dried before the withered leaves are rolled and crushed to allow the polyphenol oxidase to catalyze the oxidation, leading to conversion of catechins to theaflavins and thearubigins [7]. The partly oxidized oolong tea represents an intermediate between black and green tea [1].
Fig. 1.
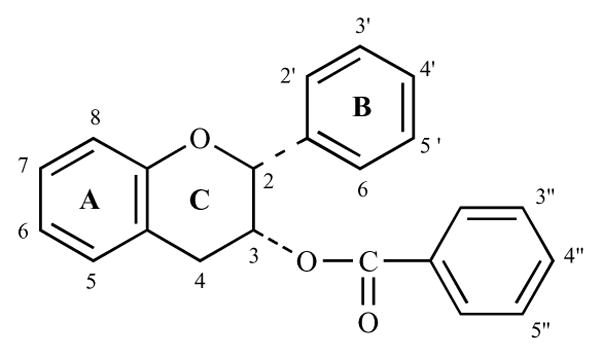
Chemical structure of catechin backbone.
Fig. 2.

Chemical structures of catechins.
A typical tea beverage, prepared in a proportion of 1 g of tea leaves to 100 ml of boiling water in a 3-minute brew, usually contains 250–350 mg of dry materials that are comprised of 30–42% catechins and 3–6% caffeine [1]. In green tea, catechins account for 80% to 90% of total flavonoids, with EGCG being the most abundant catechin (48–55%) followed by EGC (9–12%), ECG (9–12%) and EC (5–7%) [1,8]. The catechin content of green tea is affected by several factors such as drying condition, degree of fermentation, preparation of the infusion, and decaffeination. The catechin content of black tea is only 20–30%, whereas theaflavins and thearubigins represent about 10 and 50–60% of total flavonoids, respectively [1]. The bioavailability of green tea catechins in humans is an important variable for evaluation their biological activity within the target tissues. In humans, the plasma catechin concentration was notably increased 2 to 4 hours following the consumption of green tea [3]. However, the bioavailability of catechins is relatively lower, with reportedly plasma catechin (EGCG and EGC) concentrations accounting for only 0.2% to 2% of the ingested amount in healthy people [9]. The plasma total catechin levels after ingestion of a single, large dose of green tea range from 0.6 to 1.8 μM [10]. A study comparing the pharmacokinetics of pure EGC, ECG, and EGCG in healthy volunteers reported that average peak plasma concentrations after ingestion a 1.5 mM each of EGC, ECG and EGCG as a single bonus were 5.0 μM, 3.1 μM, and 1.3 μM, respectively [11], indicating that bioavailability differs markedly among catechins and that EGCG may be less bioavailable than other green tea catechins in humans. Existing evidence suggests that absorbed catechins are subjected to extensive biotransformation, including methylation, glucuronidation, sulfation and ring-fission metabolism [5]. More than 80% of the major tea catechins are found as conjugates in plasma and urine. However, these conjugates still contain intact catechol and gallate moieties and can scavenge superoxide with the same efficacy as their parent compounds [6,12,13], suggesting that the antioxidant capacity of some of the catechin metabolites is similar to that of their parent compounds [3]. Even their low bioavailability, absorbed catechins may be still sufficient to exert beneficial effects on cardiovascular parameters in vivo [14]. Consumption of green tea or its extract is considered safe, as no significant side effect was observed in various clinical trials [15]. Recently, green tea has drawn wide attention for its potentially beneficial effects on human health. Several studies reveal that green tea may exert protective effects against cardiovascular disease (CVD) and various types of cancers [2,3,5,16]. Tea catechins exert a variety of physiological actions, which may be primarily responsible for the health benefits of green tea. They possess antioxidant, anti-inflammatory, anti-hypertensive, anti-diabetic, anti-mutagenic, anti-bacterial and anti-viral effects [2,3,5,16]. Hence these small molecules in the tea extracts have gained considerable attention and are an active area of scientific enquiry for their possible health benefits. CVD is the leading cause of morbidity and mortality that accounts for an estimated 40% of all deaths [17]. The incidence of CVD continues to rise worldwide and there are more than one-fourth of the U.S. populations (about 70 million individuals) who live with CVD [17]. The beneficial effects of green tea on cardiovascular health are well documented [18–21]. Several epidemiological studies reported the positive relationship between green tea intake and reduced CVD risk [22–24]. In a large population-based (11-year follow-up, 1995–2005) study involving over 40,000 middle-aged Japanese, those who drank over two cups (about 17 ounces) of green tea per day reduced their risk of death from CVD by 22 to 33 %, compared to those who drank less than a half-cup of green tea daily [23]. In a cross-sectional study, the consumption of 120–599 ml of green tea per day for at least one year reduced the risk of developing hypertension by 46% and consuming more than 600 ml per day was shown to reduce the risk by 65%, compared to those subjects consuming less than 120 ml per day [25]. An inverse relationship between green tea consumption (> 2 cups/day) and coronary atherosclerosis was also documented in subjects undergoing coronary angiography [22]. While it was reported that there was no inverse association between green tea intake and coronary artery disease, intake of green tea was found to be inversely associated with myocardial infarction [26]. Consumption of 8 g of powdered green tea per day for two weeks enhances flow mediated blood vessel dilation in chronic smokers [27], suggesting that green tea consumption may prevent future cardiovascular events in smokers.
The cardioprotective effect of green tea was also demonstrated in numerous animal studies [28–33]. Green tea (1.7 mg catechin/day/mouse for 14 weeks) [30] and EGCG (10mg/kg for 21 and 42 days) [28] were shown to reduce the development of atherosclerosis and progression of evolving atherosclerotic lesions in hypercholesterolemic apolipoprotein E-deficient mice. Consumption of green tea in drinking water (containing 3.5g catechins/L) for two weeks attenuated the blood pressure in stroke-prone spontaneously hypertensive rats [31]. Recently, we have reported that green tea treatment (300mg/kg body weight) for 4 weeks can impede cardiac dysfunction in diabetic rats by improving oxidative stress and lipid profile [32,33]. In vitro studies provide evidence that catechins might be the major component in green tea that exert the cardioprotective effect [19]. Indeed, studies showed that supplementation of EGCG acutely improved brachial artery flow mediated dilation in subjects with endothelial dysfunction [34]. In another study, EC administration improved flow mediated dilation of the bronchial artery in healthy adults [35]. The cardiovascular heath benefits of tea consumption have been previously reviewed [19–21,36,37]. This review provides the current developments in green tea catechins and vascular health with special emphasis on recent studies exploring the mechanisms involved in the cardioprotective effect of green tea catechins, which may provide better understanding of the fundamental roles of tea catechins in vascular health. In addition, the possible relationship of chemical structure of catechins with some of their biological activities is also discussed.
MECHANISMS OF THE CARDIOPROTECTIVE EFFECTS OF GREEN TEA CATECHINS
CVD is multifactorial involving oxidative stress [38], abnormalities in lipid metabolism [39], disturbances in vascular tone [40], platelet aggregation [41], inflammation [42] and proliferation of vascular cells [43]. Catechins have been reported to beneficially impact the parameters associated with vascular dysfunction, including lipoprotein oxidation, blood platelet aggregation, vascular inflammation, vascular smooth muscle cell (VSMC) proliferation, altered lipid profile and vascular reactivity. Besides being as anti-oxidants, catechins exert their biological effects by modulating some cellular signaling pathways that lead to reduction of inflammation, platelet aggregation, and an elevation of vascular reactivity [18–21].
Antioxidant Effect
Accumulating evidence indicates that oxidative stress, a pathogenic outcome from overproduction of reactive oxygen species (ROS), play an important role in vascular damage and progression of various vascular disease, including atherosclerosis, ischemic heart disease, hypertension, cardiomyopathies, cardiac hypertrophy and congestive heart failure [38]. Indeed, increased production of ROS and attenuated antioxidant defense system have been demonstrated throughout the vascular pathogenesis [38,44,45]. The high concentrations of ROS produce deleterious effects on both vascular and cardiac myocytes [38]. ROS impair cardiovascular function by inducing VSMC proliferation [46,47], endothelial dysfunction [48,49], cardiac apoptosis and/or necrosis [50,51]. The sources for the ROS production during CVD are uncoupling of mitochondrial electron transport, pro-inflammatory cytokines and induction of oxidative enzymes such as inducible nitric oxide synthase (iNOS) and xanthine oxidase (XO) [52].
Tea catechins have been largely studied for their antioxidant capacities and are considered as important antioxidants [2,53]. The antioxidant effects of catechins are presumed to play a major role in mediating the cardioprotective role of tea [19], although emerging evidence shows that catechins also have antioxidant-independent vascular effects, which will be discussed elsewhere in this review. Green tea catechins can exert both direct and indirect antioxidant effects on cardiovascular system [19,54]. Catechins present antioxidant activity through scavenging ROS, chelating redox active transition- metal ions, inhibiting redox sensitive transcription factors, inhibiting pro-oxidant enzymes and inducing antioxidant enzymes [3,19,55]
Human and animals studies have reported that green tea and its catechins can decrease the biomarkers of oxidative stress [5] and lipid peroxidation [56] and increase the plasma antioxidant capacity [57–59]. EGCG treatment increases the antioxidant capacity in local vascular tissue and systemic circulation in apolipoprotein E null mice [28], a mouse model of human atherosclerosis. In addition, EGCG dose-dependently protects human plasma against oxidation, with 10 μM concentration reducing oxidation by 68% [60]. The rise in plasma antioxidant capacity peaks about 1 to 2 hours after tea ingestion and declines quickly thereafter [5], which could be due to rapid absorption, metabolism and excretion of tea catechins [3]. Catechins can stabilize ROS and prevent oxidative injury by directly scavenging free radicals [60–62]. The free radical scavenging mechanisms of catechins may involve the delocalization of electrons, formation of intra- and intermolecular hydrogen bonds, rearrangements of molecules and chelation of metals that may be involved in oxidation [63,64]. Because of the number and arrangement of their phenolic hydroxyl groups, catechins are excellent electron donors and efficient scavengers of free radicals such as superoxide anions, singlet oxygen, nitric oxide (NO) and peroxynitrite [3,5,60,65] The scavenging capacity of catechin molecules depends on the number of ortho-dihydroxyl and ortho-hydroxyketol groups, C2-C3 double bonds, concentration, solubility, the accessibility of the active group to the oxidant, and the stability of the reaction product [2,66]. Specific structural components in catechins are reported to be differentially responsible for the radical scavenging, chelation or antioxidant activity [6,61,67]. The antioxidant capacity decrease substantially with decrease in number of hydroxyl groups on the B ring [68], suggesting that the number of hydroxyl groups on the B ring contributes significantly to the hydroxyl scavenging activity of catechins. As catechins have a saturated heterocyclic ring, ROS predominantly attach to the ortho-dihydroxy site on the B ring, which results in electron delocalization and confers a higher stability to the resulting radical semiquinones [6,69],. The 3′, 4′ catechol structure on the B ring is a potent scavenger of peroxyl, superoxide, and peroxynitrite radicals [61]. The hydroxyl groups at three different rings (C5 and C7 of the A ring; C3′ and C4′ of the B ring; and C3 of the C ring) also enhance the inhibition of lipid peroxidation [70]. In addition, the ortho-trihydroxyl group in the B-ring is important for scavenging super-oxide anion, whereas the galloyl moiety is responsible for quenching the hydroxyl radicals [71]. Further, conjugation between the A and B rings provides a resonance effect of the aromatic nucleus that is important in stabilizing phenoxyl radicals [61].
