

Abstract
The development of a new method for the assembly of unsymmetrical carbazoles is reported. The strategy involves the selective intramolecular functionalization of an arene C-H bond and the formation of a new arene C-N bond. The substitution pattern of the carbazole product can be controlled by the design of the biaryl amide substrate, and the method is compatible with a variety of functional groups. The utility of the new protocol was demonstrated by the concise synthesis of three natural products from commercially available materials.
Keywords: Palladium, Catalysis, C-H Bond, C-N bond, Carbazole
Introduction
Carbazole alkaloids manifest a variety of biological activities and, hence, they are attractive synthetic targets.1.2 The indigenous application of the stem bark of the curry-leaf tree as folk medicine2 led to the discovery of the antibacterial, antifungal, and antiviral properties of this class of natural products.1,2,3,4 The anti-inflammatory properties of carbazomycin B4,5 and the antitumor properties of ellipticine derivatives6 encouraged the development of synthetic methods for the preparation of carbazoles. Carvedilol7 and carazolol8 were identified for their potential as multiple-action anti-hypertensive drugs. It was later realized that their therapeutic value might derive from the properties of the 4-alkoxycarbazole cores that act as potent antioxidants against radical-induced lipid oxidation. Subsequently, new molecules with a 4-alkoxycarbazole core have been developed for the treatment of obesity and type II diabetes.9
In addition to their use in medicinal chemistry, carbazoles have attracted increasing attention from the community of material scientists owing to their potential in photophysical and optoelectronic applications. Polyvinylcarbazole (PVK) has been extensively investigated for its use in photorefractive materials and xerography, while N-ethylcarbazole (ECz) was recognized as an effective charge-transporting functional plasticizer for PVK and other doped polymers.10 In the last decade, carbazole compounds were intensively investigated for the development of optoelectronic applications such as polymeric light-emitting diodes (PLED)10a,10c,11 and organic light-emitting devices (OLED).12,13,14 In particular, it was found that certain carbazole oligomers led to a family of host materials that were suitable for the electrophosphorescence of blue lights,12,13 a critical component for the fabrication of commercial OLED displays.
In view of these important applications, we set out to develop a metal-catalyzed synthesis of unsymmetrical carbazoles. Prior to our investigations, Du Bois, Che, and their respective co-workers had demonstrated that a metal-catalyzed sequence converting an alkyl C-H bond to a C-N bond can be quite efficient.15,16,17,18 On the basis of successful applications of direct arylation of arene C-H bonds in recent years19 and our long-standing interest in the formation of aromatic C-N bonds,20 we felt that it should be possible to realize an overall substitution reaction where an arene C-H bond is replaced with a C-N bond. In late 2005,21 our laboratory reported a new strategy for assembling simple unsymmetrical carbazoles by combining sequential palladium-catalyzed C-H functionalization and C-N bond forming reactions, although the substrate scope was limited to molecules containing functional groups that were compatible with the co-oxidant Cu(OAc)2. This method was recently adopted by Shi as a part of an elegant sequence for the synthesis of unsymmetrical carbazoles such as 4-deoxycarbazomycin B.22 To address the shortcomings of our earlier work, we decided to expand our research to address the issue of the functional group compatibility, to study the reaction mechanism, and to apply the method to the synthesis of naturally occurring carbazoles. Herein we report our progress toward achieving these goals.
Results and Discussion
Catalyst and Oxidation Systems for the Synthesis of Carbazoles
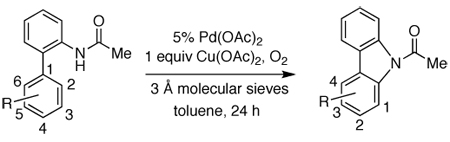
We initiated our investigation by studying the conversion of 2-acetaminobiphenyl (1) to N-acetylcarbazole (2). Substrate 1 was quantitatively converted to 2 in the presence of a stoichiometric amount of Pd(OAc)2 in toluene at 120 °C after 24 h. As shown in Table 1 (entry 1), a near quantitative yield of 2 can also be obtained with a combination of 5% Pd(OAc)2 and one equivalent of Cu(OAc)2 in an atmosphere of oxygen. No product was observed in the absence of Pd(OAc)2, suggesting that the role of Cu(OAc)2 was primarily to mediate the reoxidation of the reduced palladium species. Other commercially available palladium precatalysts (entries 4–7) were less efficient than Pd(OAc)2 in affecting the transformation. Attempts to use other reoxidants (entries 8–12), which have been reported to be effective in mediating other palladium-catalyzed oxidative coupling reactions,23 were largely unsuccessful.
Table 1.
Investigation of Palladium Precatalysts and Reoxidants
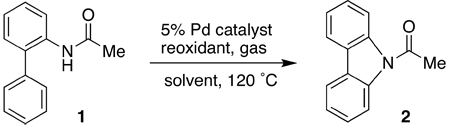 | ||||||
|---|---|---|---|---|---|---|
| Entry | Pd source | Reoxidant /equiv |
Solvent | Gas atm |
Time (h) |
Yield (%)a |
| 1 | Pd(OAc)2 | Cu(OAc)2 / 1 | toluene | O2 | 12 | 92 |
| 2 | Pd(OAc)2 | toluene | O2 | 12 | 5 | |
| 3 | Cu(OAc)2 / 1 | toluene | O2 | 12 | 0 | |
| 4 | Pd(O2CCF3)2 | Cu(OAc)2 / 1 | toluene | O2 | 24 | 70 |
| 5 | PdCl2 | Cu(OAc)2 / 1 | toluene | O2 | 24 | 21 |
| 6 | PdCl2(CH3CN)2 | Cu(OAc)2 / 1 | toluene | O2 | 24 | 27 |
| 7 | PdCl2(PPh3)2 | Cu(OAc)2 / 1 | toluene | O2 | 24 | 67b |
| 8 | Pd(OAc)2 | PPh3 / 0.2 | toluene | O2 | 12 | 5 |
| 9 | Pd(OAc)2 | pyridine / 0.2 | toluene | O2 | 12 | 1 |
| 10 | Pd(OAc)2 | PhI(OAc)2 / 0.2 | DCEc | air | 12 | 25 |
| 11 | Pd(OAc)2 | K2S2O8 / 5 | DCEd | air | 12 | 2 |
| 12 | Pd(OAc)2 | benzoquinone / 1 | toluene | air | 12 | 4 |
GC yield was determined with respect to a calibrated internal standard.
About 5–10% diacetylated amide of 2-aminobiphenyl was observed.
Slightly higher yield was obtained in dichloroethane than in methylene chloride.
The reaction was performed at 80 °C in dichloroethane; the same low yield was observed with or without bases.
