

Abstract
Recently it has been reported that phosphorylated acyclovir (ACVa) inhibits human immunodeficiency virus type 1 (HIV-1) reverse transcriptase in a cell free system. To deliver phosphorylated ACV inside cells we designed ACV monophosphorylated derivatives using ProTide technology. We found that the L-alanine derivatived ProTides show anti-HIV activity at non-cytotoxic concentrations; ester and aryl variation was tolerated. ACV ProTides with other amino acids, other than L-phenylalanine, showed no detectable activity against HIV in cell culture. The inhibitory activity of the prodrugs against herpes simplex virus (HSV) type -1, -2 and thymidine kinase-deficient HSV-1 revealed different structure-activity relationships, but was again consistent with successful nucleoside kinase bypass. Enzymatic and molecular modelling studies have been performed in order to better understand the antiviral behaviour of these compounds. ProTides showing diminished carboxypeptidase lability translated to poor anti-HIV agents and vice versa, so the assay became predictive.
Introduction
Human immunodeficiency virus (HIV) belongs to the retroviradae family and causes the acquired immunodeficiency syndrome (AIDS). A variety of different compounds have been developed for the treatment of HIV and currently 25 drugs have been approved for clinical use including: nucleoside reverse transcriptase inhibitors (NRTIs), nonnucleoside reverse transcriptase inhibitors (NNRTIs), protease inhibitors (PI), a viral fusion inhibitor (FI), a CCR-5 co-receptor inhibitor and a viral integrase (IN) inhibitor.1 Due to the rapid development of drug resistance as well as to the toxicity shown by these drugs,2 novel anti-HIV agents are needed. Diverse structures are sought to address the constant threat of viral resistance.
In this context, recently it has been reported how the anti-herpetic drug acyclovir (ACV, 1, Fig 2) inhibits HIV upon human herpesvirus (HHV) co-infection in tissue cultures.3 This activity was found to be correlated with the phosphorylation of the parent drug to the monophosphate form mediated by HHV-encoded kinase(s). HIV does not encode an enzyme that recognises ACV as a substrate for this activation (phosphorylation) step; hence the need for the HHV co-infection for activity. The subsequent phosphorylations to the di- and triphosphate derivatives may be mediated by cellular guanosine monophosphate kinase and nucleoside diphosphate kinase, respectively.4,5 In its triphosphate form, ACV inhibits HIV RT acting as a chain terminator.3 Following these results, it is evident that the anti-HIV activity can only occur upon ACV monophosphate (ACV-MP) formation which requires HHV co-infection. ACV-MP itself can not be used as efficient anti-HIV chemotherapeutic agent to by-pass the first limiting phosphorylation step because of its instability in biological media and its poor efficiency of diffusion through intact cell membranes. A suitable strategy to overcome these limitations would consist of masking the negative charges of the monophosphate with lipophilic groups. In this regard, the phosphoramidate ProTide technology has been developed and successfully applied to a range of nucleosides of antiviral and anti-cancer interest.6-9 The structural motif of this approach consists of masking the nucleoside monophosphate with an aryl moiety and an amino acid ester. Cell entry then apparently occurs by passive diffusion. Once inside the cell, the phosphoramidate prodrug is activated and converted to the monophosphorylated ACV (Fig. 1).10 The first step involves an enzymatic hydrolysis of the amino acid ester moiety mediated by an esterase- or carboxypeptidase-type enzyme followed by spontaneous cyclisation and subsequent spontaneous displacement of the aryl group and opening of the unstable ring mediated by water. The last step before release of the ACV monophosphate involves a hydrolysis of the P-N bond mediated by a phosphoramidase-type enzyme. The phosphoramidate Protide approach has been already successfully applied to ACV demonstrating its ability to by-pass the thymidine kinase deficiency of HSV-1, -2 and varicella zoster virus strains resistant to ACV.11
Fig. 2.

ACV and its ProTides.
Fig. 1.

Proposed activation pathway of the acyclovir ProTides.
In this paper, we present the synthesis and biological evaluation of a novel series of ACV ProTides (Fig. 2). The ProTide moiety has three different changeable parts: the aryl moiety, the amino acid and the ester. In the first part of this study we have chosen Lalanine as the amino acid, as it has shown previously an optimal biological profile,12 varying the other two components. For the aryl moiety we considered: phenol, naphthol and p-fluoro-phenol and as ester moiety: methyl, ethyl, n-propyl, iso-propyl, tert-butyl and benzyl and combinations thereof. All these combinations allowed us to extensively vary the LogP for these compounds and to study how this variation can influence the antiviral activity. Moreover it has been previously reported how the substitutions can influence the bioactivation of the ProTide; for example naphthol has shown an enhancement of activity against a panel of cancer cell lines for phosphoramidates6 and the tert-butyl ester showed a lack of biological activity due to the poor bioactivation of the bulky moiety. Following the results for these derivatives, different amino acids have been considered including L-valine, L-leucine, L-isoleucine, L-proline, glycine, and the non-natural D-alanine, D-valine and dimethylglycine. Moreover, some intermediateprotected (N2-DMF)-ACV ProTides have been biologically evaluated.
Chemistry
The compounds have been synthesised following the procedure reported by Uchiyama13 using tert-butylmagnesium chloride (tBuMgCl) as a coupling reagent and using THF as a solvent in most of the cases.
Aryl phosphorodichlorophosphates 26 and 27 have been synthesised coupling respectively 1-naphthol (24) or p-fluorophenol (25) with POCl3 in the presence of Et3N 1eq) (Scheme 1), while phenyl dichlorophosphate (28) was commercially available. The coupling with the appropriate amino acid ester salt (29-44) has been performed in the presence of Et3N (2 eq) (Scheme 2) giving the final product (45-65) as an oil which was, in most of the cases, purified by column chromatography.
Scheme 1.

Reagents and conditions: (i) POCl3, anhydrous TEA, anhydrous Et2O, -78 °C, 1 h then rt, overnight.
Scheme 2.

Reagents and conditions: (i) anhydrous TEA, anhydrous DCM, -78 °C, 30 min - 1 h, then rt, 30 min - 4 h.
In order to improve the solubility of ACV in THF, used as ideal solvent for the coupling reaction, the 2-amino was protected using dimethylformamide dimethyl acetal (Scheme 3). However, compound 66 is not completely soluble in THF but the solubility was improved sufficiently to carry out the reaction. The final coupling of the nucleoside was performed using an excess of the appropriate phosphorochloridate (1.50-4.00 eq) in the presence of tBuMgCl (2 eq). Due to the reactivity problem, the use of N-methylimidazole (NMI), following the Van Boom procedure14 was used for the synthesis of the L-proline (22) and glycine (23) derivatives. Moreover, a mixture of THF/pyridine (3/2) was used as a solvent to improve the solubility of N2-DMF-ACV.
Scheme 3.

Reagents and conditions: (i) dimethylformamide dimethyl acetal, anhydrous DMF, rt, 1 day; (ii) tBuMgCl, THF, rt, overnight or NMI, THF/pyridine = 3/2, rt, overnight; (iii) 1-propanol, reflux, for 18 h or 2-propanol, reflux, 24-96 h.
