

Summary
PACAP increases excitability of guinea pig cardiac neurons, an effect mediated through activation of PAC1 receptors. The signaling cascades that couple activation of the PAC1 receptor to alterations in membrane ionic conductances responsible for the PACAP effect are unknown. Intracellular recordings were made from neurons in kinase inhibitor-treated cardiac ganglia preparations to determine which of the intracellular cascades activated by PAC1 receptor stimulation mediate the PACAP effect. In control cells, long depolarizing current steps elicited 1–3 action potentials. In contrast, during application of 10 nM PACAP, depolarizing current pulses elicited multiple action potential firing (≥5 action potentials) in 79% of the neurons. Pretreatment with an adenylyl cyclase inhibitor, SQ 22536 (100 μM), suppressed the PACAP-induced increase in excitability whereas the presence of U-73122 (10 μM), a potent phospholipase C (PLC) inhibitor, had no effect. Thus, activation of adenylyl cyclase, but not PLC, was a critical step mediating the PACAP effect. Pretreatment with H-89 (1 μM), a protein kinase A inhibitor and PD 98059 (50 μM), a MEK kinase inhibitor, also significantly blunted the PACAP-induced increase in excitability. Furthermore, treatment with forskolin (5 μM), an activator of adenylyl cyclase, or exposure to the cell permeable cAMP analogue, 8-bromo-cAMP (1 mM) partially recapitulated the effect of PACAP on excitability. We conclude that the activation of signaling cascades downstream of cAMP mediate the PACAP-induced increase in cardiac neuron excitability.
Index Entries: adenylyl cyclase, cAMP, action potentials, parasympathetic neurons
Introduction
In the guinea pig, pituitary adenylate cyclase-activating polypeptide (PACAP) is present in the cholinergic preganglionic parasympathetic terminals innervating all neurons in the intrinsic cardiac ganglia (Braas et al., 1998; Calupca et al., 2000; Parsons et al., 2006). In addition, virtually all guinea pig intrinsic cardiac neurons express the PACAP-selective PAC1 receptor and application of PACAP to these neurons dramatically increases neuronal excitability (Braas et al., 1998; Tompkins et al., 2006). Most guinea pig cardiac neurons are phasic, initiating only an initial burst of 1–3 action potentials in response to long duration, suprathreshold depolarizing current pulses (Hardwick et al., 1995; Adams and Cuevas, 2004). However, following PACAP application, the neuronal response often becomes more “tonic-like” such that multiple action potentials are initiated throughout the depolarization elicited by comparable stimuli (Braas et al., 1998; Tompkins et al., 2006). The specific PAC1 receptor agonist maxidilan mimics the effect of PACAP on excitability, whereas the closely associated peptide vasointestinal polypeptide does not (Braas et al., 1998; Tompkins and Parsons, unpublished observations). These observations indicate that the increase in excitability of guinea pig cardiac neurons by PACAP is mediated solely through activation of PAC1 receptors. However, the mechanisms underlying the PAC1 receptor-mediated increase in excitability are unknown.
PAC1 receptors are members of the superfamily of seven transmembrane G-protein coupled receptors and commonly activate the adenylyl cyclase and phospholipase C (PLC) intracellular signaling pathways (Braas and May, 1999; Vaudry et al., 2000). We reasoned that if a PACAP-induced activation of the adenylyl cyclase or PLC intracellular signaling cascades mediated the PACAP-induced increase in excitability, then treatment with inhibitors of these intracellular transduction pathways should blunt the PACAP effect on excitability. Thus, in the present study, we tested whether well-established inhibitors of adenylyl cyclase and PLC suppressed the PACAP-induced increase in excitability. Our results demonstrate that activation of adenylyl cyclase is a critical mechanism underlying the PACAP-induced increase in excitability. In contrast, the PACAP-induced increase in excitability remained in the presence of a potent PLC inhibitor. The PACAP-induced increase in excitability also was suppressed in preparations treated with either H-89, a potent protein kinase A (PKA) inhibitor, or PD 98059, a MEK kinase inhibitor, suggesting that activation of these kinases also was required for the PACAP-induced increase in excitability in some cardiac neurons. Interestingly, forskolin, a potent activator of adenylyl cyclase, and 8-bromo cAMP, a cell permeable cAMP analogue only partially mimicked the PACAP-induced increase in excitability, suggesting that other mechanisms, along with a rise in cAMP and activation of additional downstream signaling cascades, must also be involved in the PACAP-induced increase in excitability.