Air could be used in the place of oxygen in the synthesis of 2 (Table 2, entry 1), although the reaction was significantly slower compared to that using oxygen (entry 2). We subsequently found that in the presence of oxygen, a catalytic amount of Cu(OAc)2 was sufficient to effect the synthesis of carbazoles. Three 2-substituted carbazoles (Note: the numbering of the rings changes from the starting biarylamides to the carbazole products) were obtained in >75% yield using only 20% Cu(OAc)2 for reactions carried out in toluene as the solvent (entries 3–5). Studies of these three cyclizations also revealed the trend: Cu(OAc)2 as low as 10% could be used in the cases where the “bottom” aryl ring is neutral or electron-rich; on the other hand, it appears that a minimum of 20% Cu(OAc)2 is necessary in the cases where the bottom ring bears an electron-withdrawing group. Because of the functional group compatibility, the need to use Cu(OAc)2 as a co-catalyst is a limitation of the method. In our search for improved reaction conditions, we found that when dimethyl sulfoxide (DMSO) was used as the solvent under an atmosphere of oxygen, no Cu(OAc)2 was necessary and a comparable yield of 2 was obtained (entry 6). Presumably in this case, DMSO is acting as a ligand to support the direct oxidation of Pd(0) to Pd(II) with oxygen.24 Protocols that employed other polar solvents such as N,N-dimethylformamide (DMF) gave poor yields of 2 (entry 7).
Table 2.
Evaluation of Reoxidants in the Synthesis of Carbazoles
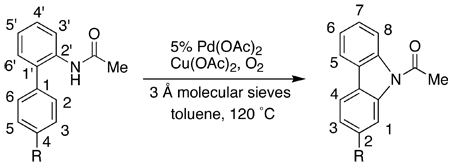 | ||||||
|---|---|---|---|---|---|---|
| Entry | R | Reoxidant /equiv |
Solvent | Gas atm |
Time (h) |
Yield (%)a |
| 1 | H | Cu(OAc)2 / 1 | toluene | air | 24 | 94 |
| 2 | H | Cu(OAc)2 / 1 | toluene | O2 | 12 | 92b |
| 3 | H | Cu(OAc)2 / 0.2 | toluene | O2 | 12 | 90 |
| 4 | OMe | Cu(OAc)2 / 0.2 | toluene | O2 | 12 | 88 |
| 5 | CF3 | Cu(OAc)2 / 0.2 | toluene | O2 | 12 | 76 |
| 6 | H | DMSO | O2 | 12 | 92b | |
| 7 | H | DMF | O2 | 12 | 11b | |
Isolated yield after silica gel chromatography.
GC yield was determined with respect to a calibrated internal standard.
Having established that acetamide based biaryl amides were efficiently transformed to the carbazoles in the presence of oxygen with either Cu(OAc)2 or when carried out in DMSO (Table 3, entries 1 and 2), we turned our attention to substrates containing other amine protecting groups. Sulfonamide-based biaryl amides underwent the cyclization just as efficiently as the corresponding acetamide (entries 3 and 4). In contrast, 2-aminobiphenyl protected as other amides such as benzamide, tert-butylcarboxycarbonylamide (BOC-amide), and pentafluoropropionamide provided much lower yields of the desired products (entries 5–10). It appeared that part of the reason for the lower yields in these cases was the cleavage of the amide under the reaction conditions prior to complete conversion to the carbazole. This was clearly observed in the case of the N-Boc substrate when the reaction was attempted in DMSO. In this case, both the 2-aminobiphenyl and free carbazole generated appeared to inhibit the palladium-catalyzed transformation. Since the acetamide group is generally easier to remove than the sulfonamide group, we focused on the formation of N-acetylcarbazoles in subsequent studies of the substrate scope. The free carbazoles can be librated in almost quantitative yield (97%) by treatment either with acid (1:5 H2SO4:MeOH) or base (DBU) at 60–80 °C (see Supporting Information).
Table 3.
Effect of Nitrogen Protecting Groups
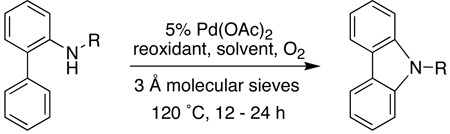 | ||||
|---|---|---|---|---|
| Entry | R | Reoxidant /equiv |
Solvent | Yield (%)a |
| 1 | Ac | Cu(OAc)2 / 1 | toluene | 92 |
| 2 | Ac | DMSO | 92 | |
| 3 | SO2Ph | Cu(OAc)2 / 1 | toluene | 81b |
| 4 | SO2Ph | DMSO | 99 | |
| 5 | BOC | Cu(OAc)2 / 1 | toluene | 34 |
| 6 | BOC | DMSO | 30 | |
| 7 | COPh | Cu(OAc)2 / 1 | toluene | <10c |
| 8 | COPh | DMSO | <10c | |
| 9 | COCF2CF3 | Cu(OAc)2 / 1 | toluene | 0 |
| 10 | COCF2CF3 | DMSO | 0 | |
Isolated yield after silica gel chromatography, average of two runs.
10% Pd(OAc)2 was used.
Yield estimated from GC analysis.
Synthesis of Substituted Biaryl Amides and 9-Acetylcarbazoles
A variety of substituted biaryl acetamide compounds were prepared in near quantitative yield from commercially available 2'-bromoacetanilide and appropriate boronic acids (Scheme 1, equation 1). The Suzuki-Miyaura coupling reaction was carried out in the presence of a bulky biaryl phosphine, 2-dicyclohexylphosphino-2',6'-dimethoxy-1,1'-biphenyl (SPhos), following a procedure recently developed in our laboratory.25 The reaction can be extended to chloroacetanilide substrates in cases where the substituted bromoacetanilide is inaccessible or costly (Scheme 1, equation 2).
Scheme 1.

Synthesis of Biaryl Acetamides by Suzuki-Miyaura Coupling
Acetylcarbazoles with various substitution patterns can be obtained in excellent yields via the palladium-catalyzed cyclization reaction (Table 4). In particular, the presence of a substituent (C3’) adjacent to the acetamide moiety in the “upper” ring did not affect the efficiency of the cyclization reaction (entries 1 and 2). This observation is significant as it allows us to conveniently assemble carbazoles in excellent yields with substitution at both positions meta to the biaryl axis, which is the substitution pattern observed in Mukonine and a number of related natural products.1,2 Furthermore, the presence of substituents at positions ortho or para to the biaryl axis did not affect the formation of the carbazole (entries 6–9). Carbazoles with substitution on both rings were also assembled in good yield (entry 10). The 2-methoxy-6-methylcarbazole core is a synthetically useful building block and has recently been elaborated to form Siamenol,26 a 3,6-disubstituted 2-hydroxycarbazole with anti-HIV activity.27
Table 4.