EGCG is the most potent tea antioxidant [2], suggesting that the ortho-trihydroxyl group and 3-gallate esters in catechins play an important role in antioxidant activity, radical scavenging and preventing oxidative destruction of biological compounds [60]. Tea catechins also protect cells against transition metal-catalyzed free radical formation by chelating iron and copper to prevent their participation in Fenton and Haber-Weiss reaction [72,73]. The ortho-3′4′-hydroxyl group of the B ring and gallate moiety of gallocatechins were reported to be the primary metal binding sites for catechins [74,75]. Though many studies report that EGCG is the most potent antioxidant among catechins, a study demonstrated that ECG is equally potent as EGCG in producing antioxidant effect in an in vitro system [76,77]. Studies indicate that the metabolities of catechins also possess similar antioxidant effects like their parent compound. (−)-Epicatechin-5-O-β-glucuronide and (+)-catechin-5-O-β-glucuronide, the metabolites of EC and catechin exhibit comparable scavenging abilities to the parent compounds [71]. Conjugation at the A ring in metabolites did not interfere with the ortho-dihydroxyl group at the 3′ and 4′ positions on the B ring [71].
While a large body of literature demonstrates the antioxidant and free radical scavenging effects of catechins as described above, several animal and human studies showed no effects of tea catechins on plasma antioxidant [53,78]. It was proposed that this could be due to insufficient concentrations of circulating catechins necessary for exerting an antioxidant effect in vivo [20]. In contrast to these positive and neutral studies, a number of studies indicate that tea catechins can act as a pro-oxidant [79,80] and induce oxidative damage through the generation of ROS [81]. Tea catechins are unstable under cell culture conditions and undergo oxidative polymerization with hydrogen peroxide [79, 82]. EC and EGCG also can generate superoxide anion and hydroxyl radicals [80]. It was suggested that the pro-oxidant activity of these catechins depends upon the hydroxyl orthodihydroxy groups on their molecules [80]. However, these reported pro-oxidant effects are only observed at pharmacological dose (>10 μM/L) of catechins [83] and under in vitro condition [79,80]. Therefore, the biological relevance of this finding is questionable.
Catechins may also reduce the oxidative stress by modulating the ROS generating enzymes such as iNOS and XO. While endothelial-derived NO from activation of constitutive NO synthase is important for maintaining vascular tone and homeostasis, higher concentrations of NO produced by iNOS from immune cells such as macrophages can cause oxidative damage. The activated macrophages to a great extent increase the simultaneous production of both NO and superoxide anions. NO reacts with free radicals, thereby producing the highly damaging peroxynitrite that can directly oxidize low density lipoprotein (LDL), resulting in irreversible damage to the cell membranes. EGCG dose-dependently inhibit the expression of iNOS in lipopolysaccharide-activated macrophages by preventing the binding of nuclear transcriptional factor-kB (NF-kB) to the iNOS gene promoter and also reduce the activity of iNOS, thereby reducing toxic NO generation [77,84]. XO-mediated metabolic pathway has been implicated as an important route in the oxidative damage to tissues, particularly after ischemia-reperfusion [85]. XO reacts with oxygen molecule, leading to the release of superoxide. In an in vitro study, catechins was shown to be the inhibitors of XO [86], and EGCG was as potent in inhibition of XO as allopurinol, a drug used to inhibit XO in gout patients [86]. This result is further corroborated by another study demonstrating that EGCG suppresses XO activity and its mediated pathway in various cultured cells [87].
Several studies have found that catechins can up-regulate anti-oxidant enzymes. Mice given 0.2% catechins in drinking water showed significantly increased activities of superoxide dismutase, catalase and glutathione peroxidase [88], which play key roles in scavenging ROS. Green tea consumption for 2 weeks was shown to induce the expression of catalase in aorta of spontaneously hypertensive rats [31]. In addition, catechins also participate in vitamin E recycling, and thus complement the functions of glutathione [89]. Furthermore, green tea was shown to increase the plasma and tissue glutathione levels in several animal studies [32,53,90].
LDL is a well recognized and the most studied risk factor for CVD. At the first stage, LDL deposits at lesion sites of the arterial wall and is subjected to oxidation when protectors such as antioxidants are depleted. Oxidized LDL then induces modifications in lipoproteins, stimulates inflammatory reactions, causes monocytes and monocyte-derived macrophages to uptake oxidized LDL, and ultimately leads to the formation of lipid-loaden foam cells and atherosclerotic plaques. These plaques protrude from the inner surface of the arteries, narrowing the lumen, and reducing blood flow that leads to coronary heart disease. Extensive studies reported that catechins can inhibit the oxidation of LDL both in vitro and in animal studies [2,20,29,37,91,92] with a potency order of EGC < EC < ECG < EGCG [93] It was also shown that EGCG alone has a lipoprotein bound antioxidant activity that is greater than that of tocopherol [56]. The addition of 2 to 20 μg/ml of EC, ECG, EGC, or EGCG to the macrophages conserved the α-tocopherol content of the LDL and delayed the onset of lipid peroxidation [89].
Effect on Lipid Profile
Hyperlipidemia, resulting from the abnormalities of lipid metabolism, is one of the major risk factors for the development of CVD. The elevated levels of plasma lipids such as fatty acids, cholesterol, phospholipids and triglycerides can lead to the development of atherosclerotic plaques [39]. The risk of CVD reduced by 2% with a 1% decrease in serum cholesterol [94] and evidence suggests that drugs with the ability to reduce plasma lipids can reduce the probability of cardiovascular death [39]. Green tea catechins affect lipid metabolism by various mechanisms and prevent the appearance of atherosclerotic plaque in various models of hyperlipidemia [30,88]. In addition, catechins influence micellular solubility, luminal lipid hydrolysis and intestinal lipid absorption [95]. Furthermore, catechins can up-regulate hepatic LDL receptor expression, thereby modulating biosynthesis, excretion and intracellular processing of lipids [20, 95–97]. While it is unclear how catechins modulates hepatic LDL receptor in vivo, one study suggests that this may be due to a reduction of cholesterol concentration in liver [98], given that LDL receptor synthesis is increased in response to decreased intracellular cholesterol levels [99].
Tea catechins were shown to reduce blood cholesterol levels and prevent the deposition and/or accumulation of cholesterol in various tissues, including liver and heart in rats with hypercholesterolemia [33,97]. In an in vitro study, EC, EGC, ECG or EGCG at 5 μM was shown to suppress intracellular lipid accumulation [100]. Studies demonstrated that tea catechins may interfere with the emulsification, hydrolysis, and micellar solubilization of lipids [101–103], suggesting that the cholesterol-lowering effect of catechins may, at least in part, is mediated by their influence on intestinal lipid absorption. Indeed, catechins were demonstrated to lower the absorption of cholesterol and triglyceride in rats [98,104]. In this regard, EGCG and ECG were more effective than EC and EGC in lowering the absorption of cholesterol [104], suggesting that the gallate ester components of catechins may be primarily responsible for inhibition of intestinal cholesterol absorption.
The micellar solubilization of hydrolyzed lipids is the critical step for the uptake and absorption of lipids by enterocytes and this process facilitates the transfer of lipids through the unstirred water layer to the enterocyte for uptake [95]. While Catechins with gallate esters were shown to decrease cholesterol absorption by forming insoluble co-precipitates of cholesterol and decreasing bile acid-induced micellar solubility [104], EC may inhibit cholesterol absorption primarily by binding to cholesterol, thereby increasing fecal excretion of cholesterol [105]. Dietary flavonoids were reported to modulate the specific transport proteins located on the brush border membranes that play a significant role in the uptake of lipids by enterocytes [106,107]. Based on these studies it was proposed that catechins may form complex with these transport proteins through hydrophobic interactions and hydrogen bonding and therefore interfere with the uptake of lipids by enterocytes [95]. As aforementioned, catechins, especially EGCG, are not readily absorbed in both animals [108] and humans [109]. Thus, catechins may interfere with the steps involved in intestinal uptake of lipids due to their presence in high concentrations in the intestinal lumen [110].. In addition, catechins may have direct inhibitory effect on cholesterol synthesis. It was recently found that green tea catechins are potent and selective inhibitors of squalene epoxidase, a likely rate limiting enzyme of cholesterol biosynthesis [111]. The presence of galloyl moiety was suggested to be important for squalene epoxidase inhibitory activity of catechins [111].
It is well recognized that LDL-cholesterol is an important risk factor for the development of CVD. Human studies showed that green tea consumption is associated with a lower ratio of LDL-cholesterol to HDL (high density lipoprotein) -cholesterol [24,112]. While it was reported that there was no change in the serum concentrations of total cholesterol, triglycerides and HDL-cholesterol on daily consumption of up to 4 cups of green tea in middle-aged men [113], the result from a similar study demonstrated the association between the consumption of more than 10 cups (1,500 ml) of green tea a day with decreased serum concentrations of total cholesterol, LDL, and triglycerides, and with an increased HDL concentration [24]. Consistent with this report, another study demonstrated that consumption of 9 or more cups of green tea per day can reduce serum cholesterol level, although serum triglycerides and HDL-cholesterol were not altered [114]. While the discrepancies of these epidemiological data could be due to various factors such as heterogeneities in study design, variations in sample size, the differences in tea preparation, production process, as well as life styles, intake of relatively high doses of catechins may be required for achieving this beneficial effect. Catechins were also reported to favorably modify lipoproteins in animal studies. EGCG reduces circulating LDL-cholesterol but promote HDL-cholesterol in rats fed with a high fat and high cholesterol diet [115]. In line with these studies, we have reported that green tea extract (300 mg/kg body weight for 4 weeks) containing 80% catechins can reduce circulating LDL-cholesterol but can increase the levels of HDL-cholesterol in diabetic rats [33]. Apolipoprotein B (ApoB) is the primary apolipoprotein component in LDL and high levels of ApoB and decreased Apolipoprotein A-1 (ApoA-1)/ApoB ratio are associated with higher risk of CVD [116]. Green tea catechins supplemented in either diet or drinking water was shown to reduce ApoB and improve the ratio of ApoA-1/ApoB [105]. Catechins also up-regulate LDL receptor, which is likely mediated through the activation of sterol regulated element binding protein-1 [117]. In addition to increasing its expression, catechins may also increase LDL receptor binding activity, which may also contribute to a hypocholesterolemic effect of green tea [117]. Combined, catechins modulate cholesterol metabolism by targeting biosynthesis, absorption, excretion of cholesterol and LDL receptor that collectively contribute to the hypocholesterolemic effect of green tea catechins.
The postprandial hypertriacylglycerolemia is a risk factor for coronary heart disease and catechins was shown to have a beneficial effect on this condition [118]. Tea catechins, particularly those with a galloyl moiety, dose dependently inhibit the activity of pancreatic lipase, thereby suppressing triacylglycerol absorption and postprandial hypertriacylglycerolemia [118]. In addition, green tea catechins at the levels achievable by typical daily intake markedly altered the physicochemical properties of a lipid emulsion by increasing its particle size and reducing the surface area [102]. These changes reduce the interaction of pancreatic lipase with fat and therefore decrease the rate of hydrolysis of fat [102]. EGCG was identified as the main catechin compound present on the lipid phase of the emulsion that is responsible for the changes in emulsion properties [101]. The hydroxyl moieties of EGCG was proposed to interact with the hydrophilic head group of phosphatidyl choline at the exterior of a lipid emulsion by forming hydrogen bonds, and such interaction may lead to formation of cross-links followed by coalescence of the emulsion droplets [101].