Methods
General Methods
Experiments were performed in vitro on atrial whole-mount preparations containing the intrinsic cardiac ganglia from Hartley guinea pigs (either sex; 250–350 g) as decribed previously (Tompkins et al., 2006; Tompkins et al., 2007). Guinea pigs were killed by halothane or isoflurane overdose followed by exsanguination using animal protocols approved by the University of Vermont Institutional Animal Care and Use Committee and methods described in the National Institutes of Health Guide for the Care and Use of Laboratory Animals. All efforts were made to minimize the number of animals used and their suffering. The heart was quickly removed and placed in cold standard Krebs solution (in mM: 121 NaCl, 5.9 KCl, 2.5 CaCl2, 1.2 MgCl2, 25 NaHCO3, 1.2 NaH2PO4, 8 glucose). The pH was maintained at 7.4 by aeration with 95% O2 - 5% CO2.
Intracellular recordings from neurons in whole mount preparations
For intracellular recordings, atrial whole-mount preparations were pinned in a 2.5 ml Sylgard-lined chamber and superfused continuously (2–3 ml/min) with a modified Krebs solution containing 10 mM HEPES buffer (33–35°C) (Hardwick et al., 1995; Hardwick et al., 1997; Braas et al., 1998; Tompkins et al., 2006; Tompkins et al., 2007). Cardiac ganglia were visualized with an inverted microscope equipped with Hoffman optics and individual cardiac neurons were impaled using high impedance borosilicate microelectrodes (2 M KCl-filled; 60–100 MΩ). Resting membrane potential and action potentials were recorded from the impaled neurons using an Axoclamp-2A amplifier coupled with a Digidata 1322A data acquisition system and pCLAMP 8 (Axon Instruments, Foster City, CA, USA). If required, hyperpolarizing current was injected through the recording electrode to electrotonically maintain the resting membrane potential between −55 and −65 mV. This ensured that action potential generation was tested at the same potential prior to and following PACAP application (Tompkins et al., 2006).
Depolarizing current steps (0.1–0.4 nA, 1 sec) were given to characterize changes in neuron excitability before and after drug application. If the number of action potentials generated during the supratheshold depolarizing steps was 4 or less, then the cell was classified as “phasic”. However, if the number of action potentials elicited was 5 or more, then the action potential firing pattern was considered “multiple firing”. Excitability curves were constructed by plotting the number of action potentials generated by increasing stimulus intensities. Results were averaged from multiple cells under each condition and fit with a straight line. The slope of the regression line was used to compare neuronal excitability between cells studied under different conditions.
Drugs
All drugs were made up as stock solutions and aliquots were added directly to the bathing solution. Because PACAP27 was determined previously to be more effective than PACAP38 on guinea pig cardiac neurons (Braas et al., 1998; Braas et al., 2004), it was used exclusively in this study and is referred to as PACAP throughout the text. All drugs were obtained from commercial sources: PACAP27 was obtained from American Peptide Co. (Sunnyvale, CA) and forskolin, 8-bromo-cAMP, 2′-amino-3′-methoxyflavone (PD 98059), 9-(tetrahydro-2′-furyl)adenine (SQ 22536) and N-[2-((p-Bromocinnamyl)amino)ethyl]-5-isoquinolinesulfonamide (H-89) were obtained from Calbiochem (La Jolla, CA). Forskolin, H-89, PD 98059, SQ 22536 and U-73122 were made up in dimethylsulfoxide (DMSO; 0.3% final concentration).
Statistical evaluation
Best-fit linear regression lines were calculated and compared between groups using GraphPad Prism (V3) statistical software (San Diego, CA). Differences were considered statistically significant if P < 0.05.