Synthesis of Substituted Carbazoles by Cyclization of Biaryl Amides
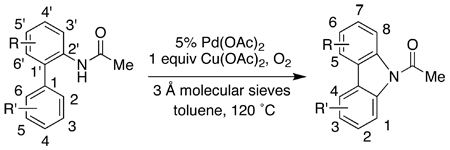 | ||||
|---|---|---|---|---|
| Entry | Substrate | Product | R | Yield (%)a |
| 1 |
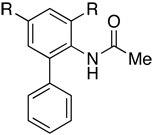
|
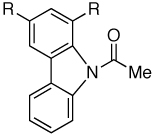 |
Me | 93 |
| 2 | F | 90 | ||
| 3 |
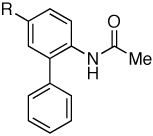
|
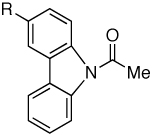 |
Me | 92 |
| 4 | F | 94 | ||
| 5 | CF3 | 88 | ||
| 6 |
|
|
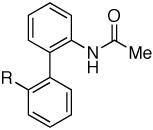
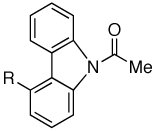
OMe | 81 |
| 7 | F | 78 | ||
| 8 | CF3 | 81 | ||
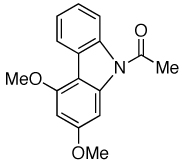
| 9 | 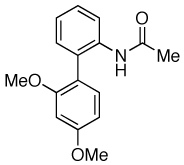 |
 |
85 | |
| 10 | 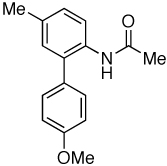 |
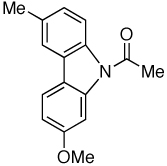 |
88 | |
Isolated yield after silica gel chromatography, average of two runs.
While all the examples in Table 4 were performed on a 0.2 mmol scale in our initial investigation, both steps in our method can be scaled up with no loss in yield, allowing efficient access to the target carbazoles in sufficient quantity from commercial reagents. For example, the biaryl amide 2-acetamino-2'-methoxybiphenyl (entry 6) could be obtained in 97% yield on a 15 mmol scale from 2'-bromoacetanilide and 2-methoxyphenylboronic acid with 1% Pd(OAc)2, 2% SPhos, and K3PO4 in toluene in 30 min at 90 °C. That product was cyclized in the presence of Pd(OAc)2, DMSO and oxygen and subsequently hydrolyzed to give 9H-4-methoxycarbazole in 86% yield in 2 steps on a 5 mmol scale. This gram-scale experiment demonstrated the practicality of our method to access carbazoles such as 9H-4-hydroxycarbazole, a major building block for a variety of therapeutically valuable candidates.7,8,9
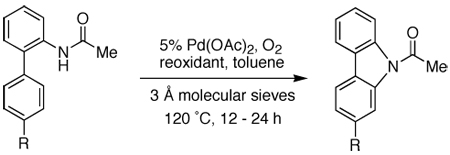
The cyclization process of the biaryl amide was compatible with a variety of functional groups. Ketone- and methyl ester-containing substrates can be used in the Cu(OAc)2 protocol (Table 5). On the other hand, biaryl amides with nitro, cyano, and even thioether groups were more efficiently cyclized in DMSO (entries 7–9). Unfortunately, attempts to extend this cyclization strategy to substrates with heteroaromatic groups in place of either aryl rings have been unsuccessful.
Table 5.
Investigation of Functional Group Compatibility
 | ||||
|---|---|---|---|---|
| Entry | R | Reoxidant/ equiv |
Solvent | Yield (%)a |
| 1 | F | Cu(OAc)2/ 1 | toluene | 87 |
| 2 | Me | Cu(OAc)2/ 1 | toluene | 98 |
| 3 | OMe | Cu(OAc)2/ 1 | toluene | 97 |
| 4 | CF3 | Cu(OAc)2/ 1 | toluene | 94 |
| 5 | COMe | Cu(OAc)2/ 1 | toluene | 86 |
| 6 | CO2Me | Cu(OAc)2/ 1 | toluene | 96 |
| 7 | SMe | DMSO | 82 | |
| 8 | NO2 | DMSO | 72b | |
| 9 | CN | DMSO | 84b | |
Isolated yield after silica gel chromatography, average of two runs.
10% Pd(OAc)2 was used.
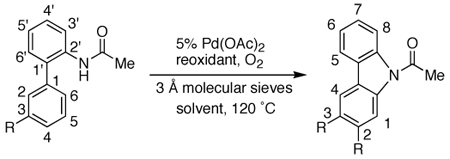
As shown in Table 4, we have been successful in obtaining 3-substituted carbazoles by derivatizing the “upper” ring (C5’) in the biaryl amide substrates (entries 3–5). Alternatively, this class of cabazoles could be obtained by derivatizing the “bottom” ring (C3). Following this approach, 3-substituted carbazoles were formed regioselectively in good to excellent yields as steric factors probably govern the regiochemical nature of the process (Table 6). The cyclization process was generally efficient using Cu(OAc)2 as the reoxidant with one exception (entries 1 and 4–7). In the case of 3-methoxycarbazole, the cyclization process appeared to be more efficient when Cu(OAc)2was replaced with DMSO (entries 2 and 3). When the “bottom” ring is substituted at both C3 and C5, the cyclization process was too slow to be synthetically useful.
Table 6.
Regioselective Synthesis of 3-substituted Carbazoles
 | ||||
|---|---|---|---|---|
| Entry | R | Reoxidant/ equiv |
Solvent | Yield (%)a |
| 1 | Me | Cu(OAc)2 / 1 | toluene | 82b |
| 2 | OMe | Cu(OAc)2 / 1 | toluene | 41c |
| 3 | OMe | DMSO | 59 | |
| 4 | CF3 | Cu(OAc)2 / 1 | toluene | 86 |
| 5 | CO2Me | Cu(OAc)2 / 1 | toluene | 99 |
| 6 | OTIPS | Cu(OAc)2 / 1 | toluene | 81 |
| 7 | TBS | Cu(OAc)2 / 1 | toluene | 98 |
Isolated yield after silica gel chromatography, average of two runs. Unless otherwise stated, the regioisomeric ratio is >25:1 (GC); only the major product is shown.
Selectivity = 18:1 (GC).
Selectivity = 16:1 (GC).
Mechanism
We previously proposed a tandem electrophilic palladation and C-N bond formation sequence to account for the intramolecular addition of the amide group to the aryl ring (Scheme 2).21,28 In such a pathway, complexation of Pd(OAc)2 to the amide moiety of the substrate (1) could form a palladium amide (3) with concomitant release of one molecule of acetic acid. Palladium amide 3 would then undergo electrophilic palladation to provide a six-membered cyclic palladium amide 4. Reductive elimination produces the carbazole product (2) and a Pd(0) species, which would be reoxidized to Pd (II) in the presence of Cu(OAc)2 or DMSO and oxygen to complete the catalytic cycle.
Scheme 2.