The deprotection of the dimethylformyl DMF derivative was initially carried out refluxing the compound in 1-propanol (Scheme 3). However, due to a transesterification during the synthesis of 2, obtaining compound 3, the solvent was changed to 2-propanol obtaining the desired compounds (2, 4-23).
All the compounds were obtained as a mixture of two diastereoisomers confirmed by the presence of two peaks in the 31P-NMR, with the exception of the glycine and dimethylglycine derivatives, due to the absence of a chiral center, and L-proline, for which we were able to isolate only one diastereoisomer.
Biological results
Anti-HSV activity
The activity of the compounds were evaluated against three different strains of HSV including HSV-1 (KOS), HSV-2 (G) and thymidine kinase-deficient HSV-1 (ACVR). Native ACV showed sub-micromolar (EC50: 0.4 μM and 0.2 μM) activity against respectively HSV-1 and HSV-2 (Table 1) but was inactive against TK-deficient HSV-1 (EC50: 50 μM). The ACV ProTides did not show increased activity against HSV-1 and HSV-2 compared to the parent compound. Only two compounds (11 and 20), respectively the p-fluorophenyl-L-Ala-OBn and the phenyl-L-Leu-OBn derivatives, showed anti-HSV activity in the sub-micromolar range, while the majority of the compounds showed an activity in the range of ca. 1-30 μM. Compound 6 having the bulky t-butyl group as ester moiety did not show any marked activity (≥ 50 μM) against the HSV strains. ACV has been evaluated against thymidine kinase-deficient HSV-1 showing a dramatic loss of activity (> 100-fold) (EC50: 50 μM). Interestingly, several of the ProTides showed significant retention of activity, demonstrating a successful by-pass of the first phosphorylation step (i.e. compounds 11, 13, 20, 21). Notably, none of the ACV ProTides showed appreciable cytostatic/cytotoxic activity, despite the potential loss of antiviral selectivity that could follow from viral nucleoside kinase bypass.
Table 1.
Anti-HSV activity for ACV and its Protides
| Antiviral Activity EC50a (μM) | Cytotoxic/Cytostatic activity (μM) | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Cpds | Aryl | Amino Acid | Ester | CLogP | HSV-1 | HSV-2 | HSV-1 TK- | MCCb (Hel) | IC50c (Hel) |
| 2 | Naph | L-Ala | Bn | 2.06 | 2±0 | 1.4±0.8 | 10±2.1 | ≥20 | 20 |
| 3 | Naph | L-Ala | nPr | 1.41 | 5.5±2.1 | 1.9±1.6 | 16±5.7 | ≥50 | 68 |
| 4 | Naph | L-Ala | Me | 0.35 | 16±5.7 | 10±2.1 | 79±29 | >50 | >100 |
| 5 | Naph | L-Ala | Et | 0.88 | 32±25 | 9.5±0.7 | 32±18 | >150 | >100 |
| 6 | Naph | L-Ala | tBu | 1.59 | >100 | 50 | >100 | >50 | >100 |
| 7 | Naph | L-Ala | iPr | 1.19 | 15±7.1 | 10±0 | ≥45 | >50 | >100 |
| 8 | Ph | L-Ala | Me | -0.82 | 20±0 | 16±5.7 | 79±29 | >100 | ND |
| 9 | Ph | L-Ala | Bn | 0.89 | 8±5.7 | 4±0 | 15±7.1 | >50 | 91 |
| 10 | Ph | L-Ala | iPr | 0.02 | 10±0 | 8.5±0.7 | 27±25 | >50 | >100 |
| 11 | p-F-Ph | L-Ala | Bn | 1.11 | 0.9±0.1 | 0.5±0 | 1.5±0.7 | >100 | ND |
| 12 | Ph | D-Ala | Bn | 0.89 | 2±0 | 1.4±0.8 | 23±16 | >100 | ND |
| 13 | Naph | DMG | Bn | 2.37 | 2.4±0 | 1.6±1.1 | 3.2±1.1 | >50 | >100 |
| 14 | Ph | DMG | Bn | 1.20 | 1.4±0.85 | 0.8±0.0 | 5.5±2.1 | >100 | ND |
| 15 | Ph | Phe | Bn | 2.31 | 17±4.2 | 8±5.7 | ≥100 | >50 | 87 |
| 16 | Ph | L-Val | Bn | 1.82 | 2±0 | 0.85±0.2 | 7.5±6.4 | >100 | ND |
| 17 | Naph | L-Val | Me | 1.28 | >100 | >100 | >100 | >100 | ND |
| 18 | Naph | L-Val | Et | 1.81 | 51±9.2 | 32±18 | 42±3.5 | >100 | ND |
| 19 | Naph | D-Val | Me | 1.28 | >100 | >100 | >100 | >100 | ND |
| 20 | Ph | L-Leu | Bn | 2.35 | 0.8±0.07 | 0.7±0 | 1.4±0.8 | >100 | ND |
| 21 | Ph | L-Ile | Bn | 2.35 | 1.1±0.4 | 1.1±0.4 | 1.4±0.8 | >100 | ND |
| 22 | Ph | L-Pro | Bn | 2.82 | >100 | >100 | >100 | >100 | >100 |
| 23 | Ph | Gly | Bn | 0.58 | 3 | 0.8 | 9 | >100 | ND |
| ACV (1) | - | - | - | -2.42 | 0.4 | 0.2 | 50 | >100 | ND |
50% Effective concentration, or compound concentration required to inhibit virus-induced cytopathicity by 50%.
Minimal cytotoxic concentration, or compound concentration required to cause a microscopically visible alteration of cell morphology.
50% Inhibitory concentration, or compound concentration required to inhibit cell proliferation by 50%.
ND = determined
Anti-HIV activity
The ACV ProTides have also been evaluated against HIV-1 and HIV-2 in CEM and against HIV-1 in MT-4 cell cultures and in HIV-infected tonsillar tissues ex vivo (Table 2).
Table 2.