Results
Cardiac neuron excitability is enhanced during bath application of 10 nM PACAP
In the initial experiments, the effect on excitability of adding 10 nM PACAP directly to the bath was established. Excitability was determined from the response to 1 sec depolarizing current pulses of increasing amplitude (Tompkins et al., 2006; Tompkins et al., 2007). In the present study, recordings were made from 23 control guinea pig cardiac neurons, each from an independent whole-mount preparation. All 23 were considered phasic cells based on the criteria described in Methods. In 21 control cells, the maximum number of action potentials elicited by any stimulus was 2. The remaining 2 cells generated a maximum of 3 action potentials. In contrast, during bath exposure to 10 nM PACAP, multiple action potentials were generated by the same depolarizing steps in 79% of the cells (Figure 1A). In 5 cardiac ganglia preparations exposed to PACAP over a recording period of 120 minutes, 11 of 14 cells produced ≥5 action potentials in response to suprathreshold voltage steps (Figure 1B). The average excitability curve for cells exposed to PACAP is shown in Figure 2.
Figure 1. PACAP increases cardiac neuron excitability.
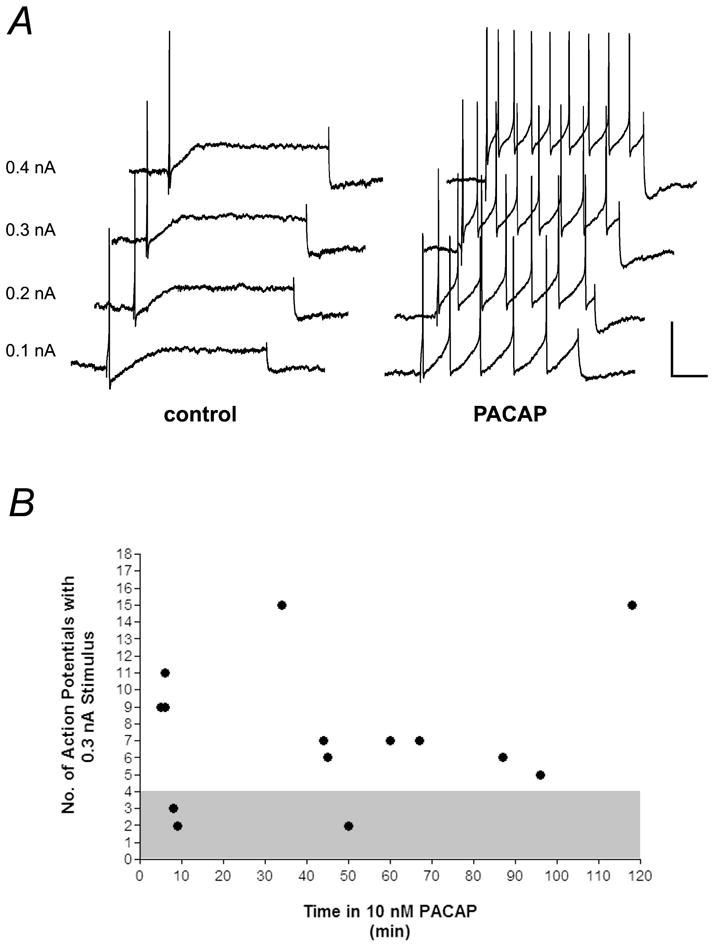
A, responses to depolarizing current pulses (1 sec) of increasing intensities (0.1 – 0.4 nA) before (control) and 5 minutes after application of 10 nM PACAP (PACAP). Cardiac neurons fire multiple action potentials in PACAP. Recordings are from the same cell. scale bar: 30 mV, 125 msec. B, During exposure to 10 nM PACAP, neuronal excitability remained elevated for up to 2 hours. Shown are the number of action potentials produced by a 1 sec 0.3 nA depolarizing current pulse in individual cells from multiple preparations. Duration of PACAP exposure shown on x-axis. Cells that fired fewer than 5 action potentials were classified as phasic (grey area).
Figure 2. Adenylyl cyclase inhibition reduces the PACAP effect on excitability.