Our Initial Proposed Pathway for Carbazole Synthesis
However, this proposed catalytic cycle appears to be inconsistent with two experimental observations. First, based on the catalytic cycle, the cyclization of 2’-acetamino-3-methoxybiphenyl was expected to be more facile than those of 2’-acetamino-3-(trifluoromethyl)biphenyl and 2’-acetamino-3-(methoxycarbonyl)biphenyl. However, the experimental data did not support this prediction (Table 6, entries 2, 4, and 5). Second, during our initial optimization studies, attempts to effect the cyclization of biaryl amide 1 at temperatures below 120 °C were met with low yields. For example, only 10% of product 2 was observed at 90 °C after 24 h, and no product was observed at 60 °C. In subsequent studies of substituent effects on the cyclization at 90 °C, we were surprised to see that among all the substrates with substituents at C4 examined, only 2’-acetamino-4-methoxybiphenyl cyclized to the carbazole in satisfactory yield, although this substrate would be expected to destabilize the proposed intermediate due to the inductive effect of the methoxy group in the mechanism suggested in Scheme 2 (Table 7, entry 5). This substituent effect became much less pronounced at 120 °C, although 2’-acetamino-4-methoxybiphenyl remained the most reactive substrate. When the biarylamide is substituted at C3, regardless of the electronic character of the C3 substituent, the carbazole was formed in about 10% yield over a 24 h period when the reaction was conducted at 90 °C (Table 7, entries 6–8).
Table 7.
Reactivity Studies at 90 °C
 | |||
|---|---|---|---|
| Entry | R | Temp (°C) |
Yield (%)a |
| 1 | H | 120 | 92 |
| 2 | 2-CF3 | 90 | 3 |
| 3 | H | 90 | 10 |
| 4 | 2-Me | 90 | 37 |
| 5 | 2-OMe | 90 | 73 |
| 6 | 3-CF3 | 90 | 7 |
| 7 | 3-Me | 90 | 14 |
| 8 | 3-OMe | 90 | 8 |
GC yield was determined relative to an internal standard.
Concerned by the discrepancy between our initial model and the data discussed above, we proposed two related catalytic pathways that could better explain the data (Scheme 3). Beginning with the formation of palladium amide 3, intramolecular cyclization could take place by different mechanisms to form two diastereomeric Pd species (5a and 5b). An insertion into the arene via a Heck-like process would form 5a, which could undergo a trans β–hydrogen elimination to provide carbazole 2.29 Alternatively, 3 could add to the arene via a Wacker-like process to provide 5b, which would then undergo a β–hydrogen elimination to provide carbazole 2. The reoxidation of the Pd(0) species to Pd(II) in the presence of Cu(OAc)2 or DMSO and oxygen would complete the catalytic cycle. To further probe these three possible pathways for the cyclization process, we undertook NMR studies of stoichiometric reactions, measurement of inter- and intramolecular kinetic isotope effects, and the examination of substituent effects on the rate of the reaction. We also note that attempts to modify the reaction conditions by the addition of acids or bases slowed down the cyclization. Despite all our efforts, however, no clear picture of the reaction mechanism emerged from these studies.
Scheme 3.

Two Alternative Pathways for Carbazole Synthesis
Target-oriented Synthesis
To demonstrate the efficiency and applicability of this method, we prepared three natural occurring carbazoles (mukonine,30 mukonidine and glycosinine) as shown in Scheme 4. Mukonidine and glycosinine were synthesized from a common acetylcarbazole intermediate 6a that was formed in 94% yield from biarylamide 5a using this method. Biarylamide 5a was obtained in 98% yield using a Suzuki-Miyaura cross coupling protocol recently developed in this lab.25 Demethylation of 6a followed by acid hydrolysis afforded mukonidine in 85% yield. Separately, reduction of the methyl ester and cleavage of the amide group of 6a followed by reoxidation of the resulting benzyl alcohol to an aldehyde provided glycosinine in 74% yield. The synthesis of mukonine followed a sequence similar to those of mukonidine and glycosinine. Biarylamide 5b was obtained in 95% yield using our protocol for a Suzuki-Miyaura cross coupling between phenylboronic acid and aryl iodide 7 (obtained in 3 steps from commercially available 4-amino-3-methoxybenzoic acid following published procedures31). Treatment of 5b with Pd(OAc)2, Cu(OAc)2, and oxygen in toluene followed by acid hydrolysis afford mukonine in 99% yield in two steps. In short, mukonidine and glycosinine were synthesized in 79% and 68% overall yield respectively in 4 steps from commercially available reagents, while mukonine was synthesized in 74% overall yield in 6 steps from commercially available reagents.
Scheme 4.

Synthesis of Three Natural Occurring Carbazole
Conclusion
The results presented in this paper describe the first applications of intramolecular conversion of an arene C-H bond to a C-N bond in the elaboration of synthetically useful molecules from commercially available and user-friendly reagents. This C-H functionalization strategy allows the assembly of carbazoles with a variety of substitution patterns and functional groups under relatively mild conditions. The utility of this method has been unambiguously demonstrated by the high efficiency displayed in the syntheses of mukonidine, glycosinine, and mukonidine. The observation that DMSO could be used in place of Cu(OAc)2 broadens the substrate scope of this reaction by increasing the number of compatible functional groups. For example, both electron-donating and electron-withdrawing groups are generally tolerated in the synthesis of these carbazoles at 120 °C. Even strong electron-withdrawing groups such as keto, ester, nitro, and cyano groups can all be incorporated into the carbazoles. Additionally, this method proves particularly effective for the synthesis of unsymmetrical carbazoles and is complementary to other syntheses of carbazole alkaloids. This feature differentiates it from known methods for the synthesis of carbazoles.19,32,33,34 For example, the introduction of substituents by traditional electrophilic aromatic substitution of 9H-carbazole is normally limited to 3 and 6 positions that are para to the NH moiety.12d,35 Our method allows efficient installation of substituents at positions ortho and para to the biaryl axis of the amide substrate and leads to carbazoles that are substituted at 2 and/or 4 positions. While the Pd-catalyzed cyclization of ortho-halo diarylamines to carbazoles generally favors 2-substituted carbazoles over 4-substituted ones,33,34 our method permits extremely efficient synthesis of 4-substituted carbazoles. Using this method, the carbazole cores substituted from C1 to C8 can be prepared. Although we have undergone a number of studies to probe the mechanism for the cyclization process, no clear picture about its detailed course has emerged from these studies.