Anti-HIV activity of the ACV ProTides and ACV.
| Antiviral Activity EC50a (μM) | Cytostatic activity (μM) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Cps | Aryl | AminoAcid | Ester | HIV-1 CEM | HIV-2 CEM | HIV-1 MT4 | Tonsil HIV | IC50b CEM | CC50c (MT4) | IC50b (MT4) |
| 2 | Naph | L-Ala | Bn | 15±14 | 8.9±6.3 | 0.8 | ND | 17 | ND | >150 |
| 3 | Naph | L-Ala | nPr | 6.6±5.6 | 24±30 | 10 | ND | 22 | ND | ND |
| 4 | Naph | L-Ala | Me | 10±7.9 | 13±6.4 | 4.7±2.1 | <0.1 | 57 | >150 | 18.7±3.2 |
| 5 | Naph | L-Ala | Et | 12±9.8 | 42±13 | 1.7±0.8 | 0.53 | 32±7.8 | >150 | 12±5.3 |
| 6 | Naph | L-Ala | tBu | >100 | >100 | >150 | N.D. | >100 | >150 | >150 |
| 7 | Naph | L-Ala | iPr | 6.2±5.4 | 12±0.71 | 5.4 | ND | 36±15 | >150 | 72.5 |
| 8 | Ph | L-Ala | Me | 17±4.6 | 26±8.5 | 15 | ND | 67±7.8 | ND | ND |
| 9 | Ph | L-Ala | Bn | 16±14 | 11±4.9 | 5.7±1.6 | 0.6 | 42±11 | >150 | 33.8±10.6 |
| 10 | Ph | L-Ala | iPr | >100 | >100 | >150 | ND | >100 | >150 | >150 |
| 11 | p-F-Ph | L-Ala | Bn | >20 | >20 | ND | ND | 76±13 | ND | ND |
| 12 | Ph | D-Ala | Bn | >250 | >250 | ND | ND | ≥250 | ND | ND |
| 13 | Naph | DMG | Bn | ≥100 | 79±30 | ND | ND | >100 | ND | ND |
| 14 | Ph | DMG | Bn | >100 | >100 | 7 | ND | >100 | >150 | >150 |
| 15 | Ph | Phe | Bn | 26±11 | 34±24 | 16 | ND | 42 | ND | ND |
| 16 | Ph | L-Val | Bn | >50 | >50 | ND | ND | ≥100 | ND | ND |
| 17 | Naph | L-Val | Me | >100 | >100 | ND | ND | >100 | ND | ND |
| 18 | Naph | L-Val | Et | >100 | >100 | ND | ND | >100 | ND | ND |
| 19 | Naph | D-Val | Me | >100 | >100 | ND | ND | >100 | ND | ND |
| 20 | Ph | L-Leu | Bn | >20 | >20 | 0.8 | ND | >20 | 17 | >150 |
| 21 | Ph | L-Ile | Bn | >20 | >20 | ND | ND | ND | ND | ND |
| 22 | Ph | L-Pro | Bn | >20 | >20 | ND | ND | >100 | ND | ND |
| 23 | Ph | Gly | Bn | >100 | >100 | ND | ND | >100 | ND | ND |
| 77* | Naph | DMG | Bn | >20 | >20 | 15 | ND | 40±2.8 | 45 | 140 |
| 78* | Ph | DMG | Bn | >100 | >100 | 70 | ND | >100 | >150 | >150 |
| 81* | Naph | L-Val | Me | >100 | >100 | > 150 | ND | >100 | >150 | >150 |
| 86* | Ph | L-Pro | Bn | >20 | >20 | 30 | ND | 45±0.0 | 90 | >150 |
| ACV (1) | - | - | - | >250 | >250 | >250 | 3.11 | >250 | >250 | >250 |
N2-DMF-ACV
50% Effective concentration, or compound concentration required to inhibit virus-induced cytopathicity by 50%.
50% Cytotoxic concentration, or compound concentration required to decrease the viability of the cell cultures by 50%.
50% Inhibitory concentration, or compound concentration required to inhibit cell proliferation by 50%.
ND = determined
While parent ACV was inactive, in most of the L-alanine derivatives (2-11), with the exception of 6, 10 and 11, showed activity (EC50) in a range of 6.2-17 μM against HIV-1 (CEM) and in a range of 8.9-42 μM against HIV-2 (CEM) (Table 2). Similar results were obtained when these compounds were applied to the HIV-1-infected MT-4 cell cultures. In these cells parental ACV was inactive whereas HIV was suppressed by all the tested compounds with EC50‘s in the range of 0.8-30 μM with the exception of 6, 10, 78 and 81 (Table 2). The antiviral activity of the ACV ProTides was confirmed in HIV-infected tonsillar tissue ex vivo. The compounds 4, 5 and 9 suppressed HIV replication with EC50‘s in a range of 0.1-0.6 μM. These findings indicate that the ACV ProTide approach can also be successfully applied to HIV-infected tissue.
No clear-cut structure-activity relationship could be observed with regard to the nature of the aryl moiety nor the alaninyl ester moiety in terms of eventual antiviral activity of the ProTide derivatives. The lack of activity obtained for the t-butyl analogue (6) is in agreement with the previous report and with the enzymatic experiment to be discussed later. All of these compounds showed an antiproliferative effect (on CEM or MT-4 cell cultures) in a range between 17 and 76 μM.
With regard to the amino acid modifications, we found that, besides of the L-alanine ProTides, only the phenylalanine derivative (15) had activity against HIV-1 (26 μM) and HIV-2 (34 μM). All other derivatives including D-alanine (12), dimethylglycine (13 and 14), L-valine (16-18), D-valine (19), L-leucine (20), L-isoleucine (21), L-proline (22) and glycine (23) did not show appreciable activity in CEM. These results are in agreement with previous reports for other nucleosides, in which the substitution of the L-alanine with different natural L-amino acids gave loss (~ 10- to 100-fold) of antiviral activity.15 However, in the case of dimethylglycine this result is quite surprising as this variation led usually to a retention of anti-HIV activity compared to the L-alanine derivatives.16
Interestingly, in MT-4 cell cultures, the amino acid modification seems to be tolerated, in fact compounds 14 (DMG) and 20 (L-Leu), showed respectively an anti-HIV-1 activity of 7 and 0.8 μM.
Moreover, in MT-4 the N2-DMF-protected ACV ProTides (77, 78 and 86) showed activity against HIV-1 (EC50’s: 15, 70 and 30 respectively) indicating that this kind of substitution may be tolerated. However, in the case of CEM, these compounds did not show any inhibitory activity.
From these results it is possible to conclude that the amino acid L-alanine is optimal for the anti-HIV activity of the ACV ProTides. Neither D-alanine, nor glycine can efficiently substitute for L-alanine nor can bulkier amino acids.
In contrast to the structural requirement for anti-HIV activities, the anti-HSV activity tolerates liberal amino acid variation. This may reflect different substrate specificities and/or different intracellular levels of the necessary activating enzymes. It should indeed be noticed that the HIV assays are performed in rapid proliferating lymphocyte cell cultures (generation time ~ 24 h) whereas the anti-herpetic assays are carried out in confluent fibroblast monolayer (non-proliferating) cell cultures. Thus, the different celltype and cell-cycle conditions between both assay models can result in different prodrug activation modalities that may explain the differential antiviral activities of the ACV ProTides.
Enzymatic Studies
The mechanism of activation of the ProTides involves a first enzymatic activation step mediated by carboxypeptidase-type enzyme(s), which hydrolyse the ester of the amino acid moiety (Fig. 1).
In order to probe the activation of the ACV ProTides to the monophosphorylated form inside cells, we performed an enzymatic study using carboxypeptidase Y following the conversion by 31P-NMR. Three different L-alanine derivatives (9, 6 and 10), the first one is active vs HIV and the second and third compounds are inactive against HIV, as well as the inactive L-valine 17 and D-valine derivatives 19 have been considered for these experiments. The assay has been carried out dissolving the compounds in d6-acetone and trizma buffer (pH = 7.6), incubating with the enzyme and recording a blank for each sample before the addition of the enzyme.