Excitability curves summarizing the effects of the adenylyl cyclase inhibitor SQ 22536 and the PLC inhibitor U-74122 on the PACAP-induced changes in neuronal firing. SQ 22536 significantly blunted the PACAP effect on neuronal excitability while the presence of U-73122 had no effect. Data are plotted as the mean number of action potentials generated with a 1 second depolarizing current pulse at multiple stimulus intensities. Data points for each treatment (error bars show SEM) were fit by linear regression and the difference in slope between treatments was analyzed. Treatments were done separately in multiple preparations.
Pretreatment with the adenylyl cyclase inhibitor SQ 22536, but not the PLC inhibitor U-73122, blocked the PACAP-induced increase in cardiac neuron excitability
As PAC1 receptors often are coupled to both adenylyl cyclase and PLC (Braas and May, 1999), the initial experiments tested whether the presence of established inhibitors of either intracellular signaling pathway suppressed the PACAP effect on excitability. To test the potential role of PLC in the PACAP effect, recordings were made from 10 neurons in three cardiac ganglia preparations treated with 10 μM U-73122. In each preparation, excitability was first determined in one cell treated for ~20 minutes with U-73122 alone and then the same cell was tested after exposure to both U-73122 and 10 nM PACAP for 5–10 minutes. All three cells exposed to U-73122 alone exhibited a phasic firing pattern in response to long duration depolarizing current pulses. However, during exposure to PACAP, the firing pattern shifted from a phasic to a multiple firing pattern in 2 of the 3 cells within 10 minutes. Recordings were then made from 7 other neurons in these 3 preparations for times up to 120 minutes in the continued presence of U-73122 and PACAP. In all, 7 of 10 neurons sampled during the 120 minute exposure to PACAP and U-73122 exhibited multiple action potential firing (Figure 3). The excitability curve determined for these 10 cells in PACAP and U-73122 (slope = 20.90 ∀ 1.4) was not different from that determined for cells exposed just to PACAP (slope = 18.14 ∀ 1.1, P = 0.17) (Figure 2). These observations indicated that activation of PLC was not required for the PACAP-induced increase in excitability.
Figure 3. Modulation of the PACAP-induced increase in neuronal excitability by second messenger inhibitors.

Shown are the total number of cells firing multiple action potentials (≥5) in response to a long duration (1 sec) depolarizing current pulse (0.3 nA) under each treatment condition. The percentage of cells firing multiple action potentials was increased in PACAP. This effect was reduced by the presence of inhibitors of adenylyl cyclase (SQ 22536), PKA (H-89) or MEK kinase (PD 98059) but not by an inhibitor of PLC (U-73122). Increases in cAMP alone (forskolin and 8-bromo-cAMP) do not replicate the PACAP effect. Number of cells for each condition in parentheses. Control includes cells from each treatment group prior to PACAP exposure.
To test whether the PACAP-induced increase in excitability required activation of adenylyl cyclase, 3 preparations were exposed to 100 μM SQ 22536 for at least 20 minutes before adding 10 nM PACAP to the bath solution. Excitability was tested in 3 cells (1 from each preparation) exposed to only SQ 22536 and then excitability was tested in the same cells 5–10 minutes after switching to a bath solution containing SQ 22536 and 10 nM PACAP. The 3 cells treated only with SQ 22536 were phasic and remained phasic after exposure to 10 nM PACAP. Four additional cells from these 3 preparations were sampled at different times over a 120 minute recording period in a solution containing PACAP and SQ 22536. Only one of these 4 cells exhibited multiple action potential firing, the other 3 exhibited a phasic firing pattern. The excitability curve determined for the 7 cells exposed to PACAP and SQ 22536 is shown in Figure 2 (slope = 7.14 ∀ 1.3). The presence of SQ 22536 significantly suppressed the PACAP-induced increase in excitability (P = 0.0006, Figure 2)(Figure 3).