EGCG also has been shown to exert a potent inhibitory effect on pancreatic phospholipase A2, which may also be attributable to the decreased absorption of lipid because luminal phosphatidyl choline hydrolysis is critical to facilitating intestinal lipid digestion and absorption [95,103]. EGCG may interact with the surface phosphatidyl choline of a lipid emulsion and hinder the access to the substrate by phospholipase A2 [95,103]. It was also proposed that EGCG may directly interact with the enzyme protein and subsequently alter its conformation and catalytic activity [119,120]. As above mentioned, postprandial hyperglyceridemia has been shown to be an independent risk factor for CVD, the consumption of green tea with or before meals therefore may be an effective strategy to attenuate postprandial lipemia in individuals with CVD risk [121].
The synthesis of fatty acid is a key step for lipogenesis. The suppression of lipogenesis by green tea extracts has been demonstrated in experimental animals [122]. In an in vitro study it was reported that EGCG at 10 μM profoundly suppress fatty acid synthase (FAS) gene transcription through epidermal growth factor receptor/phosphoinositide-3 kinase (PI3K)/Akt/Sp-1 signal transduction pathway in breast cancer cells [123]. However, another study indicates that the phenolic group of the B ring and galloyl group in EGCG are directly involved in the irreversible inhibition of FAS [124]. The ester bond of gallate group covalently reacts with the essential group of β-ketoacyl reductase of FAS that leads to the irreversible inhibition of FAS [124]. In addition, EGCG and ECG at 5 μM were shown to inhibit the activity of rat liver acetyl CoA carboxylase, which catalyzes the carboxylation of acetyl-CoA to malonyl-CoA, the rate limiting step in fatty acid biosynthesis [125], whereas other catechins has no effect on this enzyme, suggesting that catechin skeleton and 3-ortho-gallate moiety may be both required for this inhibitory activity.
Effect on Vascular Homeostasis
Endothelial cells (ECs), which not only serve as a biological barrier separating circulating blood and peripheral tissues, but also playing a central role for maintaining vascular homeostasis. Endothelial dysfunction significantly contributes to the pathogenesis and clinical manifestation of CVD [54]. Vascular ECs maintain normal vascular function by producing various vasoactive substances that regulate vascular tone, local inflammation and angiogenesis [40]. Endothelium-derived NO, synthesized from L-arginine by endothelial isoform of NO synthase (eNOS), is not only a vasodilator, but also maintains vascular homeostasis. NO inhibits the expression of leukocyte adhesion molecules at the endothelial surface and prevents the adherence of leukocytes to ECs, thereby exerting an anti-inflammatory effect [40, 54, 126, 127]. In addition, NO prevents the platelet adhesion, platelet aggregation and inhibits the proliferation of VSMCs [54]. Thus, endothelial dysfunction and subsequent impairment of NO production is widely acknowledged as a critical step in initiation of atherogenesis and vascular pathogenesis [128]. Thus, agents that can prevent damage to ECs or restore endothelial function may have important clinical implications.
Experimental and clinical studies suggest that tea catechins can significantly improve endothelial function, thereby providing an additional beneficial effect on patients with CVD. EGCG was shown to improve endothelial function and reduce blood pressure in hypertensive rats [129]. Administration of 1 or 2 mg of EC/kg body weight rapidly improves flow-mediated dilation of the branchial artery and fingertip microvessels in healthy adults [35]. In consistent with this observation, another recent human study demonstrated that acute administration of a single 300 mg dose of EGCG can reverse endothelial dysfunction and improve brachial artery flow-mediated dilation in patients with coronary artery disease [34]. While the precise mechanism by which tea improves endothelial function is not exactly clear, recent studies provided evidence that tea catechins induce endothelium- and NO-dependent vasorelaxation as demonstrated in ex vivo studies of rabbit [35] and rat aortic tissues [78,130]. In cultured ECs, EGCG (100 μM) rapidly activates eNOS through PI3K/Akt and cAMP-dependent protein kinase A, whereas eNOS protein content is not altered [130], suggesting a possible non-genomic effect of this compound. Interestedly, a further study showed that the activation of PI3K/Akt and eNOS by EGCG in ECs requires both ROS and Fyn, a protein associates with the p85 subunit of PI3K [131]. The biological relevance of these data derived from in vitro and ex vivo studies was supported by a recent animal study demonstrating that ingestion of a diet rich in catechins increased eNOS activity, endothelium-derived NO production and cyclic guanosine-3′,5′-monophosphate (cGMP) content, a determinant of biological activity for released NO, but eNOS expression was not changed in the arterial tissues of rats [132], further suggesting that catechins may modulate endothelium-dependent vasorelaxation through eNOS/NOsignaling molecules-mediated, transcription-independent mechanism.
Catechins may also increase NO bioavailability and improve endothelial function by neutralizing ROS. ROS can react directly with NO and eliminate its biological activity [128]. The alterations in cellular redox state reduce eNOS expression, decrease the availability of essential co-factors for its activation, and uncouple eNOS from its normal dimeric configuration into a monomeric form that leads to the production of superoxide anion, rather than NO [128]. Superoxide, which is also potentially produced from NAD(P)H-dependent oxidases, XO, lipoxygenase and mitochondrial oxidases [133,134], react with NO, reducing NO bioavailability and producing peroxynitrite [135], a strong oxidant that has been implicated in vascular disease [136]. In addition, increased oxidative stress also was shown to produce critical modifications in guanylyl cyclase and therefore may reduce tissue responses to NO [18]. EGCG has been shown to potently scavenge superoxide [137], suggesting that catechins may increase the bioavailability of NO by scavenging superoxide, which may in turn contribute to endothelium-dependent vascular relaxation.
EGCG and EGC were also found to stimulate the production of prostacyclin in bovine aortic ECs [138]. This effect is likely related to the pyrogallol structure of catechins because EC and ECG that are lack of this structure in their molecules had no significant effect on prostacyclin release. Prostacyclin activates adenylyl cyclase to produce cAMP, which ultimately induces endothelium-independent vasorelaxation by activation of PKA and inhibition of intracellular [Ca2+] increase in VSMCs [139,140]. While these data suggest that catechins may simultaneously activates differential signaling pathways, leading to production of endothelium-dependent and -independent relaxing factors, a catechin concentration of ≥50 μM is required for achieving these effects, which is unlikely unattainable in vivo through nutritional supplementation. Therefore, the physiological relevance of these in vitro findings is no clear.
In contrast to above described beneficial effects of catechins on vascular homeostasis, some studies reported that catechins can exert contractile effects in rat aorta and impair endothelium-dependent vasorelaxation [141,142]. It was proposed that the inhibition of vasorelaxation by catechins could be due to inactivation of endothelium-derived NO [141]. The stereoscopic structure between the C3 group and B ring of catechins along with the hydroxyl group on C5 of the B ring is possibly responsible for this effect [141]. Indeed, substitution of gallate group of C3 attenuates such an inhibitory effect of catechins on vasorelaxation due to decreased response to soluble guanylate cyclase in VSMCs [141]. The discrepancy of these observations could be due to the differences in experimental conditions, animal species, and catechin doses used [140]. EGCG and EGC were reported to cause contraction of arteries in relatively lower concentrations (1–30 μM) whereas stimulate vasorelaxation at high doses (> 100 μM) [141]. It was suggested that the contractile and relaxant effects of catechins may be due to a biphasic behaviour of catechins, more than to a dual concentration-selective mechanism [140].
Anti-inflammatory Effect
It is now recognized that atherosclerosis is a chronic inflammatory disease and inflammation-induced monocyte adhesion to ECs followed by transmigration into the subendothelial space is one of the key events in the development of atherosclerosis [143–145]. Adhesion of leukocytes to ECs is critically regulated by both chemotactic cytokines and vascular adhesion molecules. Chemokine interleukin-8 (IL-8) and monocyte chemoattractant protein -1 (MCP-1) are demonstrated to be the key factors in the firm adhesion of monocytes to activated ECs and in monocyte recruitment into sub-endothelial lesion in atherosclerosis [146]. Various adhesion molecules that are involved in this event such as endothelial leukocyte adhesion molecule-1 (E-selectin), intercellular adhesion molecule-1 (ICAM-1) and vascular adhesion molecule-1 (VCAM-1), which are regulated by NF-kB and play a pivotal role in attracting binding and transmigration of leukocytes into sites of inflammation [146–148].
In vitro studies show that catechins can inhibit leukocyte-endothelial interaction. EGCG and ECG dose-dependently reduce cytokine-induced VCAM-1 expression and monocyte adhesion to ECs, an effect that is likely dependent on the pyrogallol group in catechins since EC and EGC were without such an effect [149]. EGCG (5–30 μM) was also shown to inhibit phorbol 12-myristate 13-acetate-induced MCP-1 mRNA and protein expression in human ECs and thereby reduce the migration of monocytes, an effect mediated through the suppression of p38 mitogen-activated protein kinase (p38) and NF-kB activation [150]. At physiological achievable concentration, EGCG was shown to inhibit neutrophil migration through cultured human endothelial monolayers [151]. EGCG and ECG also induce caspase-mediated apoptosis of monocytes, thereby suppressing undesirable immune response that may play a key role in atherogenesis [152]. In addition, EGCG was shown to suppress chemokine production and neutrophil infiltration at the inflammatory site [153].
The redox-sensitive transcription factors NF-kB and activator protein-1 (AP-1) play a critical role in mediating various oxidative stress-regulated vascular inflammation and pathogenesis. NF-kB activation plays a major role in the expression of pro-inflammatory molecules, including cytokines, chemokines and adhesion molecules [154,155]. NF-kB is associated with the cytoplasmic inhibitory protein IkBα in inactive form [156]. Cellular stimulation with NF-kB agonists results in the phosphorylation and degradation of IkBα, allowing the p50/65 heterodimers of NF-kB to translocate to nucleus and initiate expression of target genes [157–159]. Tea catechins were shown to modulate these factors [160]. Administration of EGCG during reperfusion significantly decreased IkB kinase activity, resulting in the reduction of IkBα degradation and NF-kB activity [161]. EGCG also was shown to reduce the AP-1 activity by diminishing phosphorylation of c-Jun [162], suggesting that catechins may simultaneously modulate multiple signaling pathways. In addition, EGCG can directly inhibit the phosphorylation of IkB, thereby preventing NF-kB translocation to the nucleus [163]. In consistent with these results, another study reported that administration of green tea catechins (20 mg/kg for 60 days) decreased the activity of NF-kB in murine cardiac transplants [37]. Green tea catechins were also shown to inhibit the proteolytic activity of the 26S proteosome [164] that inhibits IkB degradation, thereby resulting in the inhibition of NF-kB activation. As discussed above, many adhesion molecules and pro-inflammatory cytokines and chemokines are regulated by NF-kB, catechins may inhibit the expression of these pro-inflammatory molecules through suppression of NF-kB activity.
