

Abstract
Chromatin Immunoprecipitation (ChIP) is an important technique for studying protein-DNA interactions. Whole genome ChIP methods have enjoyed much success, but are limited in that they cannot uncover important long-range chromatin interactions. Chromosome Conformation Capture (3C) and related methods are capable of detecting remote chromatin interactions, but are tedious, have low signal-to-noise ratios, and are not genome-wide. Although the addition of ChIP to 3C (ChIP-3C) would conceivably reduce noise and increase specificity for chromatin interaction detection, there are concerns that simple mixing of the ChIP and 3C protocols would lead to high levels of false positives. In this essay, we dissect current ChIP-and 3C-based methodologies, discuss the models of specific as opposed to non-specific chromatin interactions, and suggest approaches to separate specific chromatin complexes from non-specific chromatin fragments. We conclude that the combination of sonication-based chromatin fragmentation, ChIP-based enrichment, chromatin proximity ligation and Paired-End Tag ultra-high-throughput sequencing will be a winning implementation for genome-wide, unbiased and de novo discovery of long-range chromatin interactions, which will help to establish an emerging field for studying human chromatin interactomes and genome regulation networks in 3-dimensional spaces.
Keywords: Interactome, Chromatin Interactions, Transcription Factor Binding Sites, Sequencing
Introduction
Since the publication of the human genome sequence [Lander et al., 2001; Venter et al., 2001], attention has turned towards the annotation and analysis of functional genetic elements. Besides gene coding elements, regulatory elements such as insulators, boundary elements, and transcription factor binding sites (TFBS) that control gene expression, also have great relevance to human health [Maston et al., 2006]. Uncovering the locations of regulatory elements, the interplay between regulatory elements and gene coding regions, and the mechanisms by which regulatory elements act to mediate gene transcription is of critical importance for understanding how regulatory elements can impact human health. An important aspect of regulatory elements is that they are the recognition sites for protein factors to bind in the human genome to carry out regulatory functions [Maston et al., 2006]. Regulatory sites that are far apart in terms of genomic distance could functionally interact in 3D conformation, mediated by protein factors [West and Fraser, 2005]. Therefore, the study of protein-DNA interactions and the long-range interactions between regulatory sites, collectively called the chromatin interactome, illuminates important aspects of genome biology.
Such endeavors are largely dependent on the technologies to assay the complicated organization of human chromatin interactomes. The two primary technologies are Chromatin Immunoprecipitation (ChIP) [Kuo and Allis, 1999] and Chromosome Conformation Capture (3C) [Dekker et al., 2002]. ChIP is a popular method for identification of transcription factor binding site locations in the genome and 3C is designed to identify long-range chromatin interactions. Although ChIP is robust for studying protein-DNA interactions and the recent ChIP-sequencing strategy is highly effective for genome-wide identification of transcription factor binding sites [Barski et al., 2007; Johnson et al., 2007; Wei et al., 2006], it only provides linear information of protein binding sites along chromosomes. While 3C is capable of analyzing long-range chromatin interactions, the data interpretation is complicated by high levels of background noise, and therefore, has to rely on intricate controls for accurate characterization of chromatin interactions, making the assay extremely difficult to perform. Besides, 3C has limited detection scope. Even higher-throughput variants such as 4C [Ling et al., 2006; Simonis et al., 2006; Wurtele and Chartrand, 2006; Zhao et al., 2006] and 5C [Dostie et al., 2006] (Figure 1) are not genome-wide. Hence, a highly robust and global strategy for investigating higher-order chromatin structures is needed to understand mechanisms for the remote control of transcription regulation in 3D nuclear space.
Figure 1. Schematic comparison of 3C, 4C, 5C, ChIP-3C, and ChIA-PET.
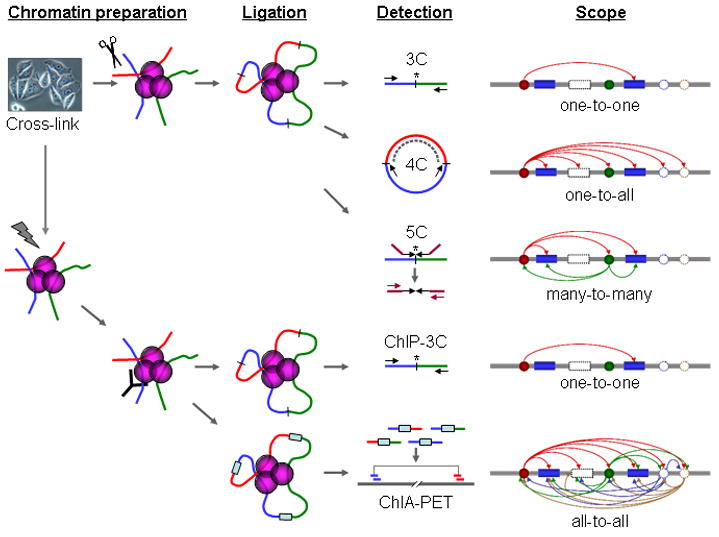
This schematic shows the differences between the (top to bottom) 3C, 4C, 5C, ChIP-3C and ChIA-PET methodologies. The ChIA-PET method can be implemented with two possible sources of material: ChIP-enriched and non ChIP-enriched sonicated chromatin.
In this essay, we dissect the current ChIP-and 3C-based methodologies, discuss the nature of chromatin interactions, and suggest new approaches that are highly specific, effective, genome-wide, and de novo for the analysis of the human chromatin interactome.