Experimental Section
General Procedure for the Synthesis of Carbazoles in the Presence of anhydrous Cu(OAc)2 (Protocol A)
An oven-dried Schlenk tube (15 mL in volume) was cooled under vacuum and Ar. Biarylamide (0.2 mmol), Pd(OAc)2 (2.3 mg, 0.01 mmol, 5%), and powdered molecular sieves (40 mg, activated 3Å) were added under air. The tube was evacuated and refilled with Ar. Anhydrous Cu(OAc)2 (36.3 mg, 0.2 mmol) was loaded into the Schlenk tube in a nitrogen-filled glovebox. Under a positive Ar pressure, toluene (2 mL) was added via syringe. The reaction mixture was sonicated and degassed under a weak vacuum and refilled with O2 from the double manifold (this sequence was carried out three times). The sealed Schlenk tube was lowered into an oil bath at 120 °C and stirred for 12–24 h. After cooling to room temperature, distilled water (1 mL), ammonium hydroxide solution (3 mL, 30%), and EtOAc (2 mL) were added to the reaction mixture. The organic phase was separated, and the aqueous phase was further extracted with EtOAc (3 × 2 mL). The combined organic layers were dried over anhydrous Na2SO4 and filtered through Celite, and concentrated. The residue was purified by silica gel chromatography using a mixture of hexanes and EtOAc to provide the desired carbazole. A representative example is as follows.
9-Acetyl-2-acetylcarbazole
Following Protocol A (12 h), an off-white powder was obtained after silica gel chromatography (gradient; hexanes:EtOAc, 95:5 to 60:40), yielding 44 mg (87%, mp 110 °C). 1H NMR (500 MHz, CDCl3) d 8.91 (s, 1H), 8.22 (d, J = 8.4 Hz, 1H), 8.12–7.98 (m, 3H), 7.57 (t, J = 7.9 Hz, 1H), 7.44 (t, J = 7.5 Hz, 1H), 2.95 (s, 3H), 2.74 (s, 3H). 13C NMR (125 MHz, CDCl3) d 198.04, 170.31, 140.08, 138.70, 136.22, 130.46, 128.96, 125.61, 124.24, 124.19, 121.01, 119.84, 116.92, 116.50, 28.07, 27.19. IR (KBr plate, CDCl3) ν 3004, 1693, 1678, 1418, 1368, 1326, 1304, 1229, 1020, 749. Anal. Calcd. for C16H13NO2: C, 76.48; H, 5.21. Found: C, 76.30; H, 5.25.
General Procedure for the Synthesis of Carbazoles in Dimethyl Sulfoxide (DMSO) (Protocol C)
An oven-dried Schlenk tube (15 mL in volume) was cooled under vacuum or Ar. Biarylamide (0.2 mmol), Pd(OAc)2 (2.3 to 4.6 mg, 0.01 to 0.02 mmol, 5 to 10 %), and powdered molecular sieves (40 mg, activated 3Å) were added under air. The tube was evacuated and refilled with Ar. Under a positive Ar pressure, DMSO (2 mL) was added via syringe. The reaction mixture was sonicated and degassed under a weak vacuum and refilled with O2 from the double manifold (this sequence was carried out three times). The sealed Schlenk tube was lowered into an oil bath at 120 °C and stirred for 24 h. After cooling to room temperature, distilled water (5 mL) and EtOAc (3 mL) were added to the reaction mixture. The organic phase was separated, and the aqueous phase was further extracted with EtOAc (3 × 3 mL). The combined organic layers were dried over anhydrous Na2SO4, filtered through Celite, and concentrated. The residue was purified by silica gel chromatography using a mixture of hexanes and EtOAc to provide the desired carbazole. A representative example is as follows.
9-Acetyl-2-methylthiocarbazole
Following Protocol C (5% Pd(OAc)2, 24 h), a white powder was obtained after silica gel chromatography (gradient; hexanes:EtOAc, 95:5 to 90:10), yielding 43.0 mg (84%, mp 79 °C). 1H NMR (500 MHz, CDCl3) δ 8.27 (s, 1H), 8.09 (d, J = 8.4 Hz, 1H), 7.95 (d, J = 7.7 Hz, 1H), 7.89 (d, J = 8.2 Hz, 1H), 7.47 (t, J = 7.8 Hz, 1H), 7.39 (t, J = 7.5 Hz, 1H), 7.32 (dd, J = 8.1, 1.5 Hz, 1H), 2.89 (s, 3H), 2.61 (s, 3H). 13C NMR (125 MHz, CDCl3) δ 170.31, 139.57, 138.59, 138.34, 127.20, 126.53, 124.05, 123.97, 122.94, 120.03, 119.90, 116.11, 114.73, 28.02, 16.75. IR (KBr plate, CDCl3) ν 2919, 1693, 1596, 1435, 1411, 1367, 1325, 1303, 1272, 1213, 1189, 1020, 765, 747. Anal. Calcd. for C15H13NOS: C, 70.56; H, 5.13. Found: C, 70.40; H, 5.17.
Supplementary Material
Experimental procedures and spectral data for all new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
Figure 1.

Therapeutically Valuable Carbazole Alkaloids
ACKNOWLEDGMENT
We thank Johnson & Johnson (Focused Giving Grant), the National Institutes of Health (GM58160), and the National Cancer Institute (Cancer Training Grant CA 09112) for supporting this research. G.B. thanks the Deutscher Akademischer Austauschdienst (DAAD) for support through a postdoctoral fellowship. N.Z. thanks the National Institutes of Health for a postdoctoral fellowship (F32-GM074407).