While these data are interesting, most of results achieved in these studies used catechin concentrations that are far from physiologically achievable levels (0.6 – 1.8 μM) in both humans and animals through dietary means. Therefore, it is largely unclear if dietary supplementation of catechins has an anti-inflammatory effect in vivo as observed in these in vitro studies, although catechin metabolites in vivo may also potentially exert the anti-inflammatory effects as suggested by their inhibition of human monocyte-ECs interaction in vitro [165]. However, although in vivo studies are very limited, and particularly, investigations of whether green tea extract supplementation is beneficial on vascular inflammation are lacking, one recent human study showed that green tea consumption for 4 weeks in smokers reduces C-reactive protein, one of the most important markers of inflammation that is associated with an increased risk of cardiovascular events [166], indicating an anti-inflammatory effect of catechins in humans [167]. In line with this result, the same human trial indicated that drinking 4 cups of green tea for 4 weeks reduces plasma levels of soluble P-selectin, an endothelial adhesion molecule implicated in the process of atherogenesiss [167]. These data from human studies suggest that it may not be necessary to achieve the plasma concentrations that high so that to exert a beneficial, anti-inflammatory effect. It is also possible that the anti-inflammatory effects achieved in vivo by physiological levels of green tea extracts are cumulative or are secondary action whereby catechins alter oxidative stress and lipid profile.
Anti-Proliferative Effect
One of the major events involved in the development of atherosclerosis is the proliferation and migration of VSMCs to the sub-endothelial space [43]. Studies showed that tea catechins can inhibit the proliferation of VSMCs induced by advanced glycation end products [168,169] and platelet-derived growth factor (PDGF) [170]. While how catechins inhibit VSMC proliferation is not exactly clear, EGCG treatment has been shown to arrest VSMCs in G1 phase of the cell cycle by down-regulating the important cell cycle regulators such as cyclins/cyclin-dependent kinases [171]. This catechin effect on VSMC proliferation appears dependent on galloyl structure and mediated by suppressing protein tyrosine kinase (PTK), JNK1 and c-jun expression [172]. It is presently unknown how EGCG inhibits PTK and JNK1 which mediate growth factor-induced growth [172]. Interestedly, recent studies showed that EGCG can incorporate into cell membranes to interrupt the binding of PDGF to its receptor that ultimately leads to the inhibition of cell proliferation [173]. This finding suggests that EGCG may inhibit PTK and JNK1 by interfering with growth factor receptors, given that both kinases are the downstream targets of PDGF receptor-initiated signaling.
The key event involved in the development of atherosclerosis and post-angioplasty vascular remodeling is the migration of VSMCs from the tunica media to the sub-endothelial region. Matrix metalloproteinases (MMPs) are a family of zinc-dependent endoproteinases that are responsible for degrading extracellular matrix proteins [174] and therefore play an essential role in cell migration and tissue remodeling. In the process of atherosclerosis, MMP activity is elevated concomitant with increased infiltration of inflammatory cells, VSMC migration and proliferation [175]. Of the three main groups of MMPs [176], MMP-2 (type IV collagenase, gelatinase A) and MMP-9 (type V collagenase, gelatinase B) play an important role in angiogenesis [177]. Catechins (30–50 μM) have been shown to prevent VSMC migration by inhibiting MMP-2 expression, an effect that may be mediated by direct inhibition of MMP-1 [178]. This result is consistent with another study finding that ECG and EGCG dose-dependently inhibit matrix protein degradation and VSMC invasion by reducing the gelatinolytic activity of MMP-2 [179]. In agreement with these findings, exposure of VSMCs to EGCG (20 – 80 μg/ml) also prevented tumor necrosis factor-α-induced expression of MMP-9 which is critically involved in the progression of atherosclerotic lesions [171]. While these data showing the inhibitory effects of catechins on VSMC migration and proliferation through counteracting MMP activity suggest a potential anti-atherogenic action of these compounds, and meanwhile, provide a mechanism underlying the anti-atherosclerotic action of green tea extracts as observed in both human [22] and animal [28] studies, the biological relevance of these in vitro findings, which were obtained only at pharmacological doses of catechins, remain to be determined.
Anti-Platelet Activity and Anti-Thrombotic Effect
Platelet activation and aggregation also play an integral role in the development of CVD. The alterations in platelet sensitivity and platelets-vessel wall interaction are associated with the development of cardiovascular events. In the presence of vascular endothelial injury, platelets rapidly aggregate to form hemostatic plugs and arterial thrombi which could trigger the acute vascular events such as myocardial infarction [180]. Indeed, there is extensive evidence that anti-platelet therapy can reduce CVD risk [181].
Both in vitro and in vivo studies have reported the beneficial effect of tea catechins on platelet aggregation [170,182]. Catechins were reported to dose- dependently inhibit various stimuli-induced in vitro platelet aggregation of humans [183] and animals [184] without changing the coagulation parameters such as activated partial thromboplastin time, prothrombin time, and thrombin time [183], suggesting that the anti-thrombotic action of catechins may be due to their anti-platelet activity rather than to an anti-coagulant effect. While the in vivo study on whether catechins have an anti-platelet effect is rare, available data show that catechin supplementation improved platelet aggregation and thrombosis in diabetic rats [185]. It has been established that intracellular calcium concentration plays a crucial role in platelet aggregation and activation. Catechins inhibit intracellular calcium mobilization via activation of Ca2+-ATPase and inhibition of inositol 1,4,5-triphosphate formation in human platelets, which leads to the inhibition of fibrinogen-GPIIb/IIIa binding [182]. Data from a more recent study showed that EGCG also inhibits phospholipase Cγ2 phosphorylation, but elevates prostaglandin D2 production [184]. Additionally, ECG and EGCG may reduce the production of platelet activating factor (PAF) by inhibiting acetyl-CoA:lysoPAF acetyltransferase [186], thereby decreasing the stickiness of platelets and the probability of platelet aggregation [186]. Arachidonic acid, a plasma membrane-bound fatty acid, which release is increased in inflammatory conditions, is metabolized by platelets to generate prostaglandin, endoperoxides, and thromboxane A2, key molecules for platelet activation and aggregation [187]. Catechins can suppress arachidonic acid release [184], [188] and thromboxane A2 synthase activity [188], and therefore reduce the production of these platelet activators. Collectively, these studies suggest that catechins modulate multiple cellular targets related to platelet activation, but the anti-platelet activity of catechins may be primarily attributable to its modulation of arachidonic acid pathway and inhibition of cytoplasmic calcium increase.
CONCLUSIONS
Epidemiological data and results from many clinical and experimental studies have shown that green tea consumption may have beneficial effects on cardiovascular health. As major polyphenolic compounds in green tea, catechins may be primarily components in green tea that exert vascular protective effects. Catechins have been shown to inhibit oxidation, vascular inflammation, atherogenesis, and thrombogenesis, and favorably modulate plasma lipid profile and vascular reactivity, suggesting a wide spectrum of beneficial effects of catechins on vascular function (Table). While some of the protective effects of catechins could be partially due to the secondary action of their antioxidant effect, it appears that catechins can directly act on immune and vascular cells to modulate their functions by targeting multiple cellular pathways and transcriptional factors involved in vascular health, inflammation and diseases such as eNOS/NO, arachidonic acid metabolism and NF-kB (Fig. 3). The different chemical groups of catechins play different roles in their various biological functions. The phenolic hydroxyl groups of catechins are primarily responsible for scavenging of free radicals, inhibition of lipid peroxidation, and hydrolysis of fat, whereas the galloyl moiety is involved in chelating metal ions, inhibition of intestinal cholesterol absorption, cholesterol biosynthesis and fatty acid biosynthesis. In addition, the galloyl structure of catechins may be mainly responsible for the production of prostacyclin, reduction of VCAM-1 expression and inhibition of VSMC proliferation. These findings from substantial recent studies may provide insights into the fundamental role of green tea extracts in vascular health. Meanwhile, it should be noted that the results from many in vitro studies, aimed at elucidating the mechanisms underling various vasoprotective effects of catechins, were obtained with catechin concentrations that are well above those achievable through dietary ingestion. Therefore, the physiological relevance of these in vitro findings remains to be investigated. While some catechin metabolites are reported to be biologically active, their vascular effects are unknown. Studies in this area are also needed to determine if catechins or their metabolites are primarily responsible for health benefit of catechin supplement. In addition, further studies on determining the structure aspects of catechin actions important for normal vascular function may provide a novel perspective for the design of catechin analog compounds with the enhanced biological activity, which may lead to clinically relevant strategies to prevent and treat vascular diseases.
Table.
Biological Effects, Mechanisms, Molecular Targets and Chemical Structures Involved in the Cardioprotective Effect of Green Tea Catechins
| Cardioprotective Effects | Mechanisms | Molecular Targets | Required Chemical Structures |
|---|---|---|---|
| Antioxidant effect | Scavenge free radicals | ↓ O2•−, NO•, 1O2, ONOO− | Hydroxyl and gallate |
| Chelate metal ions | Chelate copper and iron | Hydroxyl and gallate | |
| ⊖ redox active transcription factors | ⊖ NF-kB, AP-1 | Unknown | |
| ⊖ pro-oxidant enzymes | ⊖ iNOS, XO | Unknown | |
| Up regulate antioxidant enzymes | ↑ catalase, GPx, SOD | Unknown | |
| Sparing of antioxidants | Sparing tocopherol | Unknown | |
| Improvement of lipid metabolism | ⊖ cholesterol synthesis | ⊖ squalene epoxidase | Gallate |
| ↑ Cholesterol excretion | Bind to cholesterol | Gallate | |
| ↓ Cholesterol absorption | Bind to cholesterol | Gallate | |
| ↓ Triglycerides | ⊖ phospholipase | Hydroxyl | |
| ↓ Fatty acid synthesis | ⊖ acetyl CoA carboxylase ⊖ β-ketoacyl reductase of FAS |
Gallate Gallate |
|
| Improvement of endothelial function | NO-dependent vasodilation | ↑ eNOS | Unknown |
| NO-independent vasodilation | ↑ prostacyclin ↑ cytosolic cAMP, cGMP |
Gallate Unknown |
|
| Anti-inflammatory effect | ⊖ Adhesion of leucocytes to ECs | ↓ VCAM, MCP-1 | Gallate |
| + Apoptosis of monocytes | ↑ caspase 8 and 9 | Unknown | |
| Anti-proliferative effect | ⊖ VSMC growth | ⊖ PCNA ⊖ PDGF |
Gallate Unknown |
| ⊖ VSMC migration | ⊖ MMP-2 | Unknown | |
| Anti-platelet and Anti-thrombiotic effects | ⊖ platelet aggregation | ⊖ cytoplasmic Ca2+ release ⊖ PAF |
Unknown Unknown |
| ⊖ Thrombosis | ⊖ Thromboxane A2 synthase Scavenge free radicals |
Unknown Hydroxyl and gallate |
↑, increase; ↓, decrease; ⊖, inhibition; +, activation.
Fig. 3.