Chromatin Immunoprecipitation (ChIP)
ChIP is the most widely used method to determine protein-DNA associations in vivo. In this procedure, protein-DNA interactions in chromatin are covalently cross-linked by formaldehyde treatment, followed by fragmentation of the chromatin fibers, typically by physical shearing through sonication which randomly breaks chromatin into pieces of a few hundred base pairs, followed by immunoprecipitation of the protein-bound chromatin using specific antibodies against target protein factors, then reverse cross-linking, and detection [Kuo and Allis, 1999]. Enriched DNA fragments (from protein binding sites in chromatin) can then be quantified at specific loci by qPCR, or mapped in a genome-wide manner by DNA microarrays (ChIP-chip) [Buck and Lieb, 2004; Wu et al., 2006]. The most recent advance for ChIP analysis is the ChIP sequencing strategy [Wei et al., 2006]. This strategy has been further improved as ChIP-Seq [Barski et al., 2007; Johnson et al., 2007], which sequences reverse cross-linked, ChIP-enriched, fragmented chromatin by ultra-high-throughput next generation sequencing methods [Schuster, 2008].
One point to note in ChIP experiments is that the ChIP enrichment of specific protein binding sites is against the entire human genome sequence as the background. Therefore, although highly enriched sites may be enriched up to thousand-folds by ChIP, the overall useful sequence data for identifying TFBS constitute a very small portion of the total sequences generated in each experiment, reflecting high noise levels [Kim and Ren, 2006]. For example, one ChIP-seq experiment found that 77% of ChIP-Seq reads could be uniquely mapped to the genome; the rest mapped to multiple locations; only 12.8% of unique reads (<10% of total sequences) could be found in TFBS [Johnson et al., 2007]. Similar results have also been reported in other papers [Lim et al., 2007; Lin et al., 2007; Loh et al., 2006; Wei et al., 2006; Zeller et al., 2006; Zhao et al., 2007]. Improvements in ChIP protocols for high efficient purification are desirable.
Nonetheless, despite high levels of non-specific sequencing background, ChIP-PET and ChIP-Seq sequencing are robust and reliable for the identification of true TFBS because of high local signal-to-noise ratios. Most interestingly, analysis of whole genome ChIP sequencing TFBS maps has revealed that many transcription factors show complex binding patterns with relation to target genes including p53 [Wei et al., 2006], Oct4 and Nanog [Loh et al., 2006], and others. For many transcription factors, a great proportion of TFBS are far away from promoters of putative target genes. How remote regulatory elements function, if at all, is still largely unknown.
Chromosome Conformation Capture (3C)
3C is another important technology for studying regulatory elements. 3C focuses on long-range interactions between regulatory elements at 3D level of chromatin organization. Higher order chromatin structures, involving complex topological formations of chromatin into structures that bring two or more pieces of chromatin together in close spatial proximity, are thought to mediate transcriptional control and other cellular functions [Fraser and Bickmore, 2007; Woodcock, 2006]. The 3C method is based on the “proximity ligation” concept of the Nuclear Ligation Assay [Cullen et al., 1993], in which chromatin is formaldehyde cross-linked, restriction enzyme digested, and then “proximity ligated” to capture spatially related chromatin fragments, followed by detection of the newly formed ligation products using site-specific PCR (Figure 1). 3C was subsequently applied to the study of long-range chromatin interactions between the ß-globin locus and locus control regions (LCR) in mammalian cells [Tolhuis et al., 2002].
However, there are several limitations associated with the 3C method. First, 3C experiments have high noise levels. 3C data have shown that the frequency of such non-specific interaction noises is inversely proportional to the genomic distance between the two interrogated sites (the maximum distance at which non-specific interaction noise is seen at appreciable frequencies is about 100Kb) in any location of the genome. Formaldehyde cross-linking is known to give rise to a lot of non-specific noise [Kumar et al., 2007]. This non-specific noise reflects the cellular milieu, including noise from cross-linking of random chromatin interactions that “bumped” into each other at the time of formaldehyde treatment. Chromatin fibers as a type of polymer molecules obey the rules of thermodynamics, in which any two points in the linear structure would randomly collide, and the frequency of random collision is inversely proportional to the physical distance between the two sites; as such, sites that are close together on the linear genome would tend to randomly collide with each other at higher frequencies [Dekker, 2006]. Moreover, because the 3C method does not include any step to separate, nor to purify the true interactions from non-specific ones before detection, the noisy interactions will all be read. Second, because of complications by high noise levels, 3C analysis relies on a set of control experiments to distinguish real signals from noise [Dekker, 2006], which makes the protocol laborious and tedious.
The high noise levels and tedious protocol are among the reasons why despite high interest in chromatin structures, 3C does not have similar widespread adoption as ChIP-chip and ChIP-Seq). In addition, 3C methods are limited to single point interactions of previously known or hypothesized interaction sites. To overcome the limited detection scope of 3C for single interaction pairs, a number of groups have developed variants on 3C [Simonis et al., 2007], including Associated Chromatin Trap (ACT) [Ling et al., 2006], Chromosome Conformation Capture using Chip (4C) [Simonis et al., 2006], Circular Chromosome Conformation Capture (also called 4C) [Zhao et al., 2006] (Figure 1), Open-ended Chromosome Conformation Capture [Wurtele and Chartrand, 2006] and Chromosome Conformation Capture Carbon Copy (5C) [Dostie et al., 2006] (Figure 1) methods to expand the scope of detection for chromatin interactions. However, they are still constrained by their inability to provide a whole genome view of chromatin interactions. How to further our ability to study chromatin interactions in a highly efficient, low noise, genome-wide, and de novo manner remains a challenge.
ChIP-3C for more specific identification of chromatin interactions
Given such problems with 3C methods, ChIP-3C (also called ChIP-loop) has been developed by adding ChIP to the 3C protocol, both to reduce non-specific noise as well as identify chromatin interactions that are bound by specific proteins (Figure 1). ChIP-3C has yielded insights into chromatin looping as mechanisms whereby important proteins can mediate cellular functions such as gene regulation. There are a few different ChIP-3C protocols in use. One ChIP-3C protocol involves the preparation of urea ultracentrifugation-purified, restriction enzyme-digested, cross-linked chromatin, followed by ChIP enrichment, proximity ligation, reverse cross-linking to free DNA fragments from protein binding, and detection using PCR. SATB1-bound chromatin interactions have been examined using this ChIP-3C method [Cai et al., 2006], and the authors confirmed all interactions using 3C. The authors further employed RNA interference to knock down SATB1, which abrogated chromatin interactions and associated gene expression. Moreover, chromatin interactions bound by Mecp2, in particular a loop at the Dlx5-Dlx6 locus, have also been examined using this ChIP-3C protocol [Horike et al., 2005]. The authors used mouse knockout studies to demonstrate that Mecp2-null mice did not have this loop [Horike et al., 2005]. The second ChIP-3C protocol omitted the urea ultracentrifugation purification and simply combined 3C and ChIP. This method was used to uncover chromatin interactions bound by ERα [Carroll et al., 2005].