REFERENCE
- 1.(a) Knölker H-J. Top. Curr. Chem. 2005;244:115–148. [Google Scholar]; (b) Knölker H-J, Reddy KR. Chem. Rev. 2002;102:4303–4427. doi: 10.1021/cr020059j. [DOI] [PubMed] [Google Scholar]
- 2.(a) Chakraborty DP. In: The Alkaloids: Chemistry and Pharmacology. Cordell GA, editor. Vol. 44. New York: Academic; 1993. pp. 257–364. [Google Scholar]; (b) Chakraborty DP, Roy S. In: Progress in the Chemistry of Organic Natural Products. Herz W, Kirby GW, Steglich W, Tamm Ch, editors. Vol. 57. New York: Springer-Verlag; 1991. pp. 72–152. [Google Scholar]; (c) Bhattacharyya P, Chakraborty DP. In: Progress in the Chemistry of Organic Natural Products. Herz W, Grisebach H, Kirby GW, Tamm Ch, editors. Vol. 52. New York: Springer-Verlag; 1987. pp. 159–209. [Google Scholar]; (d) Chakraborty DP. In: Progress in the Chemistry of Organic Natural Products. Herz W, Grisebach H, Kirby GW, editors. Vol. 34. New York: Springer-Verlag; 1977. pp. 299–371. [Google Scholar]
- 3.(a) Krahl MP, Jäger A, Krause T, Knölker H-J. Org. Biomol. Chem. 2006;4:3215–3219. doi: 10.1039/b607792g. [DOI] [PubMed] [Google Scholar]; (b) Meragelman KM, McKee TC, Boyd MR. J. Nat. Prod. 2000;63:427–428. doi: 10.1021/np990570g. [DOI] [PubMed] [Google Scholar]; (c) TePaske MR, Gloer JB, Wicklow DT, Dowd PF. J. Org. Chem. 1989;54:4743–4746. [Google Scholar]; (d) Kondo S, Katayama M, Marumo S. J. Antibiot. 1986;39:727–730. doi: 10.7164/antibiotics.39.727. [DOI] [PubMed] [Google Scholar]; (e) Chakraborty DP, Bose PK. Experientia. 1965;21:1340. doi: 10.1007/BF02144703. [DOI] [PubMed] [Google Scholar]
- 4.Sakano K-I, Ishimaru K, Nakamura S. J. Antibiot. 1980;33:683–689. doi: 10.7164/antibiotics.33.683. [DOI] [PubMed] [Google Scholar]
- 5.(a) Crich D, Rumthao S. Tetrahedron. 2004;60:1513–1516. [Google Scholar]; (b) Hook DJ, Yacobucci JJ, O’Connor S, Lee M, Kerns E, Krishnan B, Matson J, Hesler G. J. Antibiot. 1990;43:1347–1348. doi: 10.7164/antibiotics.43.1347. [DOI] [PubMed] [Google Scholar]
- 6.(a) Guthrie RW, Brossi A, Mennona FA, Mullin JG, Kierstead RW, Grunberg E. J. Med. Chem. 1975;18:755–760. doi: 10.1021/jm00241a023. [DOI] [PubMed] [Google Scholar]; (b) Cranwell PA, Saxton JE. J. Chem. Soc. 1962:3482–3487. [Google Scholar]; (c) Woodward RB, Iacobucci GA, Hochstein FA. J. Am. Chem. Soc. 1959;81:4434–4435. [Google Scholar]
- 7.(a) Cheng H-Y, Randall CS, Holl WW, Constantinides PP, Yue T-L, Feuerstein GZ. Biochim. Biophys. Acta. 1996;1284:20–28. doi: 10.1016/0005-2736(96)00097-1. [DOI] [PubMed] [Google Scholar]; (b) Yue T-L, Cheng H-Y, Lysko PG, McKenna PJ, Feuerstein R, Gu J-L, Lysko KA, Davis LL, Feuerstein G. J. Pharmacol. Exp. Ther. 1992;263:92–98. [PubMed] [Google Scholar]
- 8.(a) Dubois EA, van den Bos JC, Doornbos T, van Doremalen PAPM, Somsen GA, Vekemans JAJM, Janssen AGM, Batink HD, Boer GJ, Pfaffendorf M, van Royen EA, van Zwieten PA. J. Med. Chem. 1996;39:3256–3262. doi: 10.1021/jm960122v. [DOI] [PubMed] [Google Scholar]; (b) Costin B, O’Donnell SR, Wanstall JC. J. Pharm. Pharmacol. 1983;35:590–592. doi: 10.1111/j.2042-7158.1983.tb04339.x. [DOI] [PubMed] [Google Scholar]; (c) Cohen ML, Ruffolo RR, Jr, Wiley KS. J. Pharmacol. Exp. Ther. 1980;215:325–331. [PubMed] [Google Scholar]; (d) Innis RB, Corrêa FMA, Snyder SH. Life Sci. 1979;24:2255–2264. doi: 10.1016/0024-3205(79)90102-4. [DOI] [PubMed] [Google Scholar]
- 9.(a) Czeskis BA, Wheeler WJ. J. Label. Compd. Radiopharm. 2005;48:407–419. [Google Scholar]; (b) Nakajima Y, Hamashima H, Washizuka K-I, Tomishima Y, Ohtake H, Imamura E, Miura T, Kayakiri H, Kato M. Bioorg. Med. Chem. Lett. 2005;15:251–254. doi: 10.1016/j.bmcl.2004.11.001. [DOI] [PubMed] [Google Scholar]; (c) Kumar R, Ramachandran U, Srinivasan K, Ramarao P, Raichur S, Chakrabarti R. Bioorg. Med. Chem. 2005;13:4279–4290. doi: 10.1016/j.bmc.2005.04.018. [DOI] [PubMed] [Google Scholar]; (d) Sum F-W, Wong V, Han S, Largis E, Mulvey R, Tillett J. Bioorg. Med. Chem. Lett. 2003;13:2191–2194. doi: 10.1016/s0960-894x(03)00387-1. [DOI] [PubMed] [Google Scholar]
- 10.(a) Morin J-F, Leclerc M, Adès D, Siove A. Macromol. Rapid Commun. 2005;26:761–778. [Google Scholar]; (b) Ostroverkhova O, Moerner WE. Chem. Rev. 2004;104:3267–3314. doi: 10.1021/cr960055c. [DOI] [PubMed] [Google Scholar]; (c) Grazulevicius JV, Strohriegl P, Pielichowski J, Pielchowski K. Prog. Polym. Sci. 2003;28:1297–1353. [Google Scholar]; (d) Zhang Y, Wada T, Sasabe H. J. Mater. Chem. 1998;8:809–828. [Google Scholar]; (e) Meerholz K, Volodin BL, Sandalphon, Kippelen B, Peyghambarian N. Nature. 1994;371:497–500. [Google Scholar]
- 11.Friend RH, Gymer RW, Holmes AB, Burroughes JH, Marks RN, Taliani C, Bradley DDC, Dos Santos DA, Brédas JL, Lögdlund M, Salaneck WR. Nature. 1999;397:121–128. [Google Scholar]