Activation of adenylyl cyclase increases cAMP production and the rise in cAMP can activate protein kinase A (PKA). Phosphorylation by PKA of proteins regulating surface membrane ionic conductances might be a mechanism downstream of cAMP generation involved in the PACAP modulation of excitability. To test this possibility, the ability of 10 nM PACAP to increase excitability was determined in 3 preparations pretreated with 1 μM H-89, a commonly used PKA inhibitor. As in the previous experiments, recordings were obtained from one cell in each preparation pretreated with only H-89 and then from the same cell 5–10 minutes after exposure to PACAP along with H-89. In 3 cells exposed for >15 minutes to H-89 alone, the firing pattern was phasic. In 1 of these 3 cells, the firing pattern was converted to “multiple firing” during exposure to PACAP. In the other two cells, the firing pattern in the presence of PACAP remained phasic. Recordings were then obtained from 10 other cells in these 3 preparations exposed for 120 minutes to 1 μM H-89 and 10 nM PACAP. In all, 10 of 13 cells retained a phasic firing pattern whereas only 3 exhibited a multiple action potential firing pattern (Figure 3). Thus, treatment with H-89 markedly reduced the PACAP-induced increase in excitability. The averaged excitability curve for the cells treated with PACAP and H-89 is shown in Figure 4. Note that for cells treated with H-89 and PACAP, the slope of the averaged excitability curve was significantly less than that determined for cells exposed to only PACAP (8.85 ∀ 0.8 vs 18.14∀ 1.1, P = 0.0004).
Figure 4. PKA and MAPK/ERK kinase signaling cascades contribute to the PACAP-induced increase in neuronal excitability.

Excitability curves summarize the effects of the PKA inhibitor H-89 and the MEK kinase inhibitor PD 98059 on the PACAP induced change in neuronal firing. Both H-89 and PD 98059 significantly blunted the PACAP effect on neuronal excitability. Data are plotted as the mean number of action potentials generated with a 1 second depolarizing current pulse at multiple stimulus intensities. Error bars show SEM. Data points for each treatment were fit by linear regression and the difference in slope between treatments was analyzed. Treatments were done separately in multiple preparations.
Pretreatment with the MEK kinase inhibitor PD 98059 also suppressed the PACAP-induced increase in cardiac neuron excitability
In prior studies, we noted that the MAPK kinase pathway contributed to the initiation of other PACAP actions on the guinea pig cardiac neurons (Braas et al., 2004). Thus, we also tested whether treatment with the MEK kinase inhibitor PD 98059 (50 μM), to disrupt the MAPK signaling cascade, diminished the ability of PACAP to increase cardiac neuron excitability. The effect of PD 98059 was studied in 3 preparations. Recordings were obtained from one cell in each of the 3 preparations during exposure to the inhibitor alone and then following exposure to PD 98059 and PACAP. Excitability was tested with depolarizing current steps of increasing intensity. All three cells exhibited a phasic firing pattern when exposed to PD 98059 alone. The same three cells continued to exhibit a phasic firing pattern after a 10 minute exposure to inhibitor and PACAP. The firing pattern was tested in 6 other cells during continued exposure to inhibitor and PACAP. Over the 120 minute recording period in inhibitor and PACAP, only 1 of the 6 cells sampled exhibited multiple action potential firing during the depolarizing current steps. The other 4 cells remained phasic. Thus, in the presence of the MEK kinase inhibitor, only 1 of 9 PACAP-treated cells generated multiple action potentials (Figure 3). The averaged excitability curve for the 9 cells treated with PACAP and PD 98059 is summarized in Figure 4. From inspection of the results in Figure 4, it is evident that the slope of the excitability curve for these cells was significantly less than that determined for the 14 cells exposed to PACAP alone (4.89 ∀ 0.6 vs 18.14 ∀ 1.1, P <0.0001).