A variant on ChIP-3C is the so-called 6C technique, which uses a cloning approach for detection, instead of site-specific PCR employed in conventional ChIP-3C. In a 6C study, the usual 3C procedure was followed, then ChIP against EZH2 was performed, and the enriched chromatin DNA fragments are cloned for analysis. The clones were screened by restriction digestion to identify clones with multiple inserts (representing products that ligated during the 3C step instead of single genomic DNA fragments). 5 clones from a total pool of 352 clones were found to contain multiple inserts. They were analyzed by sequencing and validated by 3C. RNA interference was used to knock down EZH2, which reduced or eliminated the 5 chromatin interactions [Tiwari et al., 2008]. Such a technique is de novo, but not genome-wide as only a few clones can be analyzed at a time. Cloning and screening using restriction digestion to find clones with multiple inserts is also a laborious procedure. The fact that while only a few clones were analyzed, chromatin interactions could still be validated is a testament to the usefulness of ChIP enrichment in chromatin interaction analysis methods.
Concerns regarding the combination of ChIP and 3C
Despite the number of ChIP-3C papers that have been published, there is much skepticism within the chromatin interaction community as to whether the combination of ChIP and 3C is valid. As discussed by Simonis et al., one complication of ChIP-3C is accurate quantification of interaction levels, which must take into account both ChIP enrichment of the sites as well as high levels of non-specific chromatin noise due to random collisions [Simonis et al., 2007]. The risk is that a ChIP-enriched non-specific chromatin complex could be detected at high levels, and thus deemed an interaction when it is really a false positive. Because 3C contains much noise, and does not include any steps to separate specific interactions from non-specific interactions before detection; therefore, if anyone uses ChIP to pull down specific protein-bound chromatin interactions from such 3C chromatin fragments, one would also co-precipitate the non-specific chromatin fragments that are attached to the specific interaction complexes. As such, it is likely that the use of the standard 3C protocol with the addition of ChIP just as it is would lead to high levels of false positives. To address this issue, one particular ChIP-3C protocol uses urea ultracentrifugation purification of ChIP complexes to reduce the amount of non-enriched ChIP noise [Cai et al., 2006; Horike et al., 2005].
Nevertheless, this debate does raise questions on what is the nature of non-specific chromatin interactions, how such non-specific interactions are different from true and specific interactions, and how to experimentally separate non-specific chromatin interactions from true ones. 3C-like methods do not have a way to separate them physically, and only teases out actual chromatin interactions through elaborate controls. The practical question is: can the non-specific and specific interactions be physically separated? If such a method can be found, the reduction in noise would greatly benefit the field of chromatin interactions.
The nature of specific and non-specific chromatin interactions
In finding a method to remove non-specific chromatin interactions, we first had to formulate hypotheses on the nature of specific and non-specific chromatin interactions. We presume the non-specific interactions from random collisions of chromatin fibers may brush passing each other at the periphery of chromatin structure, whereas specific chromatin interactions are tethered to each other by specific factors (Figure 2). Support for this model comes from the observation that non-specific noise increases as cross-linking time and concentrations increase [Simonis et al., 2006]. Because non-specific interactions would only be in contact at the periphery of the chromatin structure, fewer covalent formaldehyde cross-links would be able to form. Hence, non-specific interactions would probably be much weaker than specific interactions. The observation that specific interactions between two specific locations are stronger (produce more 3C ligation products that can be detected by quantitative PCR) than non-specific interactions between any two other locations in the genome that are separated by the same genomic distance, also confirm this idea [Dekker, 2006].
Figure 2. Sonication is a method for breaking up non-specific chromatin interaction complexes.
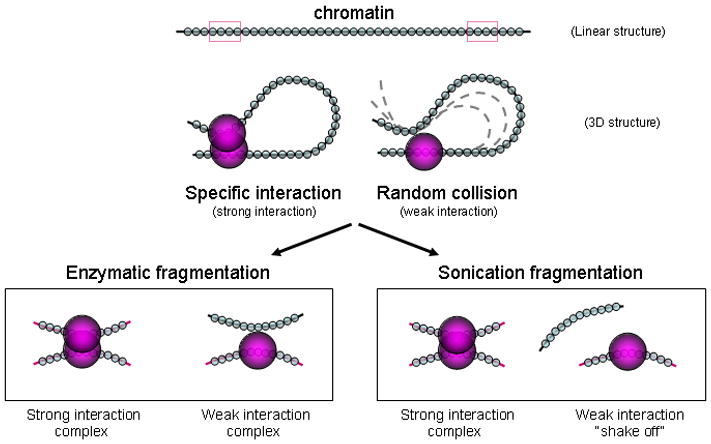
Non-specific interactions from random collisions of chromatin fibers generally involve chromatin interactions brushing past each other at the periphery of chromatin complexes, whereas specific chromatin interactions are tethered to each other by specific factors. Gentle restriction enzyme digestion would not disturb the weak non-specific interactions, but vigorous sonication could “shake off” non-specific, weak, peripheral chromatin and only retain strongly-bound core chromatin interaction complexes that involve specifically-bound chromatin interactions.
Because non-specific chromatin interactions are weak, and specific chromatin interactions are strong, we expect there must be some method for separating non-specific chromatin interactions from specific interactions. The urea ultracentrifugation method to purify chromatin complexes is one method, but it is tedious to perform. We therefore searched for a simple method of doing so.