In the case of the phenyl benzylalanine compound 9 (Fig. 3) the experiment showed a fast hydrolysis of the starting material (δP = 3.65 and 3.60) to the intermediate type 88 (Fig. 1) (δP = 4.85 and 4.70), noting the presence of the two diastereoisomers. This intermediate is then processed to a compound of type 90 (δP = 7.10) through the putative intermediate 89, which is not detected by 31P-NMR. The half-life for 9 is 17 min. In the case of the isopropyl ester analogue 10 (Fig. 4) the experiment showed a slow conversion of the starting material to 90 with a half-life of 46 h. This is ca. 150 times slower than 9. This result is in accordance with the inactivity of 10 against HIV (Table 2). Notably one of the two diasteroisomers seems to be faster converted compared to the other one.
Fig. 3.

Carboxypeptidase-mediated cleavage of compound 9, monitored by 31P NMR
Fig. 4.

Carboxypeptidase-mediated cleavage of compound 10, monitored by 31P NMR
Compound 6 the naphthyl t-butyl alanine analogue (Fig. 5) showed no conversion at all presumably due to the presence of the tert-butyl ester, which is too bulky to be processed by the enzyme. This observation is in agreement with the lack of antiviral activity for this compound.
Fig. 5.

Carboxypeptidase-mediated cleavage of compound 6, monitored by 31P NMR
In the case of the L-valine derivative 17 (Fig. 6), the experiment showed, as already demonstrated for compound 10, a slow conversion to the compound of type 90, with a half-life of 72 h. The D-valine derivative 19 (Fig. 7) is not processed due to the presence of the non-natural amino acid, which seems not to be recognised by the enzyme.
Fig. 6.
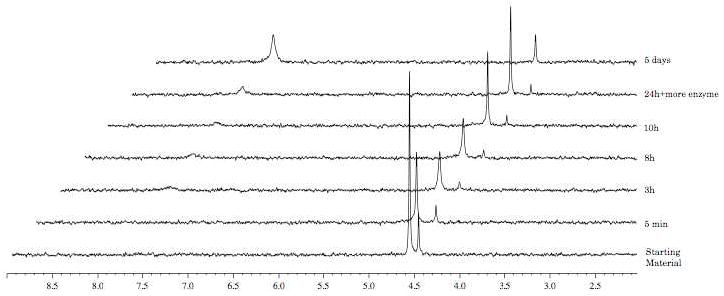
Carboxypeptidase-mediated cleavage of compound 17, monitored by 31P NMR
Fig. 7.
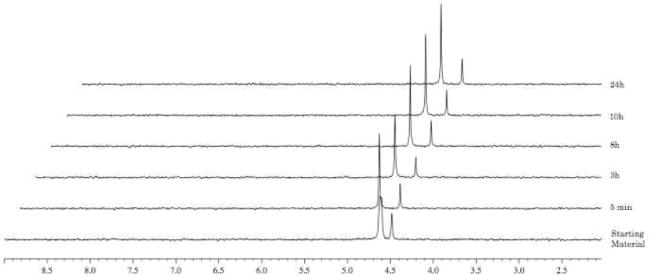
Carboxypeptidase-mediated cleavage of compound 19, monitored by 31P NMR
Also, CEM cell extracts have been prepared to examine the rate of hydrolysis of the antivirally active 9 and 4, and the inactive 6 derivatives. Whereas 9 and 4 were efficiently hydrolysed within a short time period (> 95% conversion of 9 and 65% conversion of 4 within one hour of incubation), 6 proved entirely stable after a 120 min-incubation period (Fig. 8). These observations are in agreement with the antiviral data, and demonstrate that CEM cell-associated esterases can efficiently convert methyl and benzyl esters of the ACV ProTides, but not tert-butyl esters. Tonsil extracts were also found to efficiently hydrolyse 9 and 4, with the same preference profile of 9 over 4 as found for the CEM cell extracts (data not shown).
Fig. 8.

Stability of ACV ProTides in crude CEM cell extracts as a function of incubation time.
These experiments support the need of activation of ACV ProTide in order to deliver the ACV monophosphate metabolite. The enzymatic data correlate well with the in vitro anti-HIV data and may support the role of carboxypeptidase Y in the ProTide activation in the lymphocyte cell cultures.
Stability studies of ACV ProTide
Two different stability studies of compound 9 using human serum and pH 1.0 buffer have been conducted. In the case of human serum, 9 was dissolved in DMSO and D2O and human serum was added. The experiment was conducted at 37 °C and monitored by 31PNMR. In Fig. 9 are reported 31P-NMR spectra 10 min after the addition of the serum and after 12 h. For a better resolution both original spectra and deconvoluted ones have been reported. The spectra show that the ACV ProTide is stable under these conditions. In fact, after 12 h, 56% of the compound is still present. The spectra also show the formation of the compound type 90 and the formation of a peak at δP = 1.90, which may correspond to the monophosphate form. The peak at δP = 2.25 corresponds to the human serum that in the first experiment is overlapping with the peak at at δP = 1.90.
Fig. 9.
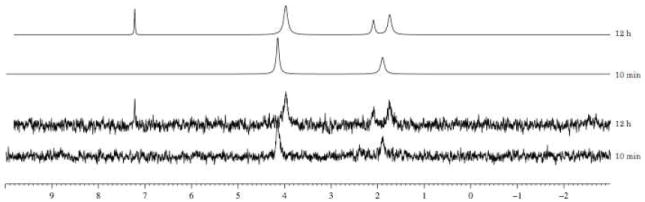
Stability of compound 9 in human serum, monitored by 31P NMR.
In the case of the stability in acid a pH of 1.0 was used. Compound 9 was dissolved in MeOD and the buffer was added. The experiment was conducted at 37 °C and monitored by 31P-NMR. The experiment show a good stability of the compound (see Fig. 12 in the supporting information) having an half-life of 11 h. Notably, the formation after 5 h of a peak at δP = -0.25, which should correspond to the monophosphate form was observed.
Molecular modelling-1: carboxypeptidase Y enzyme
To better understand the enzymatic results obtained using carboxypeptidase Y, molecular modelling studies using a crystal structure of the enzyme have been performed.17 The putative mechanism of action involves an attack from the Ser146 to the carbonyl of the ester which is coordinated with the NH from Gly52 and Gly53.18
The processed compound 9, showed a positive interaction with the active site of the enzyme for both phosphate diastereoisomers (shown only the RP diastereoisomer) (Fig. 10a). In particular the carbonyl moiety is in a suitable position for the nucleophilic attack from the catalytic Ser146, with the NH from Gly52 and Gly53 correctly placed to stabilize the tetrahedral intermediate. This result is in accordance with the enzymatic result for this compound. In the case of the inactive compound 6, the carbonyl is not in a favourable position, pointing away from Gly52 and Gly53, probably due to the presence of the bulky tert-butyl, which may influence the interaction with enzyme resulting in a poor activation (reported only the SP-diastereoisomer, Fig. 10b). The docking of compound 10, which showed a faster hydrolysis of one diastereoisomer compared to the other one, showed interesting results. In fact, the two diastereoisomers docked in a different way. The R-diastereoisomer showed a preferable position for the carbonyl moiety while in the case of the S-diastereoisomer the position of carbonyl group is different and it is not able to coordinate with the Gly52 and Gly53. This result supports the fact that one of them is faster metabolized, presumably the RP, than the other one and this is due to a different binding in the catalytic site of the enzyme. In the case of the valine derivatives, none of them showed a suitable pose in the active site of the enzyme.