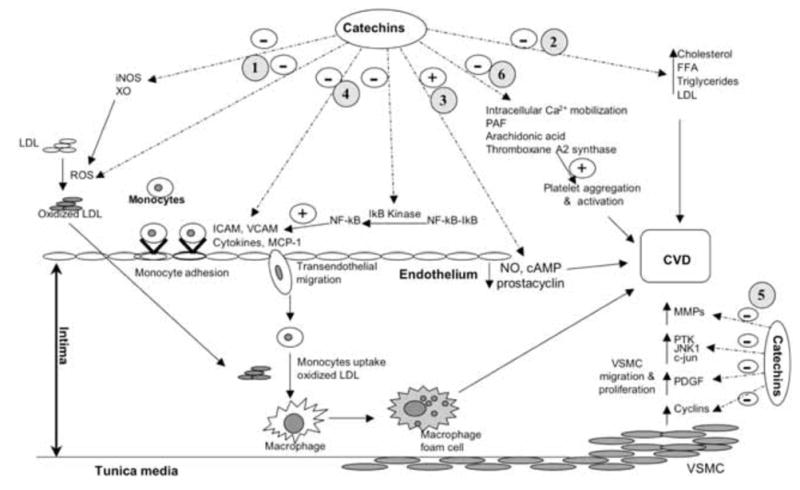
Scheme summarizing mechanisms underlying the vascular protective effects of catechin. (1) catechins scavenge free radicals and inhibits pro-oxidant enzymes, which consequently inhibit ROS induced oxidative stress and LDL oxidation; (2) catechins increases intestinal lipid excretion, inhibits cholesterol, fatty acids (FFA) and triglycerides absorption and synthesis; (3) catechins can stimulate endothelial production of NO, prostacyclin and cAMP; (4) catechins prevent adhesion of monocytes to endothelium and subsequent transendothelial migration by inhibition of NF-kB, cytokine and adhesion molecules; (5) catechins inhibit cyclins, PDGF, PTK, JNK1, c-jun and MMPs; and. (6) catechins also can reduce platelet aggregation and activation by reducing intracellular calcium mobilization, PAF and arachidonic acid release, and thromboxane A2 synthase. Consequently, modulation of these molecule events by catechins improves oxidative status, lipid profile, and vascular homeostasis whereas inhibits vascular inflammation, thrombosis, and VSMC growth and migration, thereby preventing vascular disease.
Acknowledgments
This work was supported by grants from the National Center for Complementary and Alternative Medicine of the National Institute of Health (R21AT002739 to D. Liu) and the American Heart Association Mid-Atlantic Affiliate grant (D. Liu), and by The Taishan Scholar Program (QDU-EYE) at Qingdao University, China.
ABBREVATIONS
- ADP
Adenosine diphosphate
- ApoA1
Apolipoprotein A1
- ApoB
Apolipoprotein B
- Camp
Cyclicadenosine-3′,5′-monophosphate
- cGMP
Cyclic guanosine-3′,5′-monophosphate
- CVD
Cardiovascular disease
- EC
(−)-Epicatechin
- ECG
(−)-Epicatechin gallate
- Ecs
Endothelial cells
- EGC
(−)-Epigallocatechin
- EGCG
(−)-Epigallocatechin gallate
- eNOS
Endothelial nitric oxide synthase
- FAS
Fatty acid synthase
- GPx
Glutathione peroxidase
- HDL
High density lipoprotein
- ICAM-1
Intracellular adhesion molecule-1
- IL-8
Interleukin-8
- INOS
Inducible nitric oxide synthase
- LDL
Low density lipoprotein
- MAPK
Mitogen activated protein kinase
- MCP-1
Macrophage chemoattractant protein-1
- MMP
Matrix metalloproteinase
- NF-kB
Nuclear factor- kappa B
- NO
Nitric oxide
- NO
Nitric oxide radical
- O2•−
Superoxide anion
- 1O2
Singlet oxygen
- ONOO−
Peroxynitrite radical
- PAF
Platelet activating factor
- PCNA
Proliferating cell nuclear antigen
- PDGF
Platelet derived growth factor
- PI3K
Phosphoinositide-3 kinase
- PTK
Protein tyrosine kinase
- ROS
Reactive oxygen species
- SOD
Superoxide dismutase
- SREBP-1
Sterol response element binding protein-1
- TNF-α
Tumor necrosis factor-α
- VCAM-1
Vascular cell adhesion molecule-1
- VSMC
Vascular smooth muscle cells
- XO
Xanthine oxidase
References
- 1.Balentine DA, Wiseman SA, Bouwens LC. Crit Rev Food Sci Nutr. 1997;37:693. doi: 10.1080/10408399709527797. [DOI] [PubMed] [Google Scholar]
- 2.Dufresne CJ, Farnworth ER. J Nutr Biochem. 2001;12:404. doi: 10.1016/s0955-2863(01)00155-3. [DOI] [PubMed] [Google Scholar]
- 3.Higdon JV, Frei B. Crit Rev Food Sci Nutr. 2003;43:89. doi: 10.1080/10408690390826464. [DOI] [PubMed] [Google Scholar]
- 4.Mukhtar H, Ahmad N. Am J Clin Nutr. 2000;71:1698S. doi: 10.1093/ajcn/71.6.1698S. [DOI] [PubMed] [Google Scholar]
- 5.Khan N, Mukhtar H. Life Sci. 2007;81:519. doi: 10.1016/j.lfs.2007.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rice-Evans CA, Miller NJ, Paganga G. Free Radic Biol Med. 1996;20:933. doi: 10.1016/0891-5849(95)02227-9. [DOI] [PubMed] [Google Scholar]
- 7.Crespy V, Williamson G. J Nutr. 2004;134:3431S. doi: 10.1093/jn/134.12.3431S. [DOI] [PubMed] [Google Scholar]
- 8.Shahidi F. Nahrung. 2000;44:158. doi: 10.1002/1521-3803(20000501)44:3<158::AID-FOOD158>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 9.Nakagawa K, Okuda S, Miyazawa T. Biosci Biotechnol Biochem. 1997;61:1981. doi: 10.1271/bbb.61.1981. [DOI] [PubMed] [Google Scholar]
- 10.Riemersma RA, Rice-Evans CA, Tyrrell RM, Clifford MN, Lean ME. QJM. 2001;94:277. doi: 10.1093/qjmed/94.5.277. [DOI] [PubMed] [Google Scholar]
- 11.Van Amelsvoort JM, Van Hof KH, Mathot JN, Mulder TP, Wiersma A, Tijburg LB. Xenobiotica. 2001;31:891. doi: 10.1080/00498250110079149. [DOI] [PubMed] [Google Scholar]
- 12.Lee MJ, Wang ZY, Li H, Chen L, Sun Y, Gobbo S, Balentine DA, Yang CS. Cancer Epidemiol Biomarkers Prev. 1995;4:393. [PubMed] [Google Scholar]
- 13.Harada M, Kan Y, Naoki H, Fukui Y, Kageyama N, Nakai M, Miki W, Kiso Y. Biosci Biotechnol Biochem. 1999;63:973. doi: 10.1271/bbb.63.973. [DOI] [PubMed] [Google Scholar]
- 14.Erdman JW, Jr, Balentine D, Arab L, Beecher G, Dwyer JT, Folts J, Harnly J, Hollman P, Keen CL, Mazza G, Messina M, Scalbert A, Vita J, Williamson G, Burrowes J. J Nutr. 2007;137:718S. doi: 10.1093/jn/137.3.718S. [DOI] [PubMed] [Google Scholar]
- 15.Chow HH, Cai Y, Hakim IA, Crowell JA, Shahi F, Brooks CA, Dorr RT, Hara Y, Alberts DS. Clin Cancer Res. 2003;9:3312. [PubMed] [Google Scholar]
- 16.McKay DL, Blumberg JB. J Am Coll Nutr. 2002;21:1. doi: 10.1080/07315724.2002.10719187. [DOI] [PubMed] [Google Scholar]
- 17.Centers for Disease Control and Prevention Washington, DC. Preventing Heart Disease and Stroke: Addressing the Nation’s Leading Killers 2005. 2005. [Google Scholar]
- 18.Shenouda SM, Vita JA. J Am Coll Nutr. 2007;26:366S. doi: 10.1080/07315724.2007.10719625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stangl V, Dreger H, Stangl K, Lorenz M. Cardiovasc Res. 2007;73:348. doi: 10.1016/j.cardiores.2006.08.022. [DOI] [PubMed] [Google Scholar]
- 20.Stangl V, Lorenz M, Stangl K. Mol Nutr Food Res. 2006;50:218. doi: 10.1002/mnfr.200500118. [DOI] [PubMed] [Google Scholar]
- 21.Vita JA. J Nutr. 2003;133:3293S. doi: 10.1093/jn/133.10.3293S. [DOI] [PubMed] [Google Scholar]
- 22.Sasazuki S, Kodama H, Yoshimasu K, Liu Y, Washio M, Tanaka K, Tokunaga S, Kono S, Arai H, Doi Y, Kawano T, Nakagaki O, Takada K, Koyanagi S, Hiyamuta K, Nii T, Shirai K, Ideishi M, Arakawa K, Mohri M, Takeshita A. Ann Epidemiol. 2000;10:401. doi: 10.1016/s1047-2797(00)00066-1. [DOI] [PubMed] [Google Scholar]
- 23.Kuriyama S, Shimazu T, Ohmori K, Kikuchi N, Nakaya N, Nishino Y, Tsubono Y, Tsuji I. JAMA. 2006;296:1255. doi: 10.1001/jama.296.10.1255. [DOI] [PubMed] [Google Scholar]
- 24.Imai K, Nakachi K. BMJ. 1995;310:693. doi: 10.1136/bmj.310.6981.693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yang YC, Lu FH, Wu JS, Wu CH, Chang CJ. Arch Intern Med. 2004;164:1534. doi: 10.1001/archinte.164.14.1534. [DOI] [PubMed] [Google Scholar]
- 26.Hirano R, Momiyama Y, Takahashi R, Taniguchi H, Kondo K, Nakamura H, Ohsuzu F. Am J Cardiol. 2002;90:1150. doi: 10.1016/s0002-9149(02)02787-x. [DOI] [PubMed] [Google Scholar]
- 27.Kim W, Jeong MH, Cho SH, Yun JH, Chae HJ, Ahn YK, Lee MC, Cheng X, Kondo T, Murohara T, Kang JC. Circ J. 2006;70:1052. doi: 10.1253/circj.70.1052. [DOI] [PubMed] [Google Scholar]
- 28.Chyu KY, Babbidge SM, Zhao X, Dandillaya R, Rietveld AG, Yano J, Dimayuga P, Cercek B, Shah PK. Circulation. 2004;109:2448. doi: 10.1161/01.CIR.0000128034.70732.C2. [DOI] [PubMed] [Google Scholar]
- 29.Tijburg LB, Wiseman SA, Meijer GW, Weststrate JA. Atherosclerosis. 1997;135:37. doi: 10.1016/s0021-9150(97)00139-1. [DOI] [PubMed] [Google Scholar]
- 30.Miura Y, Chiba T, Tomita I, Koizumi H, Miura S, Umegaki K, Hara Y, Ikeda M, Tomita T. J Nutr. 2001;131:27. doi: 10.1093/jn/131.1.27. [DOI] [PubMed] [Google Scholar]
- 31.Negishi H, Xu JW, Ikeda K, Njelekela M, Nara Y, Yamori Y. J Nutr. 2004;134:38. doi: 10.1093/jn/134.1.38. [DOI] [PubMed] [Google Scholar]
- 32.Babu PV, Sabitha KE, Shyamaladevi CS. Chem Biol Interact. 2006;162:114. doi: 10.1016/j.cbi.2006.04.009. [DOI] [PubMed] [Google Scholar]
- 33.Babu PVA, Sabitha KE, Shyamaladevi CS. Clin Exp Pharmacol Physiol. 2006;33:1184. doi: 10.1111/j.1440-1681.2006.04509.x. [DOI] [PubMed] [Google Scholar]