Sonication-based ChIP-3C
Our approach to separate non-specific and specific chromatin interactions in our ChIP-3C protocols is to use sonication to fragment chromatin fibers (Figure 2), which is very different from the 3C protocol that uses restriction digestion to gently fragment chromatin fibers. The use of sonication in ChIP-3C has worked well [Kumar et al., 2007; Pan et al., 2008]. Chromatin interactions bound by SATB1 and PML have been found by sonication-based ChIP-3C. RNA interference was used to knock down SATB1 and PML, and this procedure was found to change the chromatin interaction profile as well as the expression of associated genes [Kumar et al., 2007]. Sonication-based ChIP-3C was also performed on the TFF1 region [Pan et al., 2008], and could recapitulate the TFF1 chromatin interaction shown to be present previously by restriction enzyme-based ChIP-3C [Carroll et al., 2005]. Further, sonication-based ChIP-3C was used to show that ERα mediates the TFF1 interactions, by the use of RNA interference to knock down ERα which abrogated the chromatin interaction and associated gene expression [Pan et al., 2008]. In addition to the demonstrated successes of the sonication-based ChIP-3C protocols, we will further show the benefits of sonication through a theoretical argument and several lines other experimental evidence, including whole genome ChIP sequencing and mapping experiments; and our own unpublished data on sonication-based molecular interaction mapping methods.
We believe that the use of restriction enzymes to gently digest the material could retain non-specific interactions where two chromatin fragments float close to each other in the crowded cellular nucleus. We and others have used sonication to fragment chromatin. While previously underappreciated, sonication is very vigorous, and could possibly break up weak non-specific interactions (Figure 2). Moreover, chromatin is sonicated to a region of about 200-1000 bp, as opposed to 3-4 kb fragments created by 6-bp restriction enzyme cutters. A much smaller chromatin fragment would be sterically hindered from ligating to non-specific interactions that are not in very close proximity, as compared with a longer chromatin fragment which would be much more flexible. In line with this idea, a review article analyzing all 3C and related methods suggests that 6-bp restriction enzyme cutters do not give such good resolution, and recommends the use of 4-bp cutters for analysis of <10-20 kb loci [Simonis et al., 2007]. Another benefit of the use of sonication instead of restriction enzyme digestion is that incomplete digestion products can be avoided. Given that incomplete digestions can form 20-30% of a library [Simonis et al., 2007], this is a large amount of noise that could be eliminated with sonication instead of restriction enzyme digestion.
Further, adding ChIP would enrich the specific protein-bound chromatin interaction complexes and wash away the non-specific chromatin fragments that were already detached from specific chromatin complexes by sonication, hence providing an even purer chromatin DNA pool for chromatin interaction analysis. Therefore, with this composition, sonication-based ChIP-3C protocols should have much less complication by high level non-specific interaction noises than the 3C-like methods.
Therefore, in comparing restriction enzyme digested chromatin, sonicated chromatin, and sonicated plus ChIP-enriched chromatin, we would expect to see different profiles from different detection techniques, with different levels of information. Simply performing reverse cross-linking of the chromatin and sequencing all material would be similar to a ChIP-Seq experiment if ChIP-enriched chromatin were used as the input (Figure 3). Performing 3C on restriction enzyme digested chromatin would be quite noisy, whereas 3C on sonicated chromatin would be less noisy, and ChIP-3C on sonicated chromatin would be the least noisy and specific for chromatin interactions bound to the protein of interest (Figure 4). Similar to 3C, 4C on enzyme digested chromatin would be quite noisy, whereas 4C on sonicated chromatin would be less noisy, and ChIP-4C on sonicated chromatin would be the least noisy and specific for chromatin interactions bound to the protein of interest (Figure 4).
Figure 3. Sequencing from different experimental approaches.
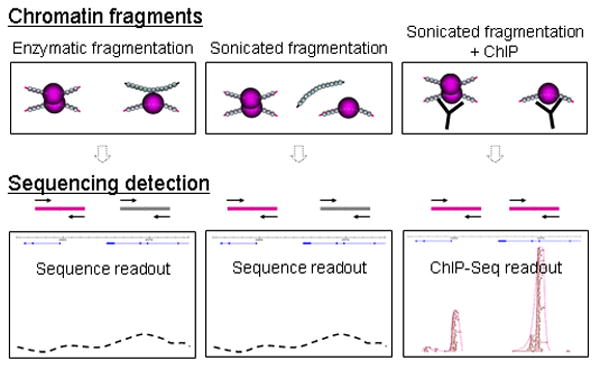
Here we show expected and actual experimental work from sequencing DNA from different experimental approaches: that of enzymatic fragmentation (left), sonicated fragmentation (center) and sonicated fragmentation with the addition of ChIP enrichment (right). Sequencing reverse cross-linked, fragmented, non ChIP-enriched chromatin would be similar to sequencing the entire genome. Sequencing reverse cross-linked, fragmented, ChIP-enriched chromatin would be similar to a ChIP-Seq experiment.
Figure 4. 3C and 4C from different experimental approaches.
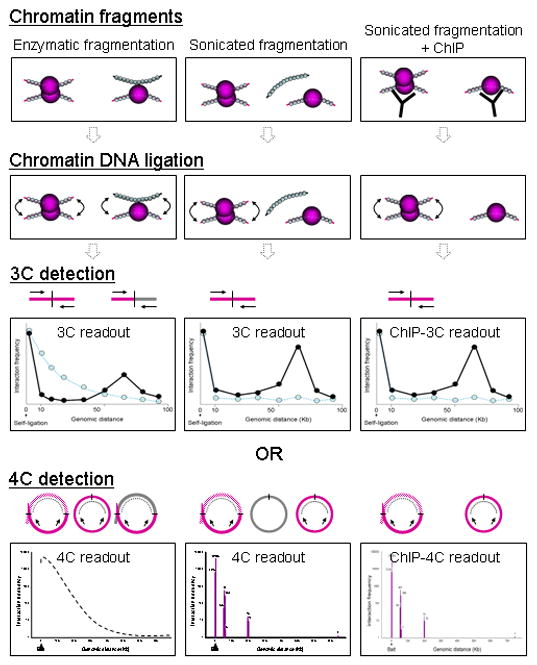
Here we show expected and actual experimental work from performing 3C or 4C on proximity-ligated material prepared by different experimental approaches: that of enzymatic fragmentation (left), sonicated fragmentation (center) and sonicated fragmentation with the addition of ChIP enrichment (right). 3C and 4C on enzyme digested chromatin would be quite noisy; 3C and 4C on sonicated chromatin would be less noisy, whilesChIP-3C and ChIP-4C on sonicated chromatin would be the least noisy and specific for chromatin interactions bound to the protein of interest. The use of sonication reduces some noise, and the further use of ChIP reduces the noise levels still further.