The potent adenylyl cyclase activator forskolin and the cell permeable cAMP analogue 8-bromo-cAMP only partially mimic the PACAP-induced increase in excitability
Suppression of the PACAP effect on excitability by SQ 22536 indicated that activation of adenylyl cyclase and generation of cAMP was a key first step in the peptide-induced increase in excitability. Additional experiments tested whether a rise in cAMP by itself could duplicate the effects of PACAP on excitability. For these experiments, we tested whether bath application of 5 μM forskolin, a potent activator of adenylyl cyclase or exposure to 1 mM 8-bromo-cAMP, a cell membrane permeable cAMP analogue, could produce a comparable increase in cardiac neuron excitability. Ten cells from 4 cardiac ganglia preparations were exposed to forskolin and 9 cells from 3 preparations were exposed to 8-bromo-cAMP. With either treatment, only 30–33 % of the cells tested exhibited a multiple firing pattern in response to the depolarizing current steps; the remainder exhibited a phasic firing pattern, throughout the 120 minute recording period (Figure 3). The averaged excitability curve for the cells exposed either to forskolin or to 8-bromo-cAMP is shown in Figure 5. The slope of the excitability curve for cells exposed to forskolin (7.40 ∀ 0.4) or 8-bromo-cAMP (10.44 ∀ 1.2) was significantly less than that for cells exposed to PACAP (P < 0.0001 and P = 0.003, respectively). Thus, a rise in cAMP by itself could only partially recapitulate the PACAP effect on excitability.
Figure 5. A rise in cAMP alone does not recapitulate the PACAP effect on excitability.

Increasing cAMP levels by treatment with either the adenylyl cyclase activator forskolin or the cell permeable cAMP analog 8-bromo-cAMP did not increase neural excitability to the level produced by PACAP. Data are plotted as the mean number of action potentials generated with a 1 second depolarizing current pulse at multiple stimulus intensities. Error bars show SEM. Data points for each treatment were fit by linear regression and the slopes were compared.
Discussion
In the present study, we determined the percentage of cells that exhibited a phasic or multiple action potential firing pattern to establish whether interruption of different intracellular signaling cascades affected the PACAP-induced increase in excitability. In many cell types, the PAC1 receptor can activate both PLC and adenylyl cyclase (Braas and May, 1999; Vaudry et al., 2000). Treatment with the adenylyl cyclase inhibitor SQ 22536 significantly suppressed the PACAP-induced increase in excitability. However, the PACAP modulation of cardiac neuron excitability remained after treatment with a well-established PLC inhibitor. Thus, the key result from this study is that the PACAP-induced increase in cardiac neuron excitability required activation of adenylyl cyclase, but not PLC. Our data indicating a lack of involvement of PLC in the effect of PACAP on guinea pig cardiac neuron excitability confirms preliminary observations reported previously (Parsons, 2004).
We found that during bath application of 10 nM PACAP, excitability was increased in 79% of the cardiac neurons using the criterion that 5 or more action potentials were initiated during the depolarizing current steps. Under the conditions of the present experiments, we anticipated that a small percentage of cardiac neurons would not be affected by PACAP as results from our previous study indicated that the EC50 for the PACAP-induced increase in excitability was ~ 20 nM (Braas et al., 1998). In addition, ~10 % of the guinea pig cardiac neurons in untreated preparations are tonic so that during long depolarizing pulses multiple action potentials are elicited (Hardwick et al., 1995; Adams and Cuevas, 2004). Therefore, it would be expected that in any drug-treated group of cells, occasionally a cardiac neuron might exhibit a tonic firing pattern even if the action of PACAP was blocked. Thus, we suggest the 1 cell that exhibited a multiple firing pattern during exposure to PACAP and SQ 22536, as well as with PACAP and PD 98059, very likely was a tonic neuron rather than a phasic cell, which had been converted to a multiple firing pattern by PACAP.
In H-89-treated ganglia, PACAP increased excitability in only a small percentage of the cardiac neurons. H-89 is commonly used to inhibit PKA. Thus, our results are consistent with activation of PKA by cAMP as a key step supporting the PACAP effect on excitability. However, the results with H-89 must be interpreted carefully. First, H-89 is not selective as it can inhibit a number of protein kinases in addition to PKA with a similar EC50 (Davies et al., 2000). Second, in initial experiments, we tested excitability in cardiac neurons exposed to higher H-89 concentrations (5 and 10 μM). When exposed to H-89 in this concentration range for more than a few minutes, most cardiac neurons had low resting membrane potentials and even when the membrane potential was electrotonically returned to −50 to −60 mV, values close to the resting membrane potential in control cells, action potential generation was compromised. In addition, when action potentials were generated, they had a reduced amplitude and were broadened. Thus, H-89 at the concentrations commonly used in cell studies (10–25 μM) apparently affected some basic electrical properties of the guinea pig cardiac neurons. Taken together, we suggest further study with other PKA inhibitors is required before the role of PKA can be established unequivocally.