- 34.Widlansky ME, Hamburg NM, Anter E, Holbrook M, Kahn DF, Elliott JG, Keaney JF, Jr, Vita JA. J Am Coll Nutr. 2007;26:95. doi: 10.1080/07315724.2007.10719590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schroeter H, Heiss C, Balzer J, Kleinbongard P, Keen CL, Hollenberg NK, Sies H, Kwik-Uribe C, Schmitz HH, Kelm M. Proc Natl Acad Sci U S A. 2006;103:1024. doi: 10.1073/pnas.0510168103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wolfram S. J Am Coll Nutr. 2007;26:373S. doi: 10.1080/07315724.2007.10719626. [DOI] [PubMed] [Google Scholar]
- 37.Sumpio BE, Cordova AC, Berke-Schlessel DW, Qin F, Chen QH. J Am Coll Surg. 2006;202:813. doi: 10.1016/j.jamcollsurg.2006.01.018. [DOI] [PubMed] [Google Scholar]
- 38.Dhalla NS, Temsah RM, Netticadan T. J Hypertens. 2000;18:655. doi: 10.1097/00004872-200018060-00002. [DOI] [PubMed] [Google Scholar]
- 39.Jain KS, Kathiravan MK, Somani RS, Shishoo CJ. Bioorg Med Chem. 2007;15:4674. doi: 10.1016/j.bmc.2007.04.031. [DOI] [PubMed] [Google Scholar]
- 40.Vita JA, Keaney JF., Jr Circulation. 2002;106:640. doi: 10.1161/01.cir.0000028581.07992.56. [DOI] [PubMed] [Google Scholar]
- 41.Ruggeri ZM. Nat Med. 2002;8:1227–34. doi: 10.1038/nm1102-1227. [DOI] [PubMed] [Google Scholar]
- 42.Zouridakis E, Avanzas P, Arroyo-Espliguero R, Fredericks S, Kaski JC. Circulation. 2004;110:1747. doi: 10.1161/01.CIR.0000142664.18739.92. [DOI] [PubMed] [Google Scholar]
- 43.Rudijanto A. Acta Med Indones. 2007;39:86. [PubMed] [Google Scholar]
- 44.Singal PK, Khaper N, Palace V, Kumar D. Cardiovasc Res. 1998;40:426. doi: 10.1016/s0008-6363(98)00244-2. [DOI] [PubMed] [Google Scholar]
- 45.Kukreja RC, Hess ML. Cardiovasc Res. 1992;26:641. doi: 10.1093/cvr/26.7.641. [DOI] [PubMed] [Google Scholar]
- 46.Griendling KK, Ushio-Fukai M. J Lab Clin Med. 1998;132:9. doi: 10.1016/s0022-2143(98)90019-1. [DOI] [PubMed] [Google Scholar]
- 47.Ushio-Fukai M, Alexander RW, Akers M, Griendling KK. J Biol Chem. 1998;273:15022. doi: 10.1074/jbc.273.24.15022. [DOI] [PubMed] [Google Scholar]
- 48.Kanani PM, Sinkey CA, Browning RL, Allaman M, Knapp HR, Haynes WG. Circulation. 1999;100:1161. doi: 10.1161/01.cir.100.11.1161. [DOI] [PubMed] [Google Scholar]
- 49.Britten MB, Zeiher AM, Schachinger V. J Intern Med. 1999;245:315. doi: 10.1046/j.1365-2796.1999.00449.x. [DOI] [PubMed] [Google Scholar]
- 50.Taimor G, Lorenz H, Hofstaetter B, Schluter KD, Piper HM. Cardiovasc Res. 1999;41:147. doi: 10.1016/s0008-6363(98)00209-0. [DOI] [PubMed] [Google Scholar]
- 51.von Harsdorf R, Li PF, Dietz R. Circulation. 1999;99:2934. doi: 10.1161/01.cir.99.22.2934. [DOI] [PubMed] [Google Scholar]
- 52.Wattanapitayakul SK, Bauer JA. Pharmacol Ther. 2001;89:187. doi: 10.1016/s0163-7258(00)00114-5. [DOI] [PubMed] [Google Scholar]
- 53.Frei B, Higdon JV. J Nutr. 2003;133:3275S. doi: 10.1093/jn/133.10.3275S. [DOI] [PubMed] [Google Scholar]
- 54.Vita JA. Am J Clin Nutr. 2005;81:292S. doi: 10.1093/ajcn/81.1.292S. [DOI] [PubMed] [Google Scholar]
- 55.Cabrera C, Artacho R, Gimenez R. J Am Coll Nutr. 2006;25:79. doi: 10.1080/07315724.2006.10719518. [DOI] [PubMed] [Google Scholar]
- 56.Vinson JA, Dabbagh YA, Serry MM, JJ J Agric Food Chem. 1995;43:2798. [Google Scholar]
- 57.Leenen R, Roodenburg AJ, Tijburg LB, Wiseman SA. Eur J Clin Nutr. 2000;54:87. doi: 10.1038/sj.ejcn.1600900. [DOI] [PubMed] [Google Scholar]
- 58.Rietveld A, Wiseman S. J Nutr. 2003;133:3285S. doi: 10.1093/jn/133.10.3285S. [DOI] [PubMed] [Google Scholar]
- 59.Erba D, Riso P, Bordoni A, Foti P, Biagi PL, Testolin G. J Nutr Biochem. 2005;16:144. doi: 10.1016/j.jnutbio.2004.11.006. [DOI] [PubMed] [Google Scholar]
- 60.Liao S, Kao YH, Hiipakka RA. Vitam Horm. 2001;62:1. doi: 10.1016/s0083-6729(01)62001-6. [DOI] [PubMed] [Google Scholar]
- 61.Heim KE, Tagliaferro AR, Bobilya DJ. J Nutr Biochem. 2002;13:572. doi: 10.1016/s0955-2863(02)00208-5. [DOI] [PubMed] [Google Scholar]
- 62.Nijveldt RJ, van Nood E, van Hoorn DE, Boelens PG, van Norren K, van Leeuwen PA. Am J Clin Nutr. 2001;74:418. doi: 10.1093/ajcn/74.4.418. [DOI] [PubMed] [Google Scholar]
- 63.van Acker SA, de Groot MJ, van den Berg DJ, Tromp MN, Donne-Op den Kelder G, van der Vijgh WJ, Bast A. Chem Res Toxicol. 1996;9:1305. doi: 10.1021/tx9600964. [DOI] [PubMed] [Google Scholar]
- 64.Morel I, Lescoat G, Cogrel P, Sergent O, Pasdeloup N, Brissot P, Cillard P, Cillard J. Biochem Pharmacol. 1993;45:13. doi: 10.1016/0006-2952(93)90371-3. [DOI] [PubMed] [Google Scholar]
- 65.Paquay JB, Haenen GR, Stender G, Wiseman SA, Tijburg LB, Bast A. J Agric Food Chem. 2000;48:5768. doi: 10.1021/jf981316h. [DOI] [PubMed] [Google Scholar]
- 66.Guo Q, Zhao B, Shen S, Hou J, Hu J, Xin W. Biochim Biophys Acta. 1999;1427:13. doi: 10.1016/s0304-4165(98)00168-8. [DOI] [PubMed] [Google Scholar]
- 67.Cao G, Sofic E, Prior RL. Free Radic Biol Med. 1997;22:749. doi: 10.1016/s0891-5849(96)00351-6. [DOI] [PubMed] [Google Scholar]
- 68.Ratty AK, Das NP. Biochem Med Metab Biol. 1988;39:69. doi: 10.1016/0885-4505(88)90060-6. [DOI] [PubMed] [Google Scholar]
- 69.Heimann WRF. Fette Seifen Anstr-Mittel. 1953;55:451. [Google Scholar]
- 70.Cholbi MR, Paya M, Alcaraz MJ. Experientia. 1991;47:195. doi: 10.1007/BF01945426. [DOI] [PubMed] [Google Scholar]
- 71.Nanjo F, Mori M, Goto K, Hara Y. Biosci Biotechnol Biochem. 1999;63:1621. doi: 10.1271/bbb.63.1621. [DOI] [PubMed] [Google Scholar]
- 72.Miller NJ, Castelluccio C, Tijburg L, Rice-Evans C. FEBS Lett. 1996;392:40. doi: 10.1016/0014-5793(96)00780-6. [DOI] [PubMed] [Google Scholar]
- 73.Mira L, Fernandez MT, Santos M, Rocha R, Florencio MH, Jennings KR. Free Radic Res. 2002;36:1199. doi: 10.1080/1071576021000016463. [DOI] [PubMed] [Google Scholar]
- 74.Hider RC, Liu ZD, Khodr HH. Methods Enzymol. 2001;335:190. doi: 10.1016/s0076-6879(01)35243-6. [DOI] [PubMed] [Google Scholar]
- 75.Kumamoto M, Sonda T, Nagayama K, Tabata M. Biosci Biotechnol Biochem. 2001;65:126. doi: 10.1271/bbb.65.126. [DOI] [PubMed] [Google Scholar]
- 76.Osada K, Takahashi M, Hoshina S, Nakamura M, Nakamura S, Sugano M. Comp Biochem Physiol C Toxicol Pharmacol. 2001;128:153. doi: 10.1016/s1532-0456(00)00192-7. [DOI] [PubMed] [Google Scholar]
- 77.Chan MM, Fong D, Ho CT, Huang HI. Biochem Pharmacol. 1997;54:1281. doi: 10.1016/s0006-2952(97)00504-2. [DOI] [PubMed] [Google Scholar]
- 78.Tijburg LB, Mattern T, Folts JD, Weisgerber UM, Katan MB. Crit Rev Food Sci Nutr. 1997;37:771. doi: 10.1080/10408399709527802. [DOI] [PubMed] [Google Scholar]
- 79.Lambert JD, Sang S, Yang CS. Chem Res Toxicol. 2007;20:583. doi: 10.1021/tx7000515. [DOI] [PubMed] [Google Scholar]
- 80.Azam S, Hadi N, Khan NU, Hadi SM. Toxicol In vitro. 2004;18:555. doi: 10.1016/j.tiv.2003.12.012. [DOI] [PubMed] [Google Scholar]
- 81.Oikawa S, Furukawaa A, Asada H, Hirakawa K, Kawanishi S. Free Radic Res. 2003;37:881. doi: 10.1080/1071576031000150751. [DOI] [PubMed] [Google Scholar]
- 82.Weisburg JH, Weissman DB, Sedaghat T, Babich H. Basic Clin Pharmacol Toxicol. 2004;95:191. doi: 10.1111/j.1742-7843.2004.pto_950407.x. [DOI] [PubMed] [Google Scholar]
- 83.Johnson MK, Loo G. Mutat Res. 2000;459:211. doi: 10.1016/s0921-8777(99)00074-9. [DOI] [PubMed] [Google Scholar]
- 84.Lin YL, Lin JK. Mol Pharmacol. 1997;52:465. [PubMed] [Google Scholar]
- 85.Sanhueza J, Valdes J, Campos R, Garrido A, Valenzuela A. Res Commun Chem Pathol Pharmacol. 1992;78:211. [PubMed] [Google Scholar]
- 86.Aucamp J, Gaspar A, Hara Y, Apostolides Z. Anticancer Res. 1997;17:4381. [PubMed] [Google Scholar]
- 87.Lin JK, Chen PC, Ho CT, Lin-Shiau SY. J Agric Food Chem. 2000;48:2736. doi: 10.1021/jf000066d. [DOI] [PubMed] [Google Scholar]
- 88.Khan SG, Katiyar SK, Agarwal R, Mukhtar H. Cancer Res. 1992;52:4050. [PubMed] [Google Scholar]