Experimental evidence from ChIP-Seq (Figure 3) and our sonication-based 4C data (Figure 4) support the hypothesis that sonication can “shake off” non-specific interactions, giving rise to more specific data. First, ChIP-Seq data show very specific binding peak results. ChIP-Seq methods commonly employ sonication to fragment the chromatin. In ChIP-Seq, formaldehyde cross-linking captures chromatin complexes, followed by sonication, and complexes are all immunoprecipitated in ChIP. Therefore, all reverse cross-linked ChIP DNA should, after mapping and sequencing, be reflected as binding density (Figure 3). This includes chromatin sequences that are not directly bound to the protein of interest, but which are involved in the complexes. As such, if weak non-specific interactions are present in chromatin complexes, they should be shown as regions of binding density in ChIP-Seq data (most ChIP-Seq protocols use sonication to fragment chromatin). In particular, if it were true that sonicated material shows high levels of non-specific interactions due to flexible polymer dynamics such that nonspecific interactions within 100 kb could occur, then we should see gradually sloping ChIP peaks. However, this is not the case, and most ChIP-Seq peaks, including ERα ChIP-Seq peaks, are very tight and narrow (Figure 3).
Second, our sonication-based 4C results are also very specific (Figure 4). In this 4C experiment, we used sonication to fragment the chromatin, which differs from the reported standard 4C protocols. Our 4C data, which is also non-ChIP enriched, shows a steep drop in the number of ligated sequences after 1kb. The number of ligated sequences remains 0 until it reaches the first interacting sequence ~50 kb away, whereupon it rises to several hundreds of sequences, indicating specific chromatin interactions. The lack of sequences between 1 to ~50 kb indicates that we do not detect any interactions despite using an unprecedented number of sequences (0.46 million sequences); hence the observations from restriction enzyme-based 3C, 4C, and 5C that distance-based, non-specific interactions up to 100 kb due to flexible polymer dynamics are present do not appear to be recapitulated when sonication is used.
Towards a whole genome method for identifying chromatin interactions
The sonication-based ChIP-3C protocol is robust, specific, and well-tested. Besides these advantages, another advantage is that sonication-based ChIP can enable the development of a method for global, de novo analysis of chromatin interactions by allowing effective noise reduction, thereby increasing signal to noise ratios to a level that current next-generation sequencing techniques can handle.
We propose a new strategy for whole genome Chromatin Interaction Analysis using Paired-End Tag sequencing (ChIA-PET) [Fullwood et al., in press]. The basic concept of ChIA-PET is to introduce a linker sequence in the junction of two DNA fragments during nuclear proximity ligation to build connectivity of DNA fragments that are tethered together by protein factors. Therefore, all linker-connected ligation products can be extracted as tag-linker-tag constructs that can be analyzed by ultra-high-throughput PET sequencing. When mapped to the reference genome, the ChIA-PET sequences are read out to detect the relationships between the two paired DNA fragments. Hence chromatin interactions captured by chromatin proximity ligation can be uncovered by ChIA-PET. As this strategy is not dependent on any specific sites for detection as 3C and 4C are, ChIA-PET has the potential to be an unbiased genome-wide approach for de novo detection of chromatin interactions (Figure 1).
We anticipate that the ChIA-PET strategy would not work well with the chromatin fragments prepared with restriction enzyme digestion due to the expected extremely high levels of noise, because the enzyme-based chromatin fragmentation would not be able to “shake off” the non-specific chromatin fragments attached to specific chromatin interactions (Figures 2 and 5). However, if the chromatin fragments are prepared using sonication, the ChIA-PET data should identify specific chromatin interactions with current sequencing capabilities as the non-specific noises are much reduced (Figure 5). In addition, the use of restriction enzymes means that the library would not be truly genome-wide, as it would be biased towards regions with the restriction enzyme sites. Also, fragments would map to restriction enzyme ends, making it difficult to apply existing methods for eliminating repeated sampling of the same sequence through removal of non-unique sequences, as well as clustering methods for identifying signals [Wei et al., 2006], as these methods require that multiple overlapping unique sequences must be found to call a signal. Further, incomplete restriction digestion, a problem in current 3C protocols [Simonis et al., 2007] would also result in high levels of sequenced noise. To reduce the complexity and background level, we propose to use sonication-based ChIP against specific protein factors to enrich the corresponding chromatin fragments before proximity ligation, in a “ChIP ChIA-PET” protocol (Figure 5). This enrichment approach would not only make the ChIA-PET sequencing practical by reducing the complexity, but also add specificity to the identified interaction points. Depending on the protein factors used for ChIP enrichment, ChIA-PET analysis can be applied to the detection of all chromatin interactions involved in a particular nuclear process. For instance, the use of general transcription factors such as RNA Polymerase II components would identify all chromatin interactions involved in transcription regulation; the use of protein factors involved in DNA replication or chromatin structure would allow identification of all chromatin interactions due to DNA replication and chromatin structural modification. More specifically, the use of specific transcription factors for ChIA-PET analysis would further reduce library complexity and add specificity, and therefore, enable examination of specific chromatin interactions mediated by particular transcription factors.