Previously, Merriam et al. (2004) demonstrated, using dissociated guineas pig cardiac neurons, that PACAP enhanced the hyperpolarization-activated, cyclic nucleotide-gated nonselective cationic conductance Ih. Forskolin had a similar effect. Thus, it was concluded that the effect of PACAP on Ih was mediated through activation of adenylyl cyclase and generation of cAMP. As Ih is a pacemaker current known to facilitate multiple action potential firing, it is very plausible that one mechanism contributing to the PACAP-induced increase in excitability is an activation of adenylyl cyclase and generation of cAMP, which then shifts the voltage dependence of activation of Ih to more positive potentials. However, Ih is present in only a subset of the guinea pig cardiac neurons (Edwards et al., 1995). Thus, although a PACAP-enhanced Ih could be a mechanism contributing to the change in excitability in some cardiac neurons, other ionic mechanisms must contribute to the enhanced excitability in cells not expressing Ih.
In addition to the canonical cAMP-PKA transduction pathway, there is a non-canonical pathway for cAMP-mediated effects, which involves cAMP activation of EPAC and subsequent activation of other intracellular cascades including the MAPK signaling pathway (Gerdin and Eiden, 2007). The MEK kinase inhibitor, PD 98059 significantly suppressed the PACAP-induced increase in excitability. This suggests that a PAC1 receptor activation of the MAPK intracellular signaling pathway also was a key pathway mediating the PACAP effect. The specific ionic conductances, which might be affected by a PACAP-induced activation of MAPK intracellular signaling remain to be determined; a question to be the subject of future studies.
Although pretreatment with an adenylyl cyclase inhibitor greatly suppressed the PACAP effect, activation of adenylyl cyclase by forskolin or application of the cell membrane permeable cAMP analogue 8-bromo-cAMP only increased excitability in a limited percentage of the cardiac neurons. Thus, other mechanisms in addition to activation of adenylyl cyclase and generation of cAMP, must be critical co-factors in mediating the PACAP-induced increase in cardiac neuron excitability. Previously, we determined that Ca2+ influx through an unidentified plasma membrane ion channel was a requisite for the PACAP-induced increase in excitability (Tompkins et al., 2006). Thus, we propose that both a rise in cAMP and Ca2+ are critical mechanisms in the initiation of the PACAP-induced increase in cardiac neuron excitability.
Acknowledgments
This work was supported by NIH grant HL65481 (RLP).