- 89.Zhu QY, Huang Y, Tsang D, Chen ZY. J Agric Food Chem. 1999;47:2020. doi: 10.1021/jf9809941. [DOI] [PubMed] [Google Scholar]
- 90.Sur-Altiner D, Yenice B. Drug Metabol Drug Interact. 2000;16:123. doi: 10.1515/dmdi.2000.16.2.123. [DOI] [PubMed] [Google Scholar]
- 91.Ishikawa T, Suzukawa M, Ito T, Yoshida H, Ayaori M, Nishiwaki M, Yonemura A, Hara Y, Nakamura H. Am J Clin Nutr. 1997;66:261. doi: 10.1093/ajcn/66.2.261. [DOI] [PubMed] [Google Scholar]
- 92.Yamanaka N, Oda O, Nagao S. FEBS Lett. 1997;401:230. doi: 10.1016/s0014-5793(96)01455-x. [DOI] [PubMed] [Google Scholar]
- 93.Miura S, Watanabe J, Tomita T, Sano M, Tomita I. Biol Pharm Bull. 1994;17:1567. doi: 10.1248/bpb.17.1567. [DOI] [PubMed] [Google Scholar]
- 94.American Heart Association. Heart and Stroke Statistical Update, 2000 2001 [Google Scholar]
- 95.Koo SI, Noh SK. J Nutr Biochem. 2007;18:179. doi: 10.1016/j.jnutbio.2006.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Lin JK, Lin-Shiau SY. Mol Nutr Food Res. 2006;50:211. doi: 10.1002/mnfr.200500138. [DOI] [PubMed] [Google Scholar]
- 97.Yang TT, Koo MW. Pharmacol Res. 1997;35:505. doi: 10.1006/phrs.1997.0176. [DOI] [PubMed] [Google Scholar]
- 98.Bursill CA, Roach PD. Lipids. 2007;42:621. doi: 10.1007/s11745-007-3077-x. [DOI] [PubMed] [Google Scholar]
- 99.Brown MS, Goldstein JL. Science. 1986;232:34. doi: 10.1126/science.3513311. [DOI] [PubMed] [Google Scholar]
- 100.Furuyashiki T, Nagayasu H, Aoki Y, Bessho H, Hashimoto T, Kanazawa K, Ashida H. Biosci Biotechnol Biochem. 2004;68:2353. doi: 10.1271/bbb.68.2353. [DOI] [PubMed] [Google Scholar]
- 101.Shishikura Y, Khokhar S, Murray BS. J Agric Food Chem. 2006;54:1906. doi: 10.1021/jf051988p. [DOI] [PubMed] [Google Scholar]
- 102.Armand M, Pasquier B, Andre M, Borel P, Senft M, Peyrot J, Salducci J, Portugal H, Jaussan V, Lairon D. Am J Clin Nutr. 1999;70:1096. doi: 10.1093/ajcn/70.6.1096. [DOI] [PubMed] [Google Scholar]
- 103.Wang S, Noh SK, Koo SI. J Nutr Biochem. 2006;17:492. doi: 10.1016/j.jnutbio.2006.03.004. [DOI] [PubMed] [Google Scholar]
- 104.Ikeda I, Imasato Y, Sasaki E, Nakayama M, Nagao H, Takeo T, Yayabe F, Sugano M. Biochim Biophys Acta. 1992;1127:141. doi: 10.1016/0005-2760(92)90269-2. [DOI] [PubMed] [Google Scholar]
- 105.Chan PT, Fong WP, Cheung YL, Huang Y, Ho WK, Chen ZY. J Nutr. 1999;129:1094. doi: 10.1093/jn/129.6.1094. [DOI] [PubMed] [Google Scholar]
- 106.Conseil G, Baubichon-Cortay H, Dayan G, Jault JM, Barron D, Di Pietro A. Proc Natl Acad Sci USA. 1998;95:9831. doi: 10.1073/pnas.95.17.9831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Leslie EM, Mao Q, Oleschuk CJ, Deeley RG, Cole SP. Mol Pharmacol. 2001;59:1171. doi: 10.1124/mol.59.5.1171. [DOI] [PubMed] [Google Scholar]
- 108.Chen L, Lee MJ, Li H, Yang CS. Drug Metab Dispos. 1997;25:1045. [PubMed] [Google Scholar]
- 109.Lee MJ, Maliakal P, Chen L, Meng X, Bondoc FY, Prabhu S, Lambert G, Mohr S, Yang CS. Cancer Epidemiol Biomarkers Prev. 2002;11:1025. [PubMed] [Google Scholar]
- 110.Wang S, Noh SK, Koo SI. J Nutr. 2006;136:2791. doi: 10.1093/jn/136.11.2791. [DOI] [PubMed] [Google Scholar]
- 111.Abe I, Seki T, Umehara K, Miyase T, Noguchi H, Sakakibara J, Ono T. Biochem Biophys Res Commun. 2000;268:767. doi: 10.1006/bbrc.2000.2217. [DOI] [PubMed] [Google Scholar]
- 112.Kono S, Shinchi K, Wakabayashi K, Honjo S, Todoroki I, Sakurai Y, Imanishi K, Nishikawa H, Ogawa S, Katsurada M. J Epidemiol. 1996;6:128. doi: 10.2188/jea.6.128. [DOI] [PubMed] [Google Scholar]
- 113.Tsubono Y, Tsugane S. Ann Epidemiol. 1997;7:280. doi: 10.1016/s1047-2797(97)00005-7. [DOI] [PubMed] [Google Scholar]
- 114.Kono S, Shinchi K, Ikeda N, Yanai F, Imanishi K. Prev Med. 1992;21:526. doi: 10.1016/0091-7435(92)90060-u. [DOI] [PubMed] [Google Scholar]
- 115.Chisaka T, Matsuda H, Kubomura Y, Mochizuki M, Yamahara J, Fujimura H. Chem Pharm Bull (Tokyo) 1988;36:227. doi: 10.1248/cpb.36.227. [DOI] [PubMed] [Google Scholar]
- 116.Kukita H, Hiwada K, Kokubu T. Atherosclerosis. 1984;51:261. doi: 10.1016/0021-9150(84)90173-4. [DOI] [PubMed] [Google Scholar]
- 117.Bursill C, Roach PD, Bottema CD, Pal S. J Agric Food Chem. 2001;49:5639. doi: 10.1021/jf010275d. [DOI] [PubMed] [Google Scholar]
- 118.Ikeda I, Tsuda K, Suzuki Y, Kobayashi M, Unno T, Tomoyori H, Goto H, Kawata Y, Imaizumi K, Nozawa A, Kakuda T. J Nutr. 2005;135:155. doi: 10.1093/jn/135.2.155. [DOI] [PubMed] [Google Scholar]
- 119.Hollman PC, Tijburg LB, Yang CS. Crit Rev Food Sci Nutr. 1997;37:719. doi: 10.1080/10408399709527799. [DOI] [PubMed] [Google Scholar]
- 120.Guharay J, Sengupta B, Sengupta PK. Proteins. 2001;43:75. doi: 10.1002/1097-0134(20010501)43:2<75::aid-prot1019>3.0.co;2-7. [DOI] [PubMed] [Google Scholar]
- 121.Basu A, Lucas EA. Nutr Rev. 2007;65:361. doi: 10.1301/nr.2007.aug.361-375. [DOI] [PubMed] [Google Scholar]
- 122.Kuo KL, Weng MS, Chiang CT, Tsai YJ, Lin-Shiau SY, Lin JK. J Agric Food Chem. 2005;53:480. doi: 10.1021/jf049375k. [DOI] [PubMed] [Google Scholar]
- 123.Yeh CW, Chen WJ, Chiang CT, Lin-Shiau SY, Lin JK. Pharmacogenomics J. 2003;3:267. doi: 10.1038/sj.tpj.6500192. [DOI] [PubMed] [Google Scholar]
- 124.Wang X, Song KS, Guo QX, Tian WX. Biochem Pharmacol. 2003;66:2039. doi: 10.1016/s0006-2952(03)00585-9. [DOI] [PubMed] [Google Scholar]
- 125.Watanabe J, Kawabata J, Niki R. Biosci Biotechnol Biochem. 1998;62:532. doi: 10.1271/bbb.62.532. [DOI] [PubMed] [Google Scholar]
- 126.Ignarro LJ, Buga GM, Wood KS, Byrns RE, Chaudhuri G. Proc Natl Acad Sci U S A. 1987;84:9265. doi: 10.1073/pnas.84.24.9265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Widlansky ME, Gokce N, Keaney JF, Jr, Vita JA. J Am Coll Cardiol. 2003;42:1149. doi: 10.1016/s0735-1097(03)00994-x. [DOI] [PubMed] [Google Scholar]
- 128.Hamburg NM, Vita J. In: Molecular mechanisms of atherosclerosis. Loscalzo J, editor. 2006. [Google Scholar]
- 129.Potenza MA, Marasciulo FL, Tarquinio M, Tiravanti E, Colantuono G, Federici A, Kim JA, Quon MJ, Montagnani M. Am J Physiol Endocrinol Metab. 2007;292:E1378. doi: 10.1152/ajpendo.00698.2006. [DOI] [PubMed] [Google Scholar]
- 130.Lorenz M, Wessler S, Follmann E, Michaelis W, Dusterhoft T, Baumann G, Stangl K, Stangl V. J Biol Chem. 2004;279:6190. doi: 10.1074/jbc.M309114200. [DOI] [PubMed] [Google Scholar]
- 131.Kim JA, Formoso G, Li Y, Potenza MA, Marasciulo FL, Montagnani M, Quon MJ. J Biol Chem. 2007;282:13736. doi: 10.1074/jbc.M609725200. [DOI] [PubMed] [Google Scholar]
- 132.Benito S, Lopez D, Saiz MP, Buxaderas S, Sanchez J, Puig-Parellada P, Mitjavila MT. Br J Pharmacol. 2002;135:910. doi: 10.1038/sj.bjp.0704534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.White CR, Darley-Usmar V, Berrington WR, McAdams M, Gore JZ, Thompson JA, Parks DA, Tarpey MM, Freeman BA. Proc Natl Acad Sci U S A. 1996;93:8745. doi: 10.1073/pnas.93.16.8745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Rajagopalan S, Kurz S, Munzel T, Tarpey M, Freeman BA, Griendling KK, Harrison DG. J Clin Invest. 1996;97:1916. doi: 10.1172/JCI118623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Furchgott RF. Biosci Rep. 1999;19:235. doi: 10.1023/a:1020537506008. [DOI] [PubMed] [Google Scholar]
- 136.Szabo C, Ischiropoulos H, Radi R. Nat Rev Drug Discov. 2007;6:662. doi: 10.1038/nrd2222. [DOI] [PubMed] [Google Scholar]
- 137.Alvarez E, Leiro J, Orallo F. Int Immunopharmacol. 2002;2:849. doi: 10.1016/s1567-5769(02)00032-2. [DOI] [PubMed] [Google Scholar]
- 138.Mizugaki M, Ishizawa F, Yamazaki T, Hishinuma T. Prostaglandins Other Lipid Mediat. 2000;62:157. doi: 10.1016/s0090-6980(00)00060-5. [DOI] [PubMed] [Google Scholar]
- 139.Huang Y, Zhang A, Lau CW, Chen ZY. Life Sci. 1998;63:275. doi: 10.1016/s0024-3205(98)00273-2. [DOI] [PubMed] [Google Scholar]