Fig. 10.
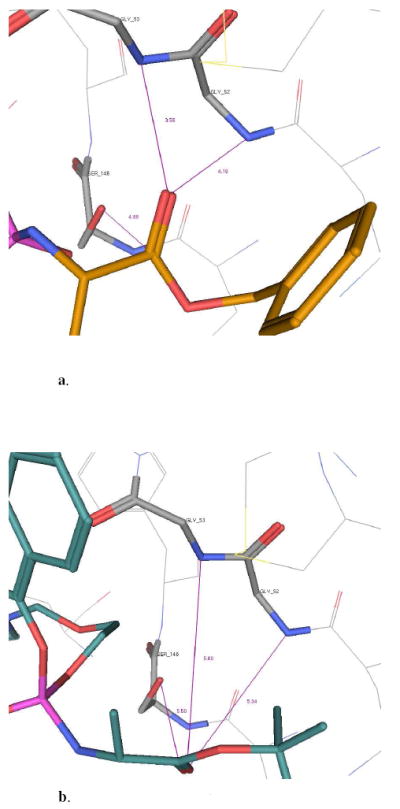
a. Docking of compounds 9 within the catalytic site of carboxypeptidase Y enzyme.
b. Docking of compounds 6 within the catalytic site of carboxypeptidase Y enzyme.
Molecular modelling-2: Human Hint enzyme
As shown in the enzymatic experiment on 9 the first step of activation proceeds well and leads to compound 90, which needs to be further converted in order to release the monophosphate form 91. The last step of the activation of the ProTide involving the cleavage of the P-N bond is not well known and it is considered to be mediated by a phosphoramidase-type enzyme called Hint, belonging to the HIT superfamily.19 A molecular modelling study using human hint enzyme 1, co-crystallized with adenosine monophosphate, has been performed in order to investigate this last step of activation.
The catalytic activity of this enzyme is due to the presence of three histidines, which interact with the substrate, and to the presence of a serine, which binds the nitrogen of the amino acid, protonating the nitrogen and favouring P-N bond cleavage (Fig. 11a). From Fig. 11b, it is clear to see how the compound binds correctly in the active site of the enzyme positioning the phosphate moiety (pink) in a suitable position for the cleavage of the P-N bond. This experiment suggests that the last step of the activation to release ACV-MP may proceed well in the case of ACV alaninyl phosphate in vivo, supporting the biological data.
Fig. 11.
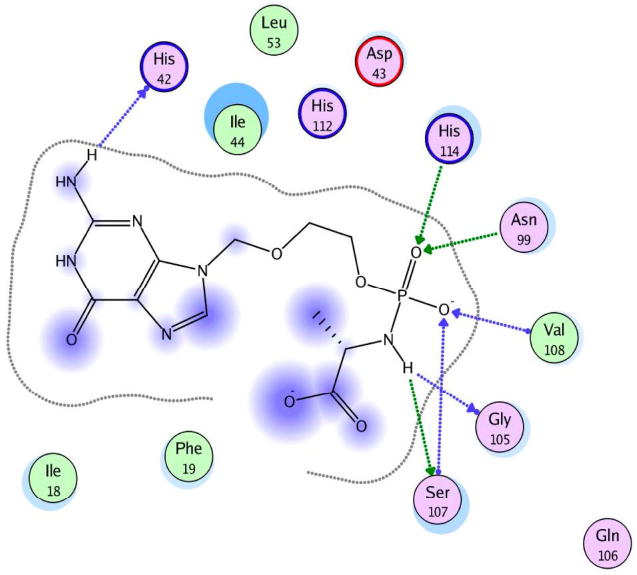
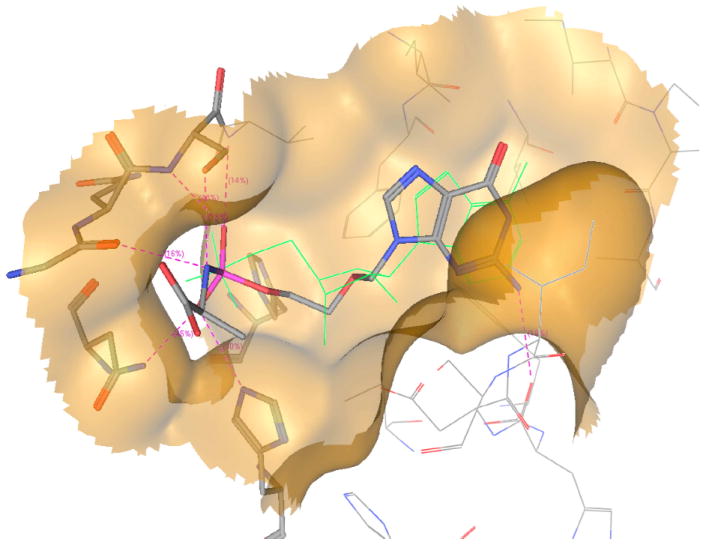
a. interactions of 90 (L-Ala) with the active site of human Hint-1.
b. Docking of compound 90 (L-Ala) ACV-MP phosphoramidate within the catalytic site of human HINT (I) enzyme
Conclusion
A series of 22 acyclovir ProTides has been reported. These compounds as well as the parent ACV have been biologically evaluated against HSV-1 and 2 and against HIV-1 and 2.
In the case of HSV-1 and 2 the compounds did not show any improvement of activity compare to the parent. However, these compounds retained activity against the TKdeficient HSV-1 strain while ACV showed a loss on activity. These results showed a successful thymidine-kinase by-pass.
In the case of HIV-1 and 2, ACV did not show any activity, while the ProTides show a good activity in a range of 6.2-17 μM (HIV-1, CEM), 8.9-42 μM (HIV-2, CEM) and 0.8-30 μM (HIV-1, MT-4).
The variation of the amino acid moiety seems to be tolerated in the case of HSV. In the case of HIV, this variation is less tolerated, showing good results only in the case of the L-alanine derivatives and phenylalanine derivative. In the MT-4 cell cultures, dimethylglycine and L-leucine are tolerated. These differences on activity may be due to a different substrate specificities and/or different intracellular levels of enzyme necessary for the activation of these compounds.