Figure 5. ChIA-PET from different experimental approaches.
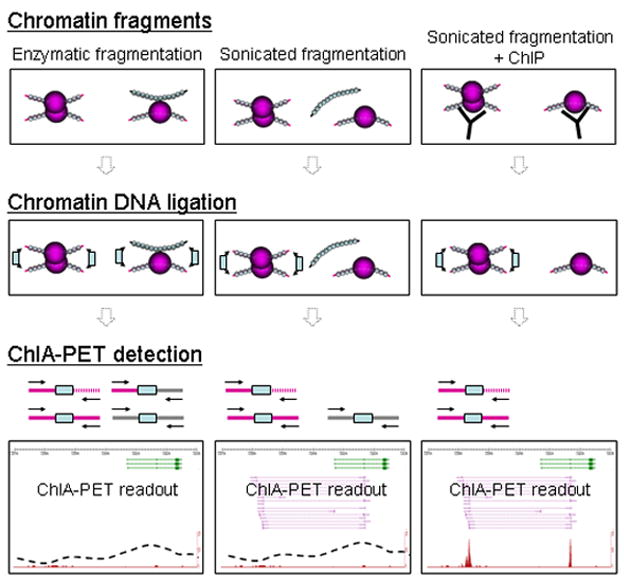
Here we show expected and actual experimental work from performing paired end sequencing on proximity-ligated chromatin prepared by different experimental approaches: that of enzymatic fragmentation (left), sonicated fragmentation (center) and sonicated fragmentation with the addition of ChIP enrichment (right). The use of sonication reduces some noise, and the further use of ChIP reduces the noise levels still further, allowing for the identification of true chromatin interaction signals. In addition, the use of ChIP ChIA-PET gives specific information regarding the TFBS and chromatin interactions between TFBS.
Our preliminary experimental data has demonstrated that ChIA-PET can generate PET sequences that identify TFBS and interactions between remote binding sites. With further development and optimization of the ChIA-PET prototype protocol, with or without ChIP enrichment, we expect this whole genome approach will become very robust for studying chromatin organization.
Future perspectives
Going forward, we believe that the incorporation of sonication-based chromatin fragmentation and ChIP-based enrichment into methods for the detection of chromatin interactions will greatly extend our knowledge of functional organization in chromatin structures and epigenomics in 3D space. With further improvements, eventually ChIA-PET, with its ability to both identify TFBS as well as chromatin interactions between these binding sites, could replace ChIP-Seq as the method of choice for studying protein-chromatin interactions and revealing entire chromatin interactomes. With much higher sequencing capabilities, ChIA-PET may be used directly on non-ChIP-enriched chromatin samples to identify all chromatin interactions in one experiment, where much greater sequencing depth can compensate for somewhat higher noise levels and greater library complexity, in order to open up still more vistas for whole genome chromatin interaction sequencing.
In conclusion, sonication-based chromatin fragmentation, ChIP-based enrichment, proximity ligation, Paired-End Tags and ultra-high-throughput sequencing methods that constitute the ChIA-PET approach will be a winning combination for genome-wide, unbiased and de novo discovery of long-range chromatin interactions, which will help to establish an emerging field for studying chromatin interacomes and regulation networks in 3D.
Acknowledgments
The authors are supported by A*STAR of Singapore. In addition, M.J.F. is supported by an A*STAR National Science Scholarship. Y.R is supported by NIH ENCODE grants R01HG003521-01 awarded to Y.R, as well as part of R01HG004456-01, and part of U54 HG004557-01.
References
- Barski A, Cuddapah S, Cui K, Roh TY, Schones DE, Wang Z, Wei G, Chepelev I, Zhao K. High-resolution profiling of histone methylations in the human genome. Cell. 2007;129:823–37. doi: 10.1016/j.cell.2007.05.009. [DOI] [PubMed] [Google Scholar]
- Buck MJ, Lieb JD. ChIP-chip: considerations for the design, analysis, and application of genome-wide chromatin immunoprecipitation experiments. Genomics. 2004;83:349–60. doi: 10.1016/j.ygeno.2003.11.004. [DOI] [PubMed] [Google Scholar]
- Cai S, Lee CC, Kohwi-Shigematsu T. SATB1 packages densely looped, transcriptionally active chromatin for coordinated expression of cytokine genes. Nat Genet. 2006;38:1278–88. doi: 10.1038/ng1913. [DOI] [PubMed] [Google Scholar]
- Carroll JS, Liu XS, Brodsky AS, Li W, Meyer CA, Szary AJ, Eeckhoute J, Shao W, Hestermann EV, Geistlinger TR, Fox EA, Silver PA, Brown M. Chromosome-wide mapping of estrogen receptor binding reveals long-range regulation requiring the forkhead protein FoxA1. Cell. 2005;122:33–43. doi: 10.1016/j.cell.2005.05.008. [DOI] [PubMed] [Google Scholar]
- Cullen KE, Kladde MP, Seyfred MA. Interaction between transcription regulatory regions of prolactin chromatin. Science. 1993;261:203–6. doi: 10.1126/science.8327891. [DOI] [PubMed] [Google Scholar]
- Dekker J. The three ‘C’ s of chromosome conformation capture: controls, controls, controls. Nat Methods. 2006;3:17–21. doi: 10.1038/nmeth823. [DOI] [PubMed] [Google Scholar]
- Dekker J, Rippe K, Dekker M, Kleckner N. Capturing chromosome conformation. Science. 2002;295:1306–11. doi: 10.1126/science.1067799. [DOI] [PubMed] [Google Scholar]