- 140.Alvarez E, Campos-Toimil M, Justiniano-Basaran H, Lugnier C, Orallo F. Br J Pharmacol. 2006;147:269. doi: 10.1038/sj.bjp.0706507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Sanae F, Miyaichi Y, Kizu H, Hayashi H. Life Sci. 2002;71:2553. doi: 10.1016/s0024-3205(02)02080-5. [DOI] [PubMed] [Google Scholar]
- 142.Shen JZ, Zheng XF, Wei EQ, Kwan CY. Clin Exp Pharmacol Physiol. 2003;30:88. doi: 10.1046/j.1440-1681.2003.03796.x. [DOI] [PubMed] [Google Scholar]
- 143.Mayer K, Merfels M, Muhly-Reinholz M, Gokorsch S, Rosseau S, Lohmeyer J, Schwarzer N, Krull M, Suttorp N, Grimminger F, Seeger W. Am J Physiol Heart Circ Physiol. 2002;283:H811. doi: 10.1152/ajpheart.00235.2002. [DOI] [PubMed] [Google Scholar]
- 144.Chen JW, Chen YH, Lin FY, Chen YL, Lin SJ. Arterioscler Thromb Vasc Biol. 2003;23:1559. doi: 10.1161/01.ATV.0000089012.73180.63. [DOI] [PubMed] [Google Scholar]
- 145.Carluccio MA, Siculella L, Ancora MA, Massaro M, Scoditti E, Storelli C, Visioli F, Distante A, De Caterina R. Arterioscler Thromb Vasc Biol. 2003;23:622. doi: 10.1161/01.ATV.0000062884.69432.A0. [DOI] [PubMed] [Google Scholar]
- 146.Gerszten RE, Garcia-Zepeda EA, Lim YC, Yoshida M, Ding HA, Gimbrone MA, Jr, Luster AD, Luscinskas FW, Rosenzweig A. Nature. 1999;398:718. doi: 10.1038/19546. [DOI] [PubMed] [Google Scholar]
- 147.Gerszten RE, Lim YC, Ding HT, Snapp K, Kansas G, Dichek DA, Cabanas C, Sanchez-Madrid F, Gimbrone MA, Jr, Rosenzweig A, Luscinskas FW. Circ Res. 1998;82:871. doi: 10.1161/01.res.82.8.871. [DOI] [PubMed] [Google Scholar]
- 148.Kevil CG, Patel RP, Bullard DC. Am J Physiol Cell Physiol. 2001;281:C1442. doi: 10.1152/ajpcell.2001.281.5.C1442. [DOI] [PubMed] [Google Scholar]
- 149.Ludwig A, Lorenz M, Grimbo N, Steinle F, Meiners S, Bartsch C, Stangl K, Baumann G, Stangl V. Biochem Biophys Res Commun. 2004;316:659. doi: 10.1016/j.bbrc.2004.02.099. [DOI] [PubMed] [Google Scholar]
- 150.Hong MH, Kim MH, Chang HJ, Kim NH, Shin BA, Ahn BW, Jung YD. Life Sci. 2007;80:1957. doi: 10.1016/j.lfs.2007.02.024. [DOI] [PubMed] [Google Scholar]
- 151.Hofbauer R, Frass M, Gmeiner B, Handler S, Speiser W, Kapiotis S. Wien Klin Wochenschr. 1999;111:278. [PubMed] [Google Scholar]
- 152.Kawai K, Tsuno NH, Kitayama J, Okaji Y, Yazawa K, Asakage M, Hori N, Watanabe T, Takahashi K, Nagawa H. J Allergy Clin Immunol. 2004;113:1211. doi: 10.1016/j.jaci.2004.02.044. [DOI] [PubMed] [Google Scholar]
- 153.Takano K, Nakaima K, Nitta M, Shibata F, Nakagawa H. J Agric Food Chem. 2004;52:4571. doi: 10.1021/jf0355194. [DOI] [PubMed] [Google Scholar]
- 154.Blackwell TS, Christman JW. Am J Respir Cell Mol Biol. 1997;17:3. doi: 10.1165/ajrcmb.17.1.f132. [DOI] [PubMed] [Google Scholar]
- 155.Ghosh S, May MJ, Kopp EB. Annu Rev Immunol. 1998;16:225. doi: 10.1146/annurev.immunol.16.1.225. [DOI] [PubMed] [Google Scholar]
- 156.Gerritsen ME, Williams AJ, Neish AS, Moore S, Shi Y, Collins T. Proc Natl Acad Sci USA. 1997;94:2927. doi: 10.1073/pnas.94.7.2927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Read MA, Whitley MZ, Williams AJ, Collins T. J Exp Med. 1994;179:503. doi: 10.1084/jem.179.2.503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Brown K, Gerstberger S, Carlson L, Franzoso G, Siebenlist U. Science. 1995;267:1485. doi: 10.1126/science.7878466. [DOI] [PubMed] [Google Scholar]
- 159.Ouchi N, Kihara S, Arita Y, Okamoto Y, Maeda K, Kuriyama H, Hotta K, Nishida M, Takahashi M, Muraguchi M, Ohmoto Y, Nakamura T, Yamashita S, Funahashi T, Matsuzawa Y. Circulation. 2000;102:1296. doi: 10.1161/01.cir.102.11.1296. [DOI] [PubMed] [Google Scholar]
- 160.Khan N, Afaq F, Saleem M, Ahmad N, Mukhtar H. Cancer Res. 2006;66:2500. doi: 10.1158/0008-5472.CAN-05-3636. [DOI] [PubMed] [Google Scholar]
- 161.Yang F, Oz HS, Barve S, de Villiers WJ, McClain CJ, Varilek GW. Mol Pharmacol. 2001;60:528. [PubMed] [Google Scholar]
- 162.Aneja R, Hake PW, Burroughs TJ, Denenberg AG, Wong HR, Zingarelli B. Mol Med. 2004;10:55. doi: 10.2119/2004-00032.aneja. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Nomura M, Ma W, Chen N, Bode AM, Dong Z. Carcinogenesis. 2000;21:1885. doi: 10.1093/carcin/21.10.1885. [DOI] [PubMed] [Google Scholar]
- 164.Nam S, Smith DM, Dou QP. J Biol Chem. 2001;276:13322. doi: 10.1074/jbc.M004209200. [DOI] [PubMed] [Google Scholar]
- 165.Koga T, Meydani M. Am J Clin Nutr. 2001;73:941. doi: 10.1093/ajcn/73.5.941. [DOI] [PubMed] [Google Scholar]
- 166.Ridker PM, Rifai N, Rose L, Buring JE, Cook NR. N Engl J Med. 2002;347:1557. doi: 10.1056/NEJMoa021993. [DOI] [PubMed] [Google Scholar]
- 167.Lee W, Min WK, Chun S, Lee YW, Park H, Lee DH, Lee YK, Son JE. Clin Biochem. 2005;38:84. doi: 10.1016/j.clinbiochem.2004.09.024. [DOI] [PubMed] [Google Scholar]
- 168.Hofmann CS, Sonenshein GE. Faseb J. 2003;17:702. doi: 10.1096/fj.02-0665fje. [DOI] [PubMed] [Google Scholar]
- 169.Ouyang P, Peng WL, Lai WY, Xu AL. Di Yi Jun Yi Da Xue Xue Bao. 2004;24:975. [PubMed] [Google Scholar]
- 170.Sachinidis A, Skach RA, Seul C, Ko Y, Hescheler J, Ahn HY, Fingerle J. FASEB J. 2002;16:893. doi: 10.1096/fj.01-0799fje. [DOI] [PubMed] [Google Scholar]
- 171.Kim CH, Moon SK. Arch Biochem Biophys. 2005;435:264. doi: 10.1016/j.abb.2004.12.022. [DOI] [PubMed] [Google Scholar]
- 172.Lu LH, Lee SS, Huang HC. Br J Pharmacol. 1998;124:1227. doi: 10.1038/sj.bjp.0701912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Weber AA, Neuhaus T, Skach RA, Hescheler J, Ahn HY, Schror K, Ko Y, Sachinidis A. FASEB J. 2004;18:128. doi: 10.1096/fj.03-0007fje. [DOI] [PubMed] [Google Scholar]
- 174.Kleiner DE, Stetler-Stevenson WG. Cancer Chemother Pharmacol. 1999;43 Suppl:S42. doi: 10.1007/s002800051097. [DOI] [PubMed] [Google Scholar]
- 175.George SJ. Expert Opin Investig Drugs. 2000;9:993. doi: 10.1517/13543784.9.5.993. [DOI] [PubMed] [Google Scholar]
- 176.Deguchi JO, Aikawa M, Tung CH, Aikawa E, Kim DE, Ntziachristos V, Weissleder R, Libby P. Circulation. 2006;114:55. doi: 10.1161/CIRCULATIONAHA.106.619056. [DOI] [PubMed] [Google Scholar]
- 177.Raymond J, Lebel V, Ogoudikpe C, Metcalfe A, Chagnon M, Robledo O. J Vasc Surg. 2004;40:1190. doi: 10.1016/j.jvs.2004.09.023. [DOI] [PubMed] [Google Scholar]
- 178.El Bedoui J, Oak MH, Anglard P, Schini-Kerth VB. Cardiovasc Res. 2005;67:317. doi: 10.1016/j.cardiores.2005.03.017. [DOI] [PubMed] [Google Scholar]
- 179.Maeda K, Kuzuya M, Cheng XW, Asai T, Kanda S, Tamaya-Mori N, Sasaki T, Shibata T, Iguchi A. Atherosclerosis. 2003;166:23. doi: 10.1016/s0021-9150(02)00302-7. [DOI] [PubMed] [Google Scholar]
- 180.Jagroop IA, Kakafika AI, Mikhailidis DP. Curr Pharm Des. 2007;13:1669. doi: 10.2174/138161207780831383. [DOI] [PubMed] [Google Scholar]
- 181.Zusman RM, Chesebro JH, Comerota A, Hartmann JR, Massin EK, Raps E, Wolf PA. Clin Cardiol. 1999;22:559. doi: 10.1002/clc.4960220905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Kang WS, Chung KH, Chung JH, Lee JY, Park JB, Zhang YH, Yoo HS, Yun YP. J Cardiovasc Pharmacol. 2001;38:875. doi: 10.1097/00005344-200112000-00009. [DOI] [PubMed] [Google Scholar]
- 183.Kang WS, Lim IH, Yuk DY, Chung KH, Park JB, Yoo HS, Yun YP. Thromb Res. 1999;96:229. doi: 10.1016/s0049-3848(99)00104-8. [DOI] [PubMed] [Google Scholar]
- 184.Jin YR, Im JH, Park ES, Cho MR, Han XH, Lee JJ, Lim Y, Kim TJ, Yun YP. J Cardiovasc Pharmacol. 2008;51:45. doi: 10.1097/FJC.0b013e31815ab4b6. [DOI] [PubMed] [Google Scholar]
- 185.Yang JA, Choi JH, Rhee SJ. J Nutr Sci Vitaminol (Tokyo) 1999;45:337. doi: 10.3177/jnsv.45.337. [DOI] [PubMed] [Google Scholar]
- 186.Sugatani J, Fukazawa N, Ujihara K, Yoshinari K, Abe I, Noguchi H, Miwa M. Int Arch Allergy Immunol. 2004;134:17. doi: 10.1159/000077529. [DOI] [PubMed] [Google Scholar]
- 187.Vezza R, Mezzasoma AM, Venditti G, Gresele P. Thromb Haemost. 2002;87:114. [PubMed] [Google Scholar]
- 188.Son DJ, Cho MR, Jin YR, Kim SY, Park YH, Lee SH, Akiba S, Sato T, Yun YP. Prostaglandins Leukot Essent Fatty Acids. 2004;71:25. doi: 10.1016/j.plefa.2003.12.004. [DOI] [PubMed] [Google Scholar]