

Abstract
Trivalent arsenite (As3+) is a known human carcinogen that is also capable of inducing apoptotic cell death. Increased production of reactive oxygen species is thought to contribute to both the carcinogenic and cytotoxic effects of As3+. Glutathione (GSH) constitutes a vital cellular defense mechanism against oxidative stress. The rate-limiting enzyme in GSH biosynthesis is glutamate-cysteine ligase (GCL), a heterodimeric holoenzyme composed of a catalytic (GCLC) and a modifier (GCLM) subunit. In this study, we demonstrate that As3+ coordinately upregulates Gclc and Gclm mRNA levels in a murine hepatocyte cell line resulting in increased GCL subunit protein expression, holoenzyme formation and activity. As3+ increased the rate of transcription of both the Gclm and Gclc genes and induced the post-transcriptional stabilization of Gclm mRNA. The antioxidant N-acetylcysteine abolished As3+-induced Gclc expression and attenuated induction of Gclm. As3+ induction of Gclc and Gclm was also differentially regulated by the MAPK signaling pathways and occurred independent of the Nrf1/2 transcription factors. These findings demonstrate that distinct transcriptional and post-transcriptional mechanisms mediate the coordinate induction of the Gclc and Gclm subunits of GCL in response to As3+ and highlight the potential importance of the GSH antioxidant defense system in regulating As3+-induced responses in hepatocytes.
Keywords: arsenite, arsenic, glutamate cysteine ligase, GCL, GCLC, GCLM, glutathione, GSH, hepatocyte, Nrf2, gene transcription
INTRODUCTION
Inorganic arsenic, as trivalent arsenite (As3+) or pentavalent arsenate (As5+), is a known human carcinogen associated with the development of cancers of the skin, bladder, lung, and kidney [1]. Chronic arsenic exposure is also associated with numerous liver pathologies, including hepatocellular carcinoma (HCC), liver angiosarcoma, hepatomegaly, fibrosis, and cirrhosis [2, 3]. As3+ exhibits clear dose-dependent effects, with low levels promoting cell proliferation and transformation and higher concentrations inducing apoptosis [4]. In this regard, chronic exposure to sub-toxic doses of As3+ induces malignant transformation in multiple in vitro cell models, including rat liver epithelial cells [5], human bladder urothelial cells [6], human prostate cells [7], human keratinocytes [8, 9], and mouse epidermal cells [10]. In contrast, acute exposure to high concentrations of As3+ induces apoptotic cell death, which provides the molecular basis for the therapeutic use of arsenic trioxide (ATO) in the treatment of acute promyelocytic leukemia [11]. While the molecular mechanisms mediating As3+-induced transformation and apoptosis have not been fully elucidated, increased production of reactive oxygen species (ROS) and aberrant gene expression have been implicated in both responses [11–13]. Interestingly, As3+-induced transformation in vitro is associated with the development of cellular resistance to acute As3+ toxicity [8, 14–19], which is usually accompanied by an up-regulation of the glutathione (GSH) antioxidant defense system [8, 15, 19, 20]. GSH plays an important role in arsenic detoxication and intracellular GSH levels can dictate cellular sensitivity to As3+-induced apoptosis [21]. Cells deficient in GSH biosynthesis or depleted of cellular GSH levels exhibit dramatically increased sensitivity to As3+-induced apoptosis [11, 22, 23], while enhanced cellular GSH levels promote resistance to As3+-induced apoptosis [11, 21]. Both acute and chronic As3+ exposure up-regulate various antioxidant enzymes that likely serve a cytoprotective adaptive mechanism against As3+-induced oxidative stress, including enzymes of the GSH antioxidant defense system.
Mammalian cells possess a number of antioxidant defense mechanisms to counteract the deleterious effects of oxidative stress associated with exposure to environmental toxicants. GSH is the most abundant non-protein thiol in the cell and plays an important role in cellular defense against oxidative stress. The antioxidant and cytoprotective functions of GSH are derivative of its ability to directly react with reactive electrophiles, act as a cofactor in the reduction of both hydrogen peroxide (H2O2) and lipid peroxides mediated by glutathione peroxidases (GPx), and conjugation reactions mediated by glutathione S-transferases (GST) [24, 25]. Non-enzymatic and GPx-mediated reduction reactions lead to the oxidation of GSH and formation of glutathione disulfide (GSSG), which is readily reduced by glutathione reductase (GR) to regenerate GSH [24]. In contrast, GST-mediated conjugation reactions usually result in a net loss of GSH due to multidrug resistance-associated protein (MRP)- and/or organic anion transporting polypeptide (OATP)-mediated export of the GSH-conjugate from the cell [26]. Failure to adequately replenish these depleted cellular GSH stores quickly compromises cellular redox homeostasis and cell viability. Most cells do not import significant quantities of GSH, highlighting the importance of de novo GSH biosynthesis in maintaining intracellular GSH levels during periods of GSH deficiency.
GSH is synthesized by two sequential ATP-dependent reactions catalyzed by glutamate cysteine ligase (GCL) and glutathione synthetase [24]. GCL is the first and rate-limiting step in GSH biosynthesis [24]. GCL is a heterodimeric holoenzyme consisting of a catalytic subunit (GCLC, 73 kDa), which contributes all the enzymatic activity and contains all the substrate binding sites of GCL, and a modifier subunit (GCLM, 31 kDa), which modulates GCLC activity and affinity for substrates and inhibitors [24, 27]. GCLM increases the Vmax and Kcat of GCLC, lowers the Km for glutamate and ATP and increases the Ki for GSH feedback regulation of GCLC [24, 27]. Thus, while GCLM does not retain any catalytic activity, GCL holoenzyme formation is essential for optimal GCLC enzymatic activity under physiological concentrations of glutamate and GSH.
GSH biosynthesis can be rapidly regulated by increased expression of the GCL subunits via transcriptional, post-transcriptional, and/or translational control mechanisms or post-translational modification of pre-existing GCL protein [27, 28]. Both GCL subunits are highly inducible and the relative levels of the GCL subunits are a major determinant of cellular GCL activity and GSH biosynthetic capacity [28]. In addition to inducing glutathione reductase and glutathione S-transferase expression and activity, acute exposure to subtoxic concentrations of As3+ induces Gclc gene expression and enhances GCL enzymatic activity in human keratinocytes and rat lung epithelial cells [29–31]. However, the molecular mechanisms mediating these responses are not known and it is not clear whether As3+ coordinately induces Gclm expression. Analysis of the Gclc and Gclm promoters reveal the presence of multiple regulatory elements that have been implicated in their inducible expression, including consensus AP-1, AP-2, ARE, SP1, and NF-κB response elements [28, 32, 33]. Nrf1, Nrf2 and p45-NFE2 are members of the Cap-n-collar bZIP transcription factor family that play an important role in the expression of phase II detoxication and antioxidative enzymes [34]. Nrf1 and Nrf2 are ubiquitously expressed, while p45-NFE2 expression is restricted to hematopoietic cells [34]. Nrf1 and Nrf2 are central mediators in the constitutive and inducible expression of the Gcl subunits in response to oxidative stress through activation of antioxidant response elements (ARE) [35, 36]. Additional transcription factors have also been implicated in mediating the inducible expression of Gclc and Gclm, including members of the Jun, Fos, ATF-2, MTF-1 and CREB family of proteins [32]. Importantly, As3+ activates Nrf1/2 as well as the MAPK pathways that activate these transcription factors and regulate Gclc and Gclm gene expression [9, 30, 37–41].
In this study, we examined the molecular mechanisms and signal transduction pathways regulating As3+-induced GCL subunit expression in the TAMH murine hepatocyte cell line. While As3+ coordinately induced Gclc and Gclm gene expression, distinct transcriptional and post-transcriptional regulatory mechanisms mediated these responses. Surprisingly, while Nrf1 and Nrf2 are known to regulate both constitutive and inducible Gclc and Gclm expression, neither was required for As3+-induced Gclc or Gclm expression. Rather, our studies indicate that the p38 MAPK and ERK pathway play a prominent role in mediating As3+-induced Gcl subunit gene expression. These transcriptional events are also functionally relevant as As3+ increased GCLC and GCLM protein expression and GCL enzymatic activity. In aggregate, these studies identify many of the transcriptional and post-transcriptional mechanisms and signal transduction pathways that regulate As3+-induced Gcl subunit gene expression, GCL activity and GSH biosynthetic capacity in murine hepatocytes.
MATERIALS AND METHODS
Reagents
NaAsO2, Na2HAsO4, N-acetylcysteine (NAC), cycloheximide (CHX), buthionine sulfoximine (BSO), and tert-butylhydroquinone (tBHQ) were purchased from Sigma Chemical Co. (St. Louis, MO). Actinomycin D (ActD) was purchased from Calbiochem (La Jolla, CA). SB202190, SP600125, PD98059, and the JNK inhibitor peptide (JNKi) were purchased from Biomol (Plymouth Meeting, PA). All reagents were prepared in either sterile phosphate-buffered saline (PBS) or DMSO, except NAC, which was dissolved in media and the pH adjusted to 7.4 prior to use.
Cell culture and treatments
The murine TAMH hepatocyte cell line is an immortalized, transformed cell line derived from hepatocytes isolated from a TGF-α transgenic mouse and retains a well-differentiated hepatocytic phenotype [42]. TAMH cells were maintained in serum-free DMEM/F12 medium (Life Technologies, Grand Island, NY) supplemented with 5 μg/ml insulin, 5 μg/ml transferrin, 5 ng/ml selenium (ITS premix, Collaborative Biomedical Products, Bedford, MA), 10 mM nicotinamide, 0.1 μM dexamethasone, and 50 μg/ml gentamicin. Mouse embryo fibroblast (MEF) cell lines derived from wild-type mice and mice lacking Nrf1 or Nrf2 have been described elsewhere [43, 44]. MEF cells were maintained in DMEM medium supplemented with 15% fetal bovine serum, 1% penicillin/streptomycin, 2 mM glutamine, 0.1 mM non-essential amino acids and 150 μM β-mercaptoethanol. Cells were cultured at 37°C in a humidified 6% CO2 atmosphere and all experiments were performed at 90–100% confluency. In some experiments, cells were preincubated with various agents for 1 h prior to treatment with As3+. For mRNA half-life studies, cells were cotreated with 500 ng/ml ActD and 10 μM As3+.
RNA analysis
Cells were seeded at a density of 5 × 106 in 100 mm plates. Total RNA was isolated from cells utilizing the RNeasy Mini Kit (Qiagen Inc., Santa Clarita, CA). For Northern blot analysis, RNA (10–20 μg) was resolved on formaldehyde gels and transferred to GeneScreen Plus membranes (Dupont-New England Nuclear, Boston, MA). GCLC, GCLM, and GAPDH cDNA probes were generated as previously described [45] and radiolabeled with [α-32P]dCTP (3000 Ci/mmol; NEN) using the Random Prime Kit (Roche Molecular Biochemicals, Indianapolis, IN). Relative levels of Gclc, Gclm, and Gapdh mRNA were determined using a Storm Phosphoimager (Molecular Dynamics, Sunnyvale, CA). Relative Gclc and Gclm mRNA levels were normalized to Gapdh and presented as fold increase over untreated control. For real-time RT-PCR, total RNA (2 ug) was subjected to reverse transcription followed by real-time quantitative PCR for Gclc and β-Actin as previously described [46]. Relative Gclc mRNA levels were normalized to β-Actin and presented as fold increase over untreated control.
Nuclear run-on analysis
Cells from 150 mm plates (~1 × 107 cells) were washed once with ice-cold Solution I (10 mM Tris, pH 7.4, 150 mM KCl, 4 mM MgOAc), and then incubated in Solution I at 4°C for 5 min. Cells were then scraped and pelleted at 300 × g. Cells were lysed by gentle vortexing in Solution II (Solution I + 0.5% Nonidet P-40) for 10 min on ice. Nuclei were pelleted for 10 min at 500 × g through a 0.6 M sucrose gradient, resuspended in 100 mu;L of Solution III (40% glycerol, 50 mM MgCl2, 0.1 mM EDTA) and stored in liquid nitrogen. Thawed nuclei were mixed with 45 μL of reaction buffer (1 mM each of ATP, GTP, CTP, 100 μCi of [α-32P]UTP (3000 Ci/mmol; NEN) and incubated at 30°C for 30 min with shaking. The reaction was stopped by the addition of 800 μL of TRIzol (Life Technologies Inc., Carlsbad, CA) and RNA was isolated according to the manufacturer’s protocol. Purified 32P-labeled RNA was hybridized to Hybond nylon membrane (Amersham Biosciences, Piscataway, NJ) containing 5 μg of linearized Gclm, Gclc, and β-Actin cDNA. Membranes were hybridized at 42°C for 24 h and then washed in 2X SSC at 42°C for 30 min and in 2X SSC/0.5% SDS at 42°C for 30 min. Band densities were quantitated using a Storm Phosphoimager and the relative levels of Gclc and Gclm were normalized to β-Actin. The normalized levels of Gclc and Gclm transcribed in vitro, which are proportional to relative transcriptional rates, were compared independently and presented as fold increase over their respective untreated control values.
Western blot analysis and JNK assay
Cells were seeded at a density of 2 × 106 in 60 mm plates. Cells were lysed by either a brief sonication on ice in TES/SB buffer as described previously [47], or incubated on ice with TX lysis buffer (20 mM HEPES pH 7.4, 2 mM ETDA, 50 mM β-glycerolphosphate, 1 mM sodium orthovanadate, 10% glycerol, 1% Triton X-100, 1X protease inhibitors). Lysates were clarified by centrifugation and equal amounts of soluble protein (20–25 μg) were resolved on 10% or 12% SDS-polyacrylamide gels and transferred to PVDF membranes (Millipore, Bedford, MA). For analysis of GCL holoenzyme formation whole cell extracts were prepared in the absence of reducing equivalents and were resolved on native 10% PAGE gels in the absence of SDS. Samples for these studies were not boiled prior to gel loading and the gels were resolved in Tris/Glycine buffer at 4°C prior to transfer to PVDF membranes. Membranes were blocked in Tris-buffered saline/0.1% Tween-20 (TBST) containing 5% non-fat milk, prior to incubation with primary antibody in TBST containing 0.5% milk. Membranes were probed for GCLC, GCLM [45, 47], βActin (Sigma), JNK, p38 MAPK, ERK2 (Santa Cruz Biotechnology, Santa Cruz, CA), phospho-p38 MAPK, phospho-JNK and phospho-Erk (Cell Signaling, Beverly, MA). Anti-mouse-HRP and anti-rabbit HRP secondary antibodies (Amersham Biosciences) were used at 1:5000 in TBST containing 0.5% milk. The immunocomplex was visualized by chemoluminescence (Dupont-New England Nuclear, Boston, MA) and quantitated using NIH image 6.1 (NIH, Rockville Pike Bethesda, MD). The immunoreactive bands shown in Figures 9D and 9F were identified as monomeric GCLC and/or GCL holoenzyme based on their co-migration with recombinant murine GCLC and GCL holoenzyme protein [48]. JNK activity was measured as previously described [49].
Figure 9. As3+ increases GCL subunit protein levels, GCL activity and holoenzyme formation.
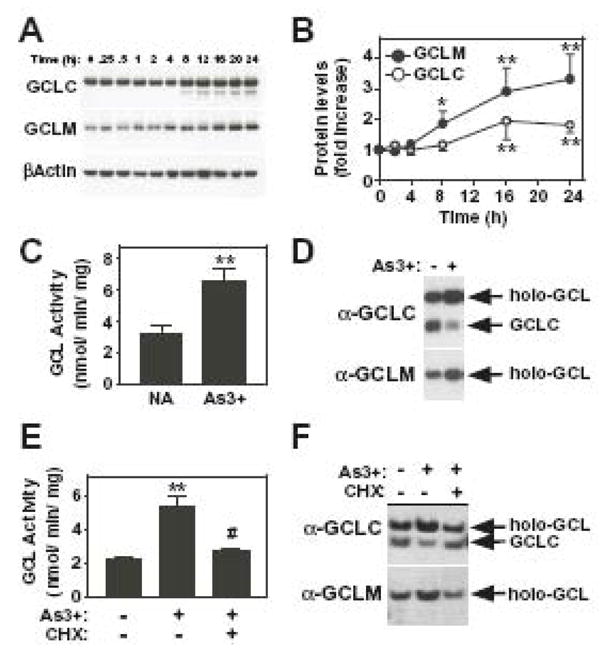
(A and B) TAMH cells were treated with As3+ (10 μM) for the time periods indicated. (A) GCLC, GCLM, and βActin protein expression were analyzed by denaturing SDS-PAGE and immunoblotting as described in the Methods section. (B) Graphical presentation of the studies described in (A). GCLC and GCLM protein expression were normalized to βActin protein levels and the data presented are averages +/− SEM of at least four experiments. (C–F) TAMH cells were preincubated in the absence or presence of cycloheximide (CHX; 1.0 μg/ml) for 1 h prior to treatment with As3+ (10 μM) for 16 h as indicated. (C and E) Cells were harvested and extracts assayed for GCL enzymatic activity as described in the Methods section. The graphical data presented are averages +/− SEM of 3–7 experiments. (D and F) GCL holoenzyme formation was analyzed by native gel electrophoresis and immunoblotting for GCLC or GCLM as described in the Methods section. *p < 0.05 and **p < 0.01, indicates a significant difference in protein expression or activity compared to untreated control. #p < 0.01, indicates a significant difference in GCL activity compared to As3+ alone.
Glutathione and GCL activity assays
Total glutathione content (GSH + GSSG) was determined by a modification of the Tietze assay [47]. Cell extracts were prepared as previously described [45, 47] and GSH levels were determined against a standard curve of GSSG and levels calculated per μg of soluble protein in the original cell extract. This value was utilized to determine the relative change in intracellular GSH levels compared to untreated samples. GCL specific activity was determined as described previously [45].
Statistical analysis
Data are presented as averages +/− SEM of at least three experiments. Statistical analysis was performed using GraphPad Prism 4 (GraphPad Software, San Diego, CA). Results were compared by two-tailed t-test or one-way ANOVA with Tukey’s post test where appropriate. Mean differences were considered significant when p < 0.05.
RESULTS
Coordinate induction of Gclc and Gclm gene expression following As3+ treatment in murine TAMH hepatocyte cells
As3+ up-regulates several enzymes associated with the GSH antioxidant defense system in various cell types [29, 31]. To determine whether As3+ induced Gcl subunit expression in hepatocytes, TAMH murine hepatocytes were treated with As3+ and steady-state mRNA levels of Gclc and Gclm were analyzed by northern blotting. Treatment of TAMH cells with 10 μM As3+ resulted in the time-dependent coordinate induction of both Gclc and Gclm (Fig. 1A). As3+ caused a transient induction in Gclc gene expression with maximum expression occurring within 4 h of treatment (~4-fold). In contrast, As3+-induced Gclm expression was more prolonged, with maximal expression occurring within 4 h (~4-fold). Induction of Gclc and Gclm by As3+ was also dose-dependent (Fig 1B). As3+ concentrations as low as 1–2 μM resulted in a 1.5–2-fold increase in Gclc and Gclm mRNA levels, while maximal induction of both subunits (3–4-fold) was observed at As3+ concentrations above 10 μM.
Figure 1. Time- and dose-dependent induction of Gclc and Gclm expression by As3+.

(A) TAMH cells were treated with As3+ (10 μM) for the time periods indicated. (B) TAMH cells were treated for 4 h with the indicated concentrations of As3+. Total RNA was isolated and relative mRNA levels were analyzed by Northern blotting as described in the Methods section. Gclc (closed circles) and Gclm (open circles) expression was normalized to Gapdh mRNA levels and the graphical data presented are averages +/− SEM of at least three experiments. +p < 0.05 and *p < 0.001, indicates a significant difference compared to untreated control.
As3+ increases the rate of Gclc and Gclm transcription
Both transcriptional and post-transcriptional mechanisms have been reported to contribute to increased steady-state levels of Gclc and Gclm mRNA [28, 50–54]. Nuclear run-on analyses were performed to determine whether As3+ increased the rate of transcription of the Gclc and/or Gclm genes. As3+ induced a time-dependent increase in the transcription rates of both Gclc and Gclm (Fig. 2). As3+ increased Gclm and Gclc transcription ~2-fold and ~3-fold, respectively, suggesting that enhanced transcription contributes to the increased steady-state Gclc and Gclm mRNA levels in As3+-treated TAMH cells.
Figure 2. As3+ increases the rate of transcription of Gclc and Gclm.
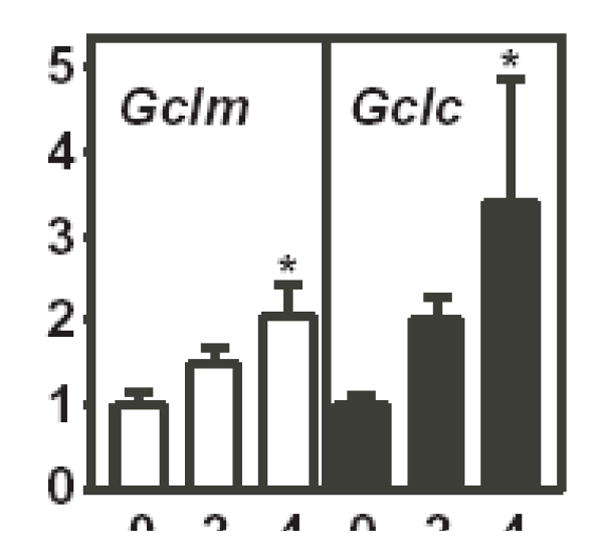
TAMH cells were treated with 10 μM As3+ for the time periods indicated. Nuclei were isolated and nuclear run-on analysis was performed as described in the Methods section. Relative rates of transcription of Gclm (open bars) and Gclc (closed bars) were quantitated and normalized to β-Actin. The data presented are averages +/− SEM of three experiments. *p < 0.05, indicates a significant difference compared to untreated control.
Differential post-transcriptional regulation of Gclc and Gclm mRNA stability by As3+
While As3+ increased the rate of Gclc and Gclm transcription, post-transcriptional effects on mRNA stability can also influence steady-state Gclc and Gclm mRNA levels [50–53]. To examine this possibility, we determined whether As3+ altered the half-life of Gclc and/or Gclm mRNA. TAMH cells were incubated with actinomycin D (500 ng/ml) in the absence or presence of As3+ (10 μM) and the time-dependent decay of Gclc and Gclm mRNA levels analyzed by Northern blotting (Fig. 3). In untreated cells, the half-lifes of the murine Gclc and Gclm mRNAs were ~4 h and ~8 h, which is consistent with previously published values [50, 52, 53]. While treatment with As3+ had little effect on the half-life of Gclc mRNA, As3+ dramatically increased the half-life of Gclm mRNA (~3-fold). These findings suggest that As3+-induced Gclc expression is the result of increased transcription, while elevated Gclm levels are mediated by both increased gene transcription and post-transcriptional stabilization of the Gclm mRNA transcript. Thus, while As3+ coordinately induces both Gcl subunits, these responses are mediated by quite distinct molecular mechanisms.
Figure 3. As3+ induces the post-transcriptional stabilization of Gclm mRNA.
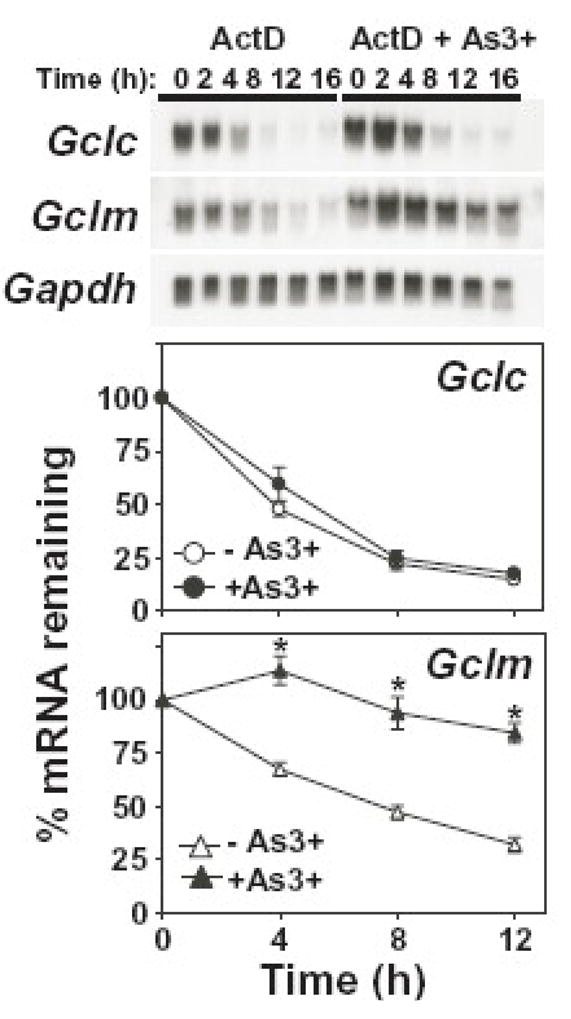
TAMH cells were treated with actinomycin D (ActD; 500 ng/ml) in the absence (open symbols) or presence (closed symbols) of As3+ (10 μM) for the time periods indicated. Relative mRNA levels were analyzed by Northern blotting. Gclc (circles; upper panel) and Gclm (triangles; lower panel) expression was normalized to Gapdh mRNA levels and the graphical data presented are averages +/− SEM of seven experiments. *p < 0.01, indicates a significant difference compared to ActD alone at that time point.
de novo protein synthesis is required for As3+-induced Gclc and Gclm expression
While transcriptional and post-transcriptional mechanisms are involved in As3+-induced Gclc and/or Gclm expression, it is unclear whether these effects are primary or secondary responses requiring de novo protein synthesis. To determine whether As3+-induced Gcl subunit expression was dependent on de novo protein synthesis, As3+-induced Gclc and Gclm expression were analyzed in the presence of the protein synthesis inhibitor cycloheximide (CHX). As shown in Figure 4, CHX abolished As3+-induced expression of both Gclc and Gclm. In contrast to previous studies conducted in glioma cells [52], CHX alone had no effect on steady-state Gclc or Gclm mRNA levels at any time point analyzed (up to 8 h; data not shown). Thus, while there are contradictory reports concerning the involvement of de novo protein synthesis in regulating the inducible expression of Gclc and Gclm [50, 52, 55–57], these findings suggest that a newly synthesized protein(s) is necessary for As3+ induction of Gclc and Gclm in the TAMH cell model.
Figure 4. de novo protein synthesis is required for As3+-induced Gclc and Gclm expression.
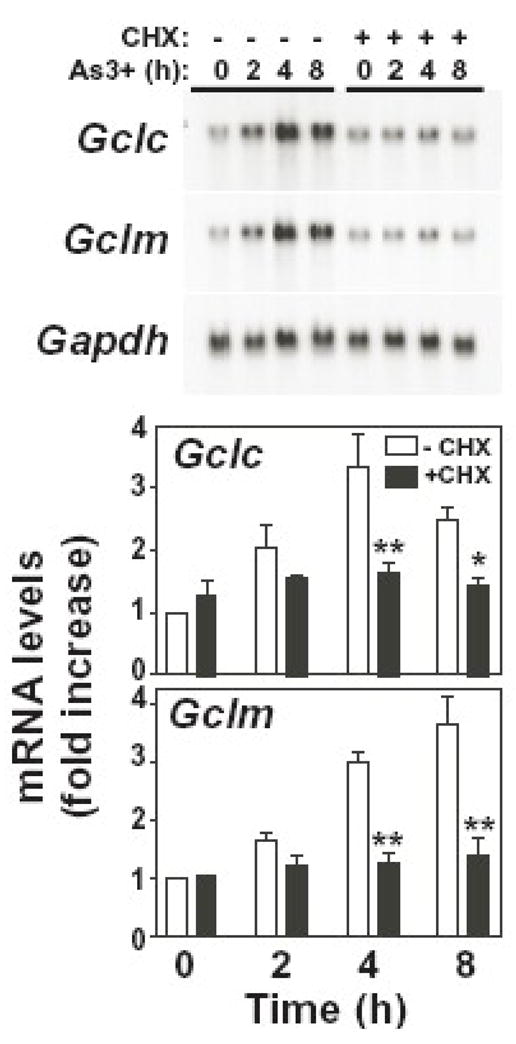
TAMH cells were preincubated in the absence (open bars) or presence (closed bars) of cycloheximide (CHX; 1.0 μg/ml) for 1 h prior to treatment with As3+ (10 μM) for the time periods indicated. Relative mRNA levels were analyzed by Northern blotting. Gclc (upper panel) and Gclm (lower panel) expression was normalized to Gapdh mRNA levels and the graphical data presented are averages +/− SEM of three experiments. *p < 0.05 and **p < 0.001, indicates a significant difference compared to As3+ alone at that time point.
Differential sensitivity of As3+-induced Gclc and Gclm expression to N-acetylcysteine
Many As3+-induced transcriptional responses are thought to be mediated by increased production of ROS [12, 58–60], which can activate transcription mediated by ARE, AP1 and NF-kB promoter elements [28]. To determine whether As3+-induced Gclc and Gclm expression was redox sensitive, we utilized the antioxidant and GSH pro-drug N-acetylcysteine (NAC). Treatment of TAMH cells with 10 mM NAC for 1 h resulted in a ~5-fold increase in intracellular GSH levels (data not shown). Cells were preincubated with NAC and the time-dependent induction of Gclc and Gclm expression in response to As3+ was assessed by northern blotting. NAC pretreatment abolished As3+-induced Gclc expression, while slightly attenuating As3+-induced Gclm expression (~30%) (Fig. 5). These findings suggest that while As3+-induced Gclc expression is highly sensitive to intracellular GSH levels, induction of Gclm is only partially regulated by alterations in cellular thiol redox homeostasis. This differential sensitivity to intracellular GSH levels provide further evidence that distinct molecular mechanisms mediate As3+-induced Gclc and Gclm expression.
Figure 5. Differential regulation of As3+-induced Gclc and Gclm expression by N-acetylcysteine.

TAMH cells were preincubated in the absence (open bars) or presence (closed bars) of N-acetylcysteine (NAC; 10 mM) for 1 h prior to treatment with As3+ (10 μM) for the time periods indicated. Relative mRNA levels were analyzed by Northern blotting. Gclc (upper panel) and Gclm (lower panel) expression was normalized to Gapdh mRNA levels and the graphical data presented are averages +/− SEM of three experiments. *p < 0.05 and **p < 0.001, indicates a significant difference compared to As3+ alone at that time point.
As3+ induces Gclc and Gclm expression independent of Nrf1 or Nrf2
The Cap-n-collar bZIP transcription factors Nrf1 and Nrf2 contribute to the basal and inducible expression of Gclc and Gclm [43, 44, 61–65]. Furthermore, As3+ has been shown to increase Nrf2 expression in multiple cell types [9, 30, 41, 66, 67]. To directly assess the putative role of Nrf1/2 in As3+-induced Gclc and Gclm expression we utilized mouse embryo fibroblasts (MEF) isolated from wild-type (WT), Nrf1−/− and Nrf2−/− mice [43, 44]. As shown in Figure 6A, As3+ treatment resulted in a similar profile of Gclc and Gclm induction in WT MEFs as observed in TAMH cells (see Fig. 1). As3+ caused a transient 3–4-fold increase in Gclc expression (maximal at 4 h), and a more prolonged 6–7-fold increase in Gclm expression (maximal at 8 h) in the WT MEF cell line. Surprisingly, As3+-induced Gclc and Gclm expression was not dramatically different in the WT, Nrf1−/−, or Nrf2−/− MEF cell lines. There was little difference in either the time course or fold-increase in As3+-induced Gclm expression in any of these cell lines. As3+-induced Gclc expression was also similar in the WT and Nrf1−/− MEF cell lines. While there was an attenuation and delay in As3+-induced Gclc expression in the Nrf2−/− MEF cell line, As3+ treatment still increased steady-state Gclc mRNA levels ~3-fold. As a positive control for Nrf-dependent transcription, tert-butylhydroquinone (tBHQ)-inducible expression of Gclc and Gclm was examined. tBHQ is the major metabolite of the phenolic antioxidant butylated hydroxyanisole (BHA) and Nrf2 is required for both BHA-induced Gclc expression in vivo [62] and tBHQ-induced Gclc and Gclm expression in MEF cells [65]. Consistent with previous studies [65], tBHQ treatment resulted in the time-dependent induction of both Gclc and Gclm in WT MEFs (Fig. 6B). Importantly, while tBHQ induction of both subunits in the Nrf1−/− MEFs was attenuated, tBHQ-induced Gclc and Gclm expression was abolished in the Nrf2−/− MEF cell line. These findings suggest that while Nrf2 is required for tBHQ-induced Gclc/Gclm expression, As3+-induced Gclc/Gclm expression occurs independent of Nrf1 and Nrf2.
Figure 6. As3+ induces Gclc and Gclm expression independent of Nrf1 or Nrf2.


MEFs from WT, Nrf1(−/−), or Nrf2(−/−) mice were treated with (A) As3+ (10 μM) or (B) tert-butylhydroquinone (tBHQ; 30 μM) for the time periods indicated. Relative mRNA levels were analyzed by Northern blotting. Gclc (open bars) and Gclm (closed bars) expression was normalized to Gapdh mRNA levels and the graphical data presented are averages +/− SEM of at least three experiments. *p < 0.05, indicates a significant difference compared to As3+-induced gene expression in WT MEFs at that time point.
The role of MAPKs in As3+-induced Gclc and Gclm expression
As3+ activates multiple MAPK pathways that have been implicated in the inducible expression of Gclc and Gclm [37–40]. In light of our demonstration that As3+-induced Gclc and Gclm expression was Nrf1/2-independent, we examined the putative role of the MAPK pathways in As3+-induced Gclc and/or Gclm expression in the TAMH cell model. To identify the MAPK pathways activated by As3+ in this cell model, TAMH cells were treated with various concentrations of As3+ for 4 h and MAPK activation assessed by immunoblot analysis utilizing phospho-specific antibodies that recognize the activated forms of JNK, p38 MAPK and ERK. As3+ treatment resulted in the dose-dependent activation of JNK, p38 MAPK and ERK (Figure 7). In the case of JNK, As3+-induced kinase activity (data not shown) correlated well with the appearance of phosphorylated JNK. Importantly, As3+ activated all three MAPK pathways at an As3+ concentration (10 uM) employed for all other experiments performed in these studies. To directly examine the involvement of these signal transduction pathways in As3+-induced Gclc/Gclm expression, we employed pharmacological inhibitors of the JNK (SP600125), p38 MAPK (SB202190) and ERK (PD98059) pathways. Cells were pretreated with these inhibitors and As3+-induced Gclc and Gclm expression was analyzed (Fig. 8A and 8B). Inhibition of the ERK pathway utilizing the MEK1/2 inhibitor PD98059 slightly attenuated As3+-induced Gclc expression (~20% reduction), but had no effect on As3+-induced Gclm expression. In contrast, the p38 MAPK inhibitor SB202190 reduced As3+-induced Gclc and Gclm expression by ~50% and ~30%, respectively. The JNK inhibitor SP600125 had little effect on As3+-induced Gclm expression. Surprisingly, SP600125 dramatically enhanced As3+-induced Gclc gene expression, increasing steady-state Gclc levels ~2-fold over that observed in response to As3+ alone. Given the potential for non-specific effects of SP600125 [54, 68], a more selective peptide JNK inhibitor (JNKi) was employed in conjunction with real-time RT-PCR to confirm the role of JNK in mediating this response. Consistent with the Northern blot findings, SP600125 was found to significantly potentiate As3+-induced Gclc expression (Fig 8C). However, JNKi had no effect on As3+-induced Gclc expression (Fig. 8C). These findings suggest that JNK is not involved in As3+-induced Gclc expression and does not mediate the superinduction of Gclc observed in the presence of SP600125. None of these inhibitors had a marked effect on basal levels of Gclm mRNA (<25% reduction), while SB202190 and PD98059 reduced basal Gclc mRNA levels by 40–50% (data not shown). Importantly, SP600125 had little effect on basal Gclc mRNA levels (data not shown), indicating that while SP600125 enhanced As3+-induced Gclc mRNA levels, it did not affect constitutive Gclc expression.
Figure 7. Dose-dependent As3+-induced MAPK activation in TAMH cells.
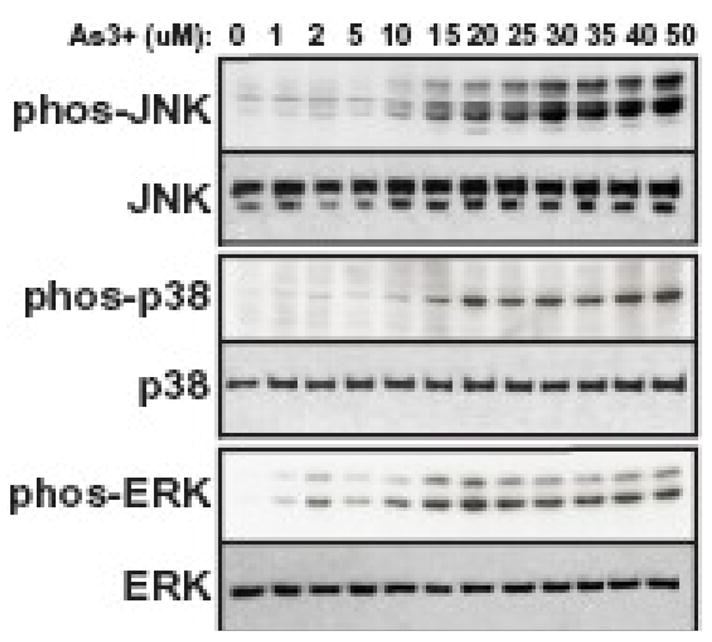
TAMH cells were treated for 4 h with the indicated concentrations of As3+ and JNK, p38 and ERK phosphorylation analyzed by immunoblotting using phospho-specific antibodies as described in the Methods section. The two bands recognized by the p-JNK and p-ERK antibodies are the p46 and p54 JNK family isoforms and p44 ERK1 and p42 ERK2, respectively. Total MAPK protein expression was assessed by immunoblotting using antibodies against JNK, p38 MAPK, and p42 ERK2. The p42 ERK2 antibody does not readily detect p44 ERK1 resulting in a single immunoreactive band.
Figure 8. Role of MAPKs in As3+-induced Gclc and Gclm expression.

TAMH cells were preincubated with SP600125 (SP; 25 μM), SB202190 (SB; 5 μM), and/or PD98059 (PD; 25 μM), or JNKi (20 uM) for 1 h prior to treatment with As3+ (10 μM) for 4 h as indicated. (A) Relative mRNA levels were analyzed by Northern blotting. (B) Gclc (upper panel) and Gclm (lower panel) expression was normalized to Gapdh mRNA levels and the graphical data presented are averages +/− SEM of at least four experiments. (C) Gclc and β-Actin mRNA levels were determined by real-time RT-PCR. Gclc mRNA levels were normalized to β-Actin and presented as fold increase over untreated control. *p < 0.001 and ^p < 0.05, indicates a significant difference compared to untreated control. #p < 0.05, indicates a significant difference compared to As3+ alone. ##p < 0.05, indicates a significant difference in the presence of SP600125.
As As3+ activated all three MAPK pathways, the potential additive effects of these protein kinase pathways on As3+-induced Gclc and Gclm expression was examined by assessing the combinatorial effects of the inhibitors described above (Fig. 8A and 8B). Various combinations of inhibitors had no greater effect on As3+-induced Gclm expression than the p38 MAPK inhibitor SB202190 alone (~30% inhibition). Analysis of Gclc mRNA levels indicated that the inhibitory effects of PD98059 and SB202190 on As3+-induced Gclc expression were additive, reducing Gclc levels to that observed in untreated cells. Interestingly, cotreatment with SB202190 abolished SP600125-induced potentiation of As3+-induced Gclc expression, resulting in reduced Gclc levels similar to that observed in cells pretreated with SB202190 alone. In contrast, cotreatment with PD98059 had no effect on the ability of SP600125 to enhance As3+-induced Gclc expression. In aggregate, these findings suggest that As3+ induction of Gclm is partially mediated by the p38 MAPK pathway, while both the p38 MAPK and ERK pathways contribute to As3+-induced Gclc expression.
As3+ increases GCLC and GCLM protein expression and GCL holoenzyme formation and enzymatic activity
While As3+ increased the steady-state mRNA levels of both Gcl subunits in TAMH cells, it was of interest to determine whether these transcriptional responses were functionally relevant. To determine whether As3+ treatment increased GCL subunit protein expression, GCLC and GCLM protein levels were examined by immunoblotting (Fig. 9A). As3+ treatment resulted in a time-dependent increase in the expression of both GCLC and GCLM. GCLC levels were increased ~2-fold after 16–24 h of As3+ treatment when normalized to βActin levels (Fig. 9B, open circles). As3+ caused an even greater increase in GCLM protein expression with a 3–4-fold induction observed after 16–24 h of treatment (Fig. 9A and 9B, closed circles). These increases in GCLC and GCLM protein levels correlated with increased GSH biosynthetic capacity as exposure to As3+ for 16 h resulted in a 2-fold increase in GCL activity (Fig. 9C). To determine whether As3+ treatment increased GCL activity by enhancing GCL holoenzyme formation, whole cell extracts were resolved by native gel electrophoresis and GCL holoenzyme detected by immunoblotting for GCLC and GCLM (Fig. 9D). Immunoblotting for GCLC revealed two immunoreactive bands of differing electrophoretic mobility (Fig. 9D, upper panel). The identity of these bands was established by their comigration with purified recombinant GCLC protein and GCL holoenzyme ([48], data now shown). The identity of the GCL holoenzyme band