- 12.(a) Müllen K, Scherf U, editors. Organic Light Emitting Devices: Synthesis, Properties, and Applications. Weinheim: Wiley-VCH; 2006. [Google Scholar]; (b) Grigalevicius S. Synth. Met. 2006;156:1–12. [Google Scholar]; (c) Brunner K, van Dijken A, Börner H, Bastiaansen JJAM, Kiggen NMM, Langeveld BMW. J. Am. Chem. Soc. 2004;126:6035–6042. doi: 10.1021/ja049883a. [DOI] [PubMed] [Google Scholar]; (d) Thomas KRJ, Lin JT, Tao Y-T, Ko C-W. J. Am. Chem. Soc. 2001;123:9404–9411. doi: 10.1021/ja010819s. [DOI] [PubMed] [Google Scholar]; (e) Baldo MA, O’Brien DF, You Y, Shoustikov A, Sibley S, Thompson ME, Forrest SR. Nature. 1998;395:151–154. [Google Scholar]
- 13.(a) Tsai M-H, Hong Y-H, Chang C-H, Su H-C, Wu C-C, Matoliukstyte A, Simokaitiene J, Grigalevicius S, Grazulevicius JV, Hsu C-P. Adv. Mater. 2007;19:862–866. [Google Scholar]; (b) Yeh S-J, Wu M-F, Chen C-T, Song Y-H, Chi Y, Ho M-H, Hsu S-F, Chen CH. Adv. Mater. 2005;17:285–289. [Google Scholar]; (c) Tokito S, Iijima T, Suzuri Y, Kita H, Tsuzuki T, Sato F. Appl. Phys. Lett. 2003;83:569–571. [Google Scholar]; (d) Holmes RJ, Forrest SR, Tung Y-J, Kwong RC, Brown JJ, Garon S, Thompson ME. Appl. Phys. Lett. 2003;82:2422–2424. [Google Scholar]; (e) Adachi C, Kwong RC, Djurovich P, Adamovich V, Baldo MA, Thompson ME, Forrest SR. Appl. Phys. Lett. 2001;79:2082–2084. [Google Scholar]
- 14.(a) Chen C-T. Chem. Mater. 2004;16:4389–4400. [Google Scholar]; (b) Lamansky S, Djurovich P, Murphy D, Abdel-Razzaq F, Lee H-E, Adachi C, Burrows PE, Forrest SR, Thompson ME. J. Am. Chem. Soc. 2001;123:4304–4312. doi: 10.1021/ja003693s. [DOI] [PubMed] [Google Scholar]; (c) Adachi C, Baldo MA, Forrest SR, Lamansky S, Thompson ME, Kwong RC. Appl. Phys. Lett. 2001;78:1622–1624. [Google Scholar]; (d) Baldo MA, Lamansky S, Burrows PE, Thompson ME, Forrest SR. Appl. Phys. Lett. 1999;75:4–6. [Google Scholar]
- 15.For recent reviews related to C-H amination, see: Dick AR, Sanford MS. Tetrahedron. 2006;62:2439–2463. ; (b) Davies HML, Long MS. Angew. Chem. Int. Ed. 2005;44:2–4. [Google Scholar]; (c) Sigman MS, Schultz MJ. Org. Biomol. Chem. 2004;2:2551–2554. doi: 10.1039/B409127M. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Stahl S. Angew. Chem. Int. Ed. 2004;43:3400–3420. doi: 10.1002/anie.200300630. [DOI] [PubMed] [Google Scholar]; (e) Müller P, Fruit C. Chem. Rev. 2003;103:2905–2919. doi: 10.1021/cr020043t. [DOI] [PubMed] [Google Scholar]
- 16.For recent reviews of other C-H functionalization or activation processes, see: Godula K, Sames D. Science. 2006;312:67–72. doi: 10.1126/science.1114731. ; (b) Sigman MS, Jensen DR. Acc. Chem. Res. 2006;39:221–229. doi: 10.1021/ar040243m. [DOI] [PubMed] [Google Scholar]; (b) Kakiuchi F, Chatani N. Adv. Synth. Catal. 2003;345:1077–1101. [Google Scholar]; (c) Kakiuchi F, Murai S. Acc. Chem. Res. 2002;35:826–834. doi: 10.1021/ar960318p. [DOI] [PubMed] [Google Scholar]; (d) Ritleng V, Sirlin C, Pfeffer M. Chem. Rev. 2002;102:1731–1769. doi: 10.1021/cr0104330. [DOI] [PubMed] [Google Scholar]; (e) Jia C, Kitamura T, Fujiwara Y. Acc. Chem. Res. 2001;34:633–639. doi: 10.1021/ar000209h. [DOI] [PubMed] [Google Scholar]; (f) Bringmann G, Menche D. Acc. Chem. Res. 2001;34:615–624. doi: 10.1021/ar000106z. [DOI] [PubMed] [Google Scholar]; (g) Dyker G. Angew. Chem. Int. Ed. 1999;38:1698–1712. doi: 10.1002/(SICI)1521-3773(19990614)38:12<1698::AID-ANIE1698>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]; (h) Shilov AE, Shul’pin GB. Chem. Rev. 1997;97:2879–2932. doi: 10.1021/cr9411886. [DOI] [PubMed] [Google Scholar]; (i) Bringmann G, Walter R, Weirich R. Angew. Chem. Int. Ed. Engl. 1990;29:977–991. [Google Scholar]
- 17.(a) Espino CG, Fiori KW, Kim M, Du Bois J. J. Am. Chem. Soc. 2004;126:15378–15379. doi: 10.1021/ja0446294. [DOI] [PubMed] [Google Scholar]; (b) Fiori KW, Fleming JJ, Du Bois J. Angew. Chem. Int. Ed. 2004;43:4349–4352. doi: 10.1002/anie.200460791. [DOI] [PubMed] [Google Scholar]; (c) Fleming JJ, Fiori KW, Du Bois J. J. Am. Chem. Soc. 2003;125:2028–2029. doi: 10.1021/ja028916o. [DOI] [PubMed] [Google Scholar]; (d) Wehn PM, Du Bois J. J. Am. Chem. Soc. 2002;124:12950–12951. doi: 10.1021/ja028139s. [DOI] [PubMed] [Google Scholar]; (e) Espino CG, Du Bois J. Angew. Chem. Int. Ed. 2001;40:598–600. [PubMed] [Google Scholar]; (f) Espino CG, Wehn PM, Chow J, Du Bois J. J. Am. Chem. Soc. 2001;123:6935–6936. [Google Scholar]
- 18.(a) Liang J-L, Yuan S-X, Huang J-S, Che C-M. J. Org. Chem. 2004;69:3610–3619. doi: 10.1021/jo0358877. [DOI] [PubMed] [Google Scholar]; (b) Liang J-L, Yuan S-X, Huang J-S, Yu W-Y, Che C-M. Angew. Chem. Int. Ed. 2002;41:3465–3468. doi: 10.1002/1521-3773(20020916)41:18<3465::AID-ANIE3465>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]; (c) Reed AA, White MC. J. Am. Chem. Soc. 2008;130:3316–3318. doi: 10.1021/ja710206u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.