- Dostie J, Richmond TA, Arnaout RA, Selzer RR, Lee WL, Honan TA, Rubio ED, Krumm A, Lamb J, Nusbaum C, Green RD, Dekker J. Chromosome Conformation Capture Carbon Copy (5C): a massively parallel solution for mapping interactions between genomic elements. Genome Res. 2006;16:1299–309. doi: 10.1101/gr.5571506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraser P, Bickmore W. Nuclear organization of the genome and the potential for gene regulation. Nature. 2007;447:413–7. doi: 10.1038/nature05916. [DOI] [PubMed] [Google Scholar]
- Horike S, Cai S, Miyano M, Cheng JF, Kohwi-Shigematsu T. Loss of silent-chromatin looping and impaired imprinting of DLX5 in Rett syndrome. Nat Genet. 2005;37:31–40. doi: 10.1038/ng1491. [DOI] [PubMed] [Google Scholar]
- Johnson DS, Mortazavi A, Myers RM, Wold B. Genome-wide mapping of in vivo protein-DNA interactions. Science. 2007;316:1497–502. doi: 10.1126/science.1141319. [DOI] [PubMed] [Google Scholar]
- Kim TH, Ren B. Genome-wide analysis of protein-DNA interactions. Annu Rev Genomics Hum Genet. 2006;7:81–102. doi: 10.1146/annurev.genom.7.080505.115634. [DOI] [PubMed] [Google Scholar]
- Kumar PP, Bischof O, Purbey PK, Notani D, Urlaub H, Dejean A, Galande S. Functional interaction between PML and SATB1 regulates chromatin-loop architecture and transcription of the MHC class I locus. Nat Cell Biol. 2007;9:45–56. doi: 10.1038/ncb1516. [DOI] [PubMed] [Google Scholar]
- Kuo MH, Allis CD. In vivo cross-linking and immunoprecipitation for studying dynamic Protein:DNA associations in a chromatin environment. Methods. 1999;19:425–33. doi: 10.1006/meth.1999.0879. [DOI] [PubMed] [Google Scholar]
- Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J, Devon K, Dewar K, Doyle M, FitzHugh W, Funke R, Gage D, Harris K, Heaford A, Howland J, Kann L, Lehoczky J, LeVine R, McEwan P, McKernan K, Meldrim J, Mesirov JP, Miranda C, Morris W, Naylor J, Raymond C, Rosetti M, Santos R, Sheridan A, Sougnez C, Stange-Thomann N, Stojanovic N, Subramanian A, Wyman D, Rogers J, Sulston J, Ainscough R, Beck S, Bentley D, Burton J, Clee C, Carter N, Coulson A, Deadman R, Deloukas P, Dunham A, Dunham I, Durbin R, French L, Grafham D, Gregory S, Hubbard T, Humphray S, Hunt A, Jones M, Lloyd C, McMurray A, Matthews L, Mercer S, Milne S, Mullikin JC, Mungall A, Plumb R, Ross M, Shownkeen R, Sims S, Waterston RH, Wilson RK, Hillier LW, McPherson JD, Marra MA, Mardis ER, Fulton LA, Chinwalla AT, Pepin KH, Gish WR, Chissoe SL, Wendl MC, Delehaunty KD, Miner TL, Delehaunty A, Kramer JB, Cook LL, Fulton RS, Johnson DL, Minx PJ, Clifton SW, Hawkins T, Branscomb E, Predki P, Richardson P, Wenning S, Slezak T, Doggett N, Cheng JF, Olsen A, Lucas S, Elkin C, Uberbacher E, Frazier M, et al. Initial sequencing and analysis of the human genome. Nature. 2001;409:860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- Lim CA, Yao F, Wong JJ, George J, Xu H, Chiu KP, Sung WK, Lipovich L, Vega VB, Chen J, Shahab A, Zhao XD, Hibberd M, Wei CL, Lim B, Ng HH, Ruan Y, Chin KC. Genome-wide mapping of RELA(p65) binding identifies E2F1 as a transcriptional activator recruited by NF-kappaB upon TLR4 activation. Mol Cell. 2007;27:622–35. doi: 10.1016/j.molcel.2007.06.038. [DOI] [PubMed] [Google Scholar]
- Lin CY, Vega VB, Thomsen JS, Zhang T, Kong SL, Xie M, Chiu KP, Lipovich L, Barnett DH, Stossi F, Yeo A, George J, Kuznetsov VA, Lee YK, Charn TH, Palanisamy N, Miller LD, Cheung E, Katzenellenbogen BS, Ruan Y, Bourque G, Wei CL, Liu ET. Whole-genome cartography of estrogen receptor alpha binding sites. PLoS Genet. 2007;3:e87. doi: 10.1371/journal.pgen.0030087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling JQ, Li T, Hu JF, Vu TH, Chen HL, Qiu XW, Cherry AM, Hoffman AR. CTCF mediates interchromosomal colocalization between Igf2/H19 and Wsb1/Nf1. Science. 2006;312:269–72. doi: 10.1126/science.1123191. [DOI] [PubMed] [Google Scholar]
- Loh YH, Wu Q, Chew JL, Vega VB, Zhang W, Chen X, Bourque G, George J, Leong B, Liu J, Wong KY, Sung KW, Lee CW, Zhao XD, Chiu KP, Lipovich L, Kuznetsov VA, Robson P, Stanton LW, Wei CL, Ruan Y, Lim B, Ng HH. The Oct4 and Nanog transcription network regulates pluripotency in mouse embryonic stem cells. Nat Genet. 2006;38:431–40. doi: 10.1038/ng1760. [DOI] [PubMed] [Google Scholar]
- Maston GA, Evans SK, Green MR. Transcriptional Regulatory Elements in the Human Genome. Annu Rev Genomics Hum Genet. 2006;7:29–59. doi: 10.1146/annurev.genom.7.080505.115623. [DOI] [PubMed] [Google Scholar]
- Pan YF, Wansa KD, Liu MH, Zhao B, Hong SZ, Tan PY, Lim KS, Borque G, Liu ET, Cheung E. Regulation of estrogen receptor-mediated long-range transcription via evolutionarily conserved distal response elements. J Biol Chem. 2008 doi: 10.1074/jbc.M802024200. [DOI] [PubMed] [Google Scholar]