Experimental Section
General
Anhydrous solvents were bought from Aldrich and used without further purification. All reactions were carried out under an argon atmosphere. Reactions were monitored with analytical TLC on silica gel 60-F254 precoated aluminium plates and visualised under UV (254 nm) and/or with 31P-NMR spectra. Column chromatography wasperformed on silica gel (35-70 μM). Proton (1H), carbon (13C) and phosphorus (31P) NMR spectra were recorded on a Bruker Avance 500 spectrometer at 25 °C. Spectra were auto-calibrated to the deuterated solvent peak and all 13C NMR and 31P NMR were proton-decoupled. High resolution mass spectra was performed as a service by Birmingham University, using electrospray (ES). CHN microanalysis were performed as a service by the School of Pharmacy at the University of London. Purity (≥95%) of all final products was assured by a combination of microanalysis, and HPLC, with additional characterization in every case by: H, C, and P NMR, and HRMS.
Standard Procedure A: synthesis of dichlorophosphates [26, 27]
To a solution of phosphorus oxychloride (1.00 mol/eq) and the appropriate substituted phenol or naphthol (1.00 mol/eq) in anhydrous diethyl ether, stirred under an argon atmosphere, and was added dropwise at -78 °C under an argon atmosphere anhydrous TEA (1.00 mol/eq). Following the addition, the reaction mixture was stirred at -78 °C for 30 min, then at room temperature overnight. Formation of the desired compound was monitored by 31P NMR. The mixture was filtered under nitrogen and the corresponding filtrate reduced to dryness to give the crude product as an oil.
Standard Procedure B: synthesis of phosphorochloridates [45-65]
To a stirred solution of the appropriate aryl dichlorophosphate 26-28 (1.00 mol/eq) and the appropriate amino acid ester salt 29-44 (1.00 mol/eq) in anhydrous DCM was added, dropwise at -78 °C under an argon atmosphere, anhydrous TEA (2.00 mol/eq). Following the addition the reaction mixture was stirred at -78 °C for 30 min-1 h, then at room temperature for 30 min - 3.5 h. Formation of the desired compound was monitored by 31P NMR. After this period the solvent was removed under reduced pressure and the residue triturated with anhydrous diethyl ether. The precipitate was filtered under nitrogen and the solution was concentrated to give an oil. Most of the aryl phosphorochloridates synthesised were purified by flash column chromatography (eluting with ethyl acetate/petroleum ether in different proportions).
Standard Procedure C: synthesis of phosphoramidates [67-85]
To a stirring suspension of N2-DMF-ACV (1.00 mol/eq) in anhydrous THF was added dropwise under an argon atmosphere tBuMgCl (2.00 mol/eq) and the reaction mixture was stirred at room temperature for 30 min. Then was added dropwise a solution of the appropriate phosphorochloridate (1.50 to 4.00 mol/eq) in anhydrous THF. The reaction mixture was stirred at room temperature overnight. The solvent was removed under reduced pressure and the residue was purified by column chromatography eluting with DCM/MeOH in different proportions.
Standard Procedure D: deprotection of N2-DMF-phosphoramidates [2-23]
A solution of 67-87 in 1-propanol or 2-propanol was stirred under reflux for 24-96 h. The solvent was then removed under reduced pressure and the residue was purified by column chromatography eluting with DCM/MeOH in different proportions. The product was usually further purified by preparative TLC or semi-preparative HPLC to give a white solid.
Synthesis of N2-DMF acyclovir (N’-(9-((2-hydroxyethoxy)methyl)-6-oxo-6,9-dihydro-1H-purin-2-yl)-N,N-dimethylformimidamide) [66]
To a suspension of 1 (1.00 g, 4.44 mmol) in dry DMF (20 mL) was added N,N-dimethylformamide dimethyl acetal (2.96 mL, 22.20 mmol) and the reaction mixture was stirred at room temperature for 1 day. After this period, the solvent was removed and the residue triturated with diethyl ether and filtered. The solid was washed with diethyl ether to give a white solid (97%, 1.20 g). 1H-NMR (DMSO, 500 MHz): δ 11.30 (1H, s, NH), 8.58 (1H, s, CHN(CH3)2), 7.94 (1H, s, H-8), 5.45 (2H, s, H-1’), 4.65 (1H, t, OH), 3.52-3.49 (4H, m, H-4’, H-5’), 3.17, 3.04 (6H, 2s, N(CH3)2).
Synthesis of 1-naphthyl dichlorophosphate [26]
Prepared according to standard procedure A, using 24 (4.00 g, 27.74 mmol) in anhydrous diethyl ether (60 mL), POCl3 (2.59 mL, 27.74 mmol) and anhydrous TEA (3.87 mL, 27.74 mmol). After 31P NMR, the solvent was removed under reduced pressure and the residue was triturated with anhydrous diethyl ether. The precipitate was filtered, and the organic phase was removed under reduced pressure to give an oil (95%, 6.91 g). 31PNMR (CDCl3, 202 MHz): δ 3.72. 1H-NMR (CDCl3, 500 MHz): δ 8.02-8.00 (1H, m, H-8), 7.81-7.80 (1H, m, H-5), 7.72-7.70 (1H, m, H-4), 7.54-7.45 (4H, m, H-2, H-3, H-6, H-7).
Synthesis of 1-naphthyl(benzoxy-L-alaninyl)-phosphorochloridate [45]
Prepared according to standard procedure B, 26 (6.91 g, 26.48 mmol), L-alanine benzyl ester tosylate 29 (9.30 g, 26.48 mmol), anhydrous TEA (7.40 mL, 52.96 mmol) in anhydrous DCM (100 mL). The reaction mixture was stirred at -78 °C for 1 h, then at room temperature for 2 h. The crude was purified by column chromatography eluting with ethyl acetate/hexane = 5/5 to give an oil (72%, 7.68 g). 31P-NMR (CDCl3, 202 MHz): δ 8.14, 7.88. 1H-NMR (CDCl3, 500 MHz): δ 7.99-7.25 (12H, m, Naph, OCH2Ph), 5.15-5.07 (2H, m, CH2Ph), 4.30-4.23 (1H, m, CHCH3), 1.49-1.46 (3H, m, CHCH3).
Synthesis of N2-DMF-acyclovir-[1-naphthyl(benzoxy-L-alaninyl)] phosphate [67]
Prepared according to Standard Procedure C, from 66 (0.30 g, 1.07 mmol) in anhydrous THF (10 mL), tBuMgCl (1.0 M THF solution, 2.14 mL, 2.14 mmol), 45 (1.31 g, 3.25 mmol) in anhydrous THF (10 mL) and the reaction mixture was stirred at room temperature overnight. The residue was purified by column chromatography, eluting with DCM/MeOH = 95/5, to give a white solid (17%, 0.12 g). 31P-NMR (MeOD, 202 MHz): δ 4.18, 3.92. 1H-NMR (MeOD, 500 MHz): δ 8.47, 8.46 (1H, 2s, NCHN(CH3)2), 8.01-7.98 (1H, m, H-8 Naph), 7.78-7.74 (2H, m, H-8, H-6 Naph), 7.56, 7.55 (1H, m, H-2 Naph), 7.41-7.12 (9H, m, Naph, OCH2Ph), 5.37-5.36 (2H, 2s, H-1’), 5.00-4.93 (2H, m, OCH2Ph), 4.14-4.06 (2H, m, H-5’), 3.96-3.88 (1H, m, CHCH3), 3.88-3.59 (2H, m, H-4’), 2.95-2.93 (6H, m, N(CH3)2), 1.20-1.17 (3H, m, CHCH3).