(a) Kalyani D, Deprez NR, Desai LV, Sanford MS. J. Am. Chem. Soc. 2005;127:7330–7331. doi: 10.1021/ja051402f. [DOI] [PubMed] [Google Scholar]; (b) Daugulis O, Zaitsev VG. Angew. Chem. Int. Ed. 2005;44:4046–4048. doi: 10.1002/anie.200500589. [DOI] [PubMed] [Google Scholar]; (c) Kakiuchi F, Matsuura Y, Kan S, Chatani N. J. Am. Chem. Soc. 2005;127:5936–5945. doi: 10.1021/ja043334n. [DOI] [PubMed] [Google Scholar]; (d) Cuny G, Bois-Choussy M, Zhu J. J. Am. Chem. Soc. 2004;126:14475–14484. doi: 10.1021/ja047472o. [DOI] [PubMed] [Google Scholar]; (e) Campeau L-C, Parisien M, Leblanc M, Fagnou K. J. Am. Chem. Soc. 2004;126:9186–9187. doi: 10.1021/ja049017y. [DOI] [PubMed] [Google Scholar]; (f) Huang Q, Fazio A, Dai G, Campo MA, Larock RC. J. Am. Chem. Soc. 2004;126:7460–7461. doi: 10.1021/ja047980y. [DOI] [PubMed] [Google Scholar]; (g) Bedford RB, Coles SJ, Hursthouse MB, Limmert ME. Angew. Chem., Int. Ed. 2003;42:112–114. doi: 10.1002/anie.200390037. [DOI] [PubMed] [Google Scholar]; (h) Terao Y, Wakui H, Satoh T, Miura M, Nomura M. J. Am. Chem. Soc. 2001;123:10407–10408. doi: 10.1021/ja016914i. [DOI] [PubMed] [Google Scholar]; (i) Ames DE, Opalko A. Tetrahedron. 1984;40:1919–1925. [Google Scholar]; (j) Åkermark B, Eberson L, Jonsson E, Pettersson E. J. Org. Chem. 1975;40:1365–1367. [Google Scholar]
- 20.For recent reviews of Pd- and Cu-catalyzed carbon-heteroatom bond formation processes, see: Corbet J-P, Mignani G. Chem. Rev. 2006;106:2651–2710. doi: 10.1021/cr0505268. ; (b) Wolfe JP, Thomas JS. Curr. Org. Chem. 2005;9:625–655. [Google Scholar]; (c) Jiang L, Buchwald SL. In: Metal-Catalyzed Cross-Coupling Reactions. 2nd ed. de Meijere A, Diederich F, editors. Vol. 2. Weinheim: Wiley-VCH; 2004. pp. 699–760. [Google Scholar]; (d) Beletskaya IP, Cheprakov AV. Coord. Chem. Rev. 2004;248:2337–2364. [Google Scholar]; (e) Ley SV, Thomas AW. Angew. Chem., Int. Ed. 2003;42:5400–5449. doi: 10.1002/anie.200300594. [DOI] [PubMed] [Google Scholar]; (f) Hartwig JF. In: Handbook of Organopalladium Chemistry for Organic Synthesis. Negishi E-I, de Meijere A, editors. Vol. 1. New York: Wiley- Interscience; 2002. pp. 1051–1096. [Google Scholar]
- 21.Tsang WCP, Zheng N, Buchwald SL. J. Am. Chem. Soc. 2005;127:14560–14561. doi: 10.1021/ja055353i. [DOI] [PubMed] [Google Scholar]
- 22.Li B-J, Tian S-L, Fang Z, Shi Z-J. Angew. Chem. Int. Ed. 2008;47:1115–1118. doi: 10.1002/anie.200704092. [DOI] [PubMed] [Google Scholar]
- 23.(a) Thu HY, Yu W-Y, Che C-M. J. Am. Chem. Soc. 2006;128:9048–9049. doi: 10.1021/ja062856v. [DOI] [PubMed] [Google Scholar]; (b) Yip K-T, Yang M, Law K-L, Zhu N-Y, Yang D. J. Am. Chem. Soc. 2006;128:3130–3131. doi: 10.1021/ja060291x. [DOI] [PubMed] [Google Scholar]; (c) Dick AR, Hull KL, Sanford MS. J. Am. Chem. Soc. 2004;126:2300–2301. doi: 10.1021/ja031543m. [DOI] [PubMed] [Google Scholar]; (d) Boele MDK, van Strijdonck GPF, de Vries AHM, Kamer PCJ, de Vries JG, van Leeuwen PWNM. J. Am. Chem. Soc. 2002;124:1586–1587. doi: 10.1021/ja0176907. [DOI] [PubMed] [Google Scholar]
- 24.(a) Steinhoff BA, Fix SR, Stahl SS. J. Am. Chem. Soc. 2002;124:766–767. doi: 10.1021/ja016806w. [DOI] [PubMed] [Google Scholar]; (b) Chen MS, White MC. J. Am. Chem. Soc. 2004;126:1346–1347. doi: 10.1021/ja039107n. [DOI] [PubMed] [Google Scholar]
- 25.(a) Barder TE, Walker SD, Martinelli JR, Buchwald SL. J. Am. Chem. Soc. 2005;127:4685–4696. doi: 10.1021/ja042491j. [DOI] [PubMed] [Google Scholar]; (b) Walker SD, Barder TE, Martinelli JR, Buchwald SL. Angew. Chem. Int. Ed. 2004;43:1871–1876. doi: 10.1002/anie.200353615. [DOI] [PubMed] [Google Scholar]
- 26.Krahl MP, Jäger A, Krause T, Knölker H-J. Org. Biomol. Chem. 2006;4:3215–3219. doi: 10.1039/b607792g. [DOI] [PubMed] [Google Scholar]
- 27.Meragelman KM, McKee TC, Boyd MR. J. Nat. Prod. 2000;63:427–428. doi: 10.1021/np990570g. [DOI] [PubMed] [Google Scholar]
- 28.Inamoto K, Saito T, Katsuno M, Sakamoto T, Hiroya K. Org. Lett. 2007;9:2931–2934. doi: 10.1021/ol0711117. [DOI] [PubMed] [Google Scholar]
- 29.For one selected example of intramolecular Heck reaction via a trans β–hydrogen elimination, see: Lautens M, Fang Y-Q. Org. Lett. 2003;5:3679–3682. doi: 10.1021/ol035354k.
- 30.Liégault B, Lee D, Huestis MP, Stuart DR, Fagnou K. J. Org. Chem. 2008;73:5022–5028. doi: 10.1021/jo800596m. [DOI] [PubMed] [Google Scholar]
- 31.Ezquerra J, Pedregal C, Lamas C. J. Org. Chem. 1996;61:5804–5812. [Google Scholar]
- 32.Campeau L-C, Parisien M, Jean A, Fagnou K. J. Am. Chem. Soc. 2006;128:581–590. doi: 10.1021/ja055819x. [DOI] [PubMed] [Google Scholar]
- 33.Liu Z, Larock RC. Tetrahedron. 2007;63:347–355. doi: 10.1016/j.tet.2006.10.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bedford RB, Betham M. J. Org. Chem. 2006;71:9403–9410. doi: 10.1021/jo061749g. [DOI] [PubMed] [Google Scholar]
- 35.(a) Zhu Z, Moore JS. J. Org. Chem. 2000;65:116–123. doi: 10.1021/jo991167h. [DOI] [PubMed] [Google Scholar]; (b) Zhu Z, Moore JS. Macromolecules. 2000;33:801–807. [Google Scholar]; (c) Buu-Hoï NP, Hoán N. J. Org. Chem. 1951;16:1327–1332. [Google Scholar]; (d) Buu-Hoï NP, Royer R. J. Org. Chem. 1951;16:1198–1205. [Google Scholar]; (e) Tucker SH. J. Chem. Soc. 1926:546–553. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Experimental procedures and spectral data for all new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.