References
- 1.Adams DJ, Cuevas J. In: Electrophysiological properties of intrinsic cardiac neurons, Basic and Clinical Neurocardiology. Armour JA, Ardell JL, editors. Oxford University Press; 2004. [Google Scholar]
- 2.Braas KM, May V. Pituitary adenylate cyclase-activating polypeptides directly stimulate sympathetic neuron neuropeptide Y release through PAC(1) receptor isoform activation of specific intracellular signaling pathways. J Biol Chem. 1999;274(39):27702–10. doi: 10.1074/jbc.274.39.27702. [DOI] [PubMed] [Google Scholar]
- 3.Braas KM, May V, Harakall SA, Hardwick JC, Parsons RL. Pituitary adenylate cyclase-activating polypeptide expression and modulation of neuronal excitability in guinea pig cardiac ganglia. J Neurosci. 1998;18(23):9766–79. doi: 10.1523/JNEUROSCI.18-23-09766.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Braas KM, Rossignol TM, Girard BM, May V, Parsons RL. Pituitary adenylate cyclase activating polypeptide (PACAP) decreases neuronal somatostatin immunoreactivity in cultured guinea-pig parasympathetic cardiac ganglia. Neuroscience. 2004;126(2):335–46. doi: 10.1016/j.neuroscience.2004.04.007. [DOI] [PubMed] [Google Scholar]
- 5.Calupca MA, Vizzard MA, Parsons RL. Origin of pituitary adenylate cyclase-activating polypeptide (PACAP)-immunoreactive fibers innervating guinea pig parasympathetic cardiac ganglia. J Comp Neurol. 2000;423(1):26–39. [PubMed] [Google Scholar]
- 6.Davies SP, Reddy H, Caivano M, Cohen P. Specificity and mechanism of action of some commonly used protein kinase inhibitors. Biochem J. 2000;351(Pt 1):95–105. doi: 10.1042/0264-6021:3510095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Edwards FR, Hirst GD, Klemm MF, Steele PA. Different types of ganglion cell in the cardiac plexus of guinea-pigs. J Physiol. 1995;486 ( Pt 2):453–71. doi: 10.1113/jphysiol.1995.sp020825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gerdin MJ, Eiden LE. Regulation of PC12 cell differentiation by cAMP signaling to ERK independent of PKA: do all the connections add up? Sci STKE. 2007;(382):pe15. doi: 10.1126/stke.3822007pe15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hardwick JC, Mawe GM, Parsons RL. Evidence for afferent fiber innervation of parasympathetic neurons of the guinea-pig cardiac ganglion. J Auton Nerv Syst. 1995;53(2–3):166–74. doi: 10.1016/0165-1838(94)00182-j. [DOI] [PubMed] [Google Scholar]
- 10.Hardwick JC, Mawe GM, Parsons RL. Tachykinin-induced activation of non-specific cation conductance via NK3 neurokinin receptors in guinea-pig intracardiac neurones. J Physiol. 1997;504 ( Pt 1):65–74. doi: 10.1111/j.1469-7793.1997.065bf.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Merriam LA, Barstow KL, Parsons RL. Pituitary adenylate cyclase-activating polypeptide enhances the hyperpolarization-activated nonselective cationic conductance, Ih, in dissociated guinea pig intracardiac neurons. Regul Pept. 2004;123(1–3):123–33. doi: 10.1016/j.regpep.2004.04.019. [DOI] [PubMed] [Google Scholar]
- 12.Parsons RL. Mammalian cardiac ganglia as local integration centers: hostochemical and electrophysiological evidence. In: Dun NJ, Machado BH, Pilowsky PM, editors. Neural mechanisms of cardiovascular regulation. Kluwer Academic Publishers; Boston: 2004. pp. 335–356. [Google Scholar]
- 13.Parsons RL, Locknar SA, Young BA, Hoard JL, Hoover DB. Presence and co-localization of vasoactive intestinal polypeptide with neuronal nitric oxide synthase in cells and nerve fibers within guinea pig intrinsic cardiac ganglia and cardiac tissue. Cell Tissue Res. 2006;323(2):197–209. doi: 10.1007/s00441-005-0074-3. [DOI] [PubMed] [Google Scholar]
- 14.Tompkins JD, Ardell JL, Hoover DB, Parsons RL. Neurally released pituitary adenylate cyclase-activating polypeptide enhances guinea pig intrinsic cardiac neurone excitability. J Physiol. 2007;582(Pt 1):87–93. doi: 10.1113/jphysiol.2007.134965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tompkins JD, Hardwick JC, Locknar SA, Merriam LA, Parsons RL. Ca2+ influx, but not Ca2+ release from internal stores, is required for the PACAP-induced increase in excitability in guinea pig intracardiac neurons. J Neurophysiol. 2006;95(4):2134–42. doi: 10.1152/jn.01077.2005. [DOI] [PubMed] [Google Scholar]
- 16.Vaudry D, Gonzalez BJ, Basille M, Yon L, Fournier A, Vaudry H. Pituitary adenylate cyclase-activating polypeptide and its receptors: from structure to functions. Pharmacol Rev. 2000;52(2):269–324. [PubMed] [Google Scholar]