Synthesis of acyclovir-[1-naphthyl(benzoxy-L-alaninyl)] phosphate [2]
A solution of 67 (0.10 g, 0.16 mmol) in 2-propanol (5 mL) was stirred under reflux for 2 days. The solvent was then removed under reduced pressure and the residue was purified by column chromatography eluting with DCM/MeOH = 96/4. The product was purified by preparative TLC (gradient elution of DCM/MeOH = 99/1, then 98/2, then 96/4) to give a white solid (35%, 0.032 g). 31P-NMR (MeOD, 202 MHz): δ 4.13, 3.96. 1H-NMR (MeOD, 500 MHz): δ 8.01-7.99 (1H, m, H-8 Naph), 7.77-7.75 (1H, m, H-6 Naph), 7.67, 7.64 (1H, 2s, H-8), 7.58-7.13 (10H, m, Naph, OCH2Ph), 5.28, 5.25 (2H, 2s, H-1’), 4.99-4.94 (2H, m, OCH2Ph), 4.12-4.06 (2H, m, H-5’), 3.97-3.93 (1H, m, CHCH3), 3.64-3.59 (2H, m, H-4’), 1.24-1.20 (3H, m, CHCH3). 13C-NMR (MeOD, 125 MHz): δ 20.32 (d, JC−P = 7.63, CHCH3), 20.43 (d, JC−P = 6.61, CHCH3), 51.76, 51.81 (2s, CHCH3), 67.20 (d, JC−P = 5.58, C-5’), 67.28 (d, JC−P = 4.91, C-5’), 67.95, 67.98 (2s, OCH2Ph), 69.34 (d, JC−P = 7.72, C-4’), 69.40 (d, JC−P = 8.14, C-4’), 73.65 (C-1’), 116.26, 116.29, 116.35, 122.69, 122.80, 125.92, 126.51, 127.20, 127.42, 127.46, 127.74, 128.81, 128.83, 129.27, 129.33, 129.52, 129.57 (C-5, C-2 Naph, C-3 Naph, C-4 Naph, C-5 Naph, C-6 Naph, C-7 Naph, C-8 Naph, C-8a Naph, OCH2Ph), 136.26, 137.23 (C-4a Naph, ‘ipso’ OCH2Ph), 139.69 (C-8), 147.98, 148.04 (‘ipso’ Naph, C-4), 152.44 (C-2), 159.39 (C-6), 174.61, 174.88 (2s, COOCH2Ph). EI MS= 615.1735 (M+Na). Anal. Calcd for C28H29N6O7P·0.5H2O: C, 55.91; H, 5.03; N, 13.97. Found: C, 55.81; H, 4.91; N, 13.78.
Antiviral activity assays
The compounds were evaluated against the following viruses: HSV-1 strain KOS, thymidine kinase-deficient (TK-) HSV-1 KOS strain resistant to ACV (ACVr), HSV-2 strain G, HIV-1 strain IIIB/Lai and HIV-2 strain ROD. The antiviral, other than anti-HIV, assays were based on inhibition of virus-induced cytopathicity or plaque formation in human embryonic lung (HEL) fibroblasts. Confluent cell cultures in microtiter 96-well plates were inoculated with 100 CCID50 of virus (1CCID50 being the virus dose required to infect 50% of the cell cultures). After a 1-2 h adsorption period, residual virus was removed, and the cell cultures were incubated in the presence of varying concentrations of the test compounds. Viral cytopathicity was recorded as soon as it reached completion in the control virus-infected cell cultures that were not treated with the test compounds. Antiviral activity was expressed as the EC50 or effective compound concentration required to reduce virus-induced cytopathicity by 50%.
Human CEM cell cultures (~3 × 105 cells mL-1) were infected with 100 CCID50 HIV- 1(IIIB) or HIV-2(ROD) per mL and seeded in 200 μL-well microtiter plates, containing appropriate dilutions of the test compounds. After 4 days of incubation at 37°C, CEM giant cell formation was examined microscopically.
MT-4 cells (1 × 104 cells per mL) were suspended in fresh culture medium and infected with 10 μL (0.7 ng of p24) of X4LAI.04 viral stock per ml of cell suspension. Infected cell suspensions were then transferred to microplate wells, mixed with 1 mL of medium containing the test compound at an appropriate dilution, and further incubated at 37°C. After 3 days, p24 production was measured in the MT-4 cell culture supernatants. The EC50 corresponded to the compound concentration required to suppress the production of p24 in the virus-infected MT-4 cell cultures by 50%. Viability in MT-4 cell cultures were evaluated using a nucleocounter automated cell counting system (Chemometec, Denmark). Total number of cells and number of dead cells in the cultures untreated and treated with ACV ProTides were enumerated using a propidium iodide-based assay according to the manufacturers’ protocol. Data were collected and analyzed using Nucleoview software (Chemometec, Denmark).
Human tonsils obtained under an IRB-approved protocol were dissected into ~2mm blocks and cultured on collagen rafts at the medium-air interface. Tissues were inoculated ex vivo with X4LAI.04 (~ 0.5 μg of p24gag per block) and treated with ACV ProTides at concentrations ranging from 0.1 to 10 μM. The culture medium was changed every 3 days, and ACV ProTides were replenished. For each compounds’ concentration HIV-1 release was quantified by measurements of p24gag accumulated over 3-day periods in the culture media bathing 18 tissue blocks. The EC50 corresponded to the compound concentration required to suppress by 50% the production of p24.
Preparation of CEM and tonsil cell extracts and analysis of ProTide conversion
Exponentially growing CEM cells or tonsil tissues were washed twice with PBS. Then, cells and tissues were suspended in PBS, and extracts were made in a Precellys-24 homogenizator (Berlin Technologies, Montigny-en-Bretonneux, France) (tonsils) or by a Hielscher-Ultrasound Technology (CEM cells) (Germany). The extracts were cleared by centrifugation (10 min, 15,000 rpm) and frozen at -20 °C before use. Ten micromolar solutions of 9, 4 and 6 were added to the crude cell and tissue extracts (100 μL) and incubated for 30, 60 and 120 min at 37 °C. At each time point, 20 μl of the incubation mixtures were withdrawn, and added to 30 μL cold methanol to precipitate the proteins. After centrifugation, the supernatants were subjected to HPLC analysis on a reverse phase C18 column (Merck) to separate the parent ACV ProTides from their hydrolysis products that may be formed during the incubation process. Data were plotted as percent of disappearance of the intact parent ACV ProTide from the incubation mixture.
Supplementary Material
Acknowledgments
Marco Derudas dedicates this work to the memory of Mrs Antonietta Derudas.
We thank Frieda De Meyer, Leentje Persoons, Vicky Broeckx, Leen Ingels and Ria Van Berwaer for excellent technical assistance with the antiviral and enzymatic assays.
We acknowledge the excellent administrative support of Ms Helen Murphy. Financial support by a grant of the K.U.Leuven (GOA no. 05/19) was provided. The work of AL, CV and LM was supported by the NICHD Intramural Program.
Footnotes
Abbreviations: ACV, acyclovir; HIV, human immunodeficiency virus; HSV, herpes simplex virus; NNRTIs, non-nucleoside reverse transcriptase inhibitors; PI, protease inhibitor; FI, fusion inhibitor; HHV, human herpes virus; ACVMP, acyclovir 5’-monophosphate; DMF, N,N-dimethylformamide;
Supporting Information Available: Preparative methods, spectroscopic and analytical data on target compounds. This material is available free of charge via the internet at http://pubs.acs.org.
References
- 1.U.S Food and drug Administration. Drugs used in the treatment of HIV infection. [5 March 2009];2008 http://www.fda.gov/oashi/aids/virals.html.
- 2.Clavel F, Hance AJ. HIV drug resistance. N Engl J Med. 2004;350:1023–1035. doi: 10.1056/NEJMra025195. [DOI] [PubMed] [Google Scholar]
- 3.Lisco A, Vanpouille C, Tchesnokov EP, Grivel J-C, Biancotto A, Brichacek B, Elliott J, Fromentin E, Shattock R, Anton P, Gorelick R, Balzarini J, McGuigan C, Derudas M, Gotte M, Schinazi RF, Margolis L. Acyclovir is activated into a HIV-1 reverse transcriptase inhibitor in herpesvirus-infected human tissues. Cell Host & Microbe. 2008;4:260–270. doi: 10.1016/j.chom.2008.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Miller WH, Miller RL. Phosphorylation of acyclovir monophosphate by GMP kinase. J Biol Chem. 1980;255:7204–7207. [PubMed] [Google Scholar]
- 5.Miller WH, Miller RL. Phosphorylation of acyclovir diphosphate by cellular enzyme. Biochem Phamacol. 1982;31:3879–3884. doi: 10.1016/0006-2952(82)90305-7. [DOI] [PubMed] [Google Scholar]
- 6.Congiatu C, McGuigan C, Jiang WG, Davies G, Mason MD. Naphthyl phosphoramidate derivatives of BVdU as potential anticancer agents: design, synthesis and biological evaluation. Nucleosides Nucleotides Nucleic Acids. 2005;24:485–489. doi: 10.1081/ncn-200061774. [DOI] [PubMed] [Google Scholar]
- 7.McGuigan C, Cahard D, Sheeka HM, De Clercq E, Balzarini J. Aryl phosphoramidate derivatives of d4T have improved anti-HIV efficacy in tissue culture and may act by the generation of a novel intracellular metabolite. J Med Chem. 1996;39:1748–1753. doi: 10.1021/jm950605j. [DOI] [PubMed] [Google Scholar]
- 8.McGuigan C, Harris SA, Daluge SM, Gudmundsson KS, McLean EW, Burnette TC, Marr H, Hazen R, Condreay LD, Johnson L, De Clercq E, Balzarini J. Application of phosphoramidate pronucleotide technology to abacavir leads to a significant enhancement of antiviral potency. J Med Chem. 2005;48:3504–3515. doi: 10.1021/jm0491400. [DOI] [PubMed] [Google Scholar]
- 9.Perrone P, Luoni GM, Kelleher MR, Daverio F, Angell A, Mulready S, Congiatu C, Rajyaguru S, Martin JA, Levêque V, Le Pogam S, Najera I, Klumpp K, Smith DB, McGuigan C. Application of the phosphoramidate ProTide approach to 4’-Azidouridine confers sub-micro-molar potency versus hepatitis C virus on an inactive nucleoside. J Med Chem. 2007;50:1840–1849. doi: 10.1021/jm0613370. [DOI] [PubMed] [Google Scholar]
- 10.Saboulard D, Naesens L, Cahard D, Salgado A, Pathirana R, Velazquez S, McGuigan C, De Clercq E, Balzarini J. Characterization of the activation pathway of phosphoramidate triester prodrugs of stavudine (d4T) and Zidovudine (AZT) Mol Pharmacol. 1999;56:693–704. [PubMed] [Google Scholar]
- 11.McGuigan C, Derudas M, Bugert JJ, Andrei G, Snoeck R, Balzarini J. Successful kinase bypass with new acyclovir phosphoramidate prodrugs. Bioorg Med Chem Lett. 2008;18:4364–4367. doi: 10.1016/j.bmcl.2008.06.069. [DOI] [PubMed] [Google Scholar]
- 12.McGuigan C, Tsang H-W, Cahard D, Turner K, Velazquez S, Salgado A, Bidois L, Naesens L, De Clercq E, Balzarini J. Phosphoramidate derivatives of d4T as inhibitors of HIV: The effect of amino acid variation. Antiviral Res. 1997;35:195–204. doi: 10.1016/s0166-3542(97)00029-6. [DOI] [PubMed] [Google Scholar]
- 13.Uchiyama M, Aso Y, Noyori R, Hayakawa Y. O-selective phosphorylation of nucleosides without N-protection. J Org Chem. 1993;58:373–379. [Google Scholar]
- 14.Van Boom JH, Burgers PMJ, Crea R, Luyten WCMM, Vink ABJ, Reese CB. Phosphorylation of nucleoside derivatives with aryl phosphoramidochloridates. Tetrahedron. 1975;31:2953–2959. [Google Scholar]
- 15.McGuigan C, Tsang H-W, Cahard D, Turner K, Velazquez S, Salgado A, Bidois L, Naesens L, De Clercq E, Balzarini J. Phosphoramidate derivatives of d4T as inhibitors of HIV: the effect of amino acid variation. Antiviral Res. 1997;35:195–204. doi: 10.1016/s0166-3542(97)00029-6. [DOI] [PubMed] [Google Scholar]
- 16.Cahard D, McGuigan C, Balzarini J. Aryloxy Phosphoramidate Triesters as Pro-Tides. Mini-Rev Med Chem. 2004;4:371–382. doi: 10.2174/1389557043403936. [DOI] [PubMed] [Google Scholar]
- 17.Endrizzi JA, Breddam K, Remington SJ. 2.8-Å Structure of yeast serine carboxypeptidase. Biochemistry. 1994;33:11106–11120. doi: 10.1021/bi00203a007. [DOI] [PubMed] [Google Scholar]
- 18.Jung G, Ueno H, Hayashi R. Carboxypeptidase Y: structural basis for protein sorting and catalytic triad. J Biochem. 1999;126:1–6. doi: 10.1093/oxfordjournals.jbchem.a022408. [DOI] [PubMed] [Google Scholar]
- 19.Brenner C. Hint, Fhit, and GaIT: function, structure, evolution, and mechanism of three branches of the histidine triad superfamily of nucleotide hydrolases and transferase. Biochemistry. 2002;41:9003–9014. doi: 10.1021/bi025942q. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.