- Schuster SC. Next-generation sequencing transforms today’s biology. Nat Methods. 2008;5:16–8. doi: 10.1038/nmeth1156. [DOI] [PubMed] [Google Scholar]
- Simonis M, Klous P, Splinter E, Moshkin Y, Willemsen R, de Wit E, van Steensel B, de Laat W. Nuclear organization of active and inactive chromatin domains uncovered by chromosome conformation capture-on-chip (4C) Nat Genet. 2006;38:1348–54. doi: 10.1038/ng1896. [DOI] [PubMed] [Google Scholar]
- Simonis M, Kooren J, de Laat W. An evaluation of 3C-based methods to capture DNA interactions. Nat Methods. 2007;4:895–901. doi: 10.1038/nmeth1114. [DOI] [PubMed] [Google Scholar]
- Tiwari VK, Cope L, McGarvey KM, Ohm JE, Baylin SB. A novel 6C assay uncovers Polycomb-mediated higher order chromatin conformations. Genome Res. 2008;18:1171–9. doi: 10.1101/gr.073452.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tolhuis B, Palstra RJ, Splinter E, Grosveld F, de Laat W. Looping and interaction between hypersensitive sites in the active beta-globin locus. Mol Cell. 2002;10:1453–65. doi: 10.1016/s1097-2765(02)00781-5. [DOI] [PubMed] [Google Scholar]
- Venter JC, Adams MD, Myers EW, Li PW, Mural RJ, Sutton GG, Smith HO, Yandell M, Evans CA, Holt RA, Gocayne JD, Amanatides P, Ballew RM, Huson DH, Wortman JR, Zhang Q, Kodira CD, Zheng XH, Chen L, Skupski M, Subramanian G, Thomas PD, Zhang J, Gabor Miklos GL, Nelson C, Broder S, Clark AG, Nadeau J, McKusick VA, Zinder N, Levine AJ, Roberts RJ, Simon M, Slayman C, Hunkapiller M, Bolanos R, Delcher A, Dew I, Fasulo D, Flanigan M, Florea L, Halpern A, Hannenhalli S, Kravitz S, Levy S, Mobarry C, Reinert K, Remington K, Abu-Threideh J, Beasley E, Biddick K, Bonazzi V, Brandon R, Cargill M, Chandramouliswaran I, Charlab R, Chaturvedi K, Deng Z, Di Francesco V, Dunn P, Eilbeck K, Evangelista C, Gabrielian AE, Gan W, Ge W, Gong F, Gu Z, Guan P, Heiman TJ, Higgins ME, Ji RR, Ke Z, Ketchum KA, Lai Z, Lei Y, Li Z, Li J, Liang Y, Lin X, Lu F, Merkulov GV, Milshina N, Moore HM, Naik AK, Narayan VA, Neelam B, Nusskern D, Rusch DB, Salzberg S, Shao W, Shue B, Sun J, Wang Z, Wang A, Wang X, Wang J, Wei M, Wides R, Xiao C, Yan C, et al. The sequence of the human genome. Science. 2001;291:1304–51. doi: 10.1126/science.1058040. [DOI] [PubMed] [Google Scholar]
- Wei CL, Wu Q, Vega VB, Chiu KP, Ng P, Zhang T, Shahab A, Yong HC, Fu Y, Weng Z, Liu J, Zhao XD, Chew JL, Lee YL, Kuznetsov VA, Sung WK, Miller LD, Lim B, Liu ET, Yu Q, Ng HH, Ruan Y. A global map of p53 transcription-factor binding sites in the human genome. Cell. 2006;124:207–19. doi: 10.1016/j.cell.2005.10.043. [DOI] [PubMed] [Google Scholar]
- West AG, Fraser P. Remote control of gene transcription. Hum Mol Genet. 2005;14(Spec No 1):R101–11. doi: 10.1093/hmg/ddi104. [DOI] [PubMed] [Google Scholar]
- Woodcock CL. Chromatin architecture. Curr Opin Struct Biol. 2006;16:213–20. doi: 10.1016/j.sbi.2006.02.005. [DOI] [PubMed] [Google Scholar]
- Wu J, Smith LT, Plass C, Huang TH. ChIP-chip comes of age for genome-wide functional analysis. Cancer Res. 2006;66:6899–902. doi: 10.1158/0008-5472.CAN-06-0276. [DOI] [PubMed] [Google Scholar]
- Wurtele H, Chartrand P. Genome-wide scanning of HoxB1-associated loci in mouse ES cells using an open-ended Chromosome Conformation Capture methodology. Chromosome Res. 2006;14:477–95. doi: 10.1007/s10577-006-1075-0. [DOI] [PubMed] [Google Scholar]
- Zeller KI, Zhao X, Lee CW, Chiu KP, Yao F, Yustein JT, Ooi HS, Orlov YL, Shahab A, Yong HC, Fu Y, Weng Z, Kuznetsov VA, Sung WK, Ruan Y, Dang CV, Wei CL. Global mapping of c-Myc binding sites and target gene networks in human B cells. Proc Natl Acad Sci U S A. 2006;103:17834–9. doi: 10.1073/pnas.0604129103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao XD, Han X, Chew JL, Liu J, Chiu KP, Choo A, Orlov YL, Sung WK, Shahab A, Kuznetsov VA, Bourque G, Oh S, Ruan Y, Ng HH, Wei CL. Whole-genome mapping of histone H3 Lys4 and 27 trimethylations reveals distinct genomic compartments in human embryonic stem cells. Cell Stem Cell. 2007;1:286–98. doi: 10.1016/j.stem.2007.08.004. [DOI] [PubMed] [Google Scholar]
- Zhao Z, Tavoosidana G, Sjolinder M, Gondor A, Mariano P, Wang S, Kanduri C, Lezcano M, Sandhu KS, Singh U, Pant V, Tiwari V, Kurukuti S, Ohlsson R. Circular chromosome conformation capture (4C) uncovers extensive networks of epigenetically regulated intra-and interchromosomal interactions. Nat Genet. 2006;38:1341–7. doi: 10.1038/ng1891. [DOI] [PubMed] [Google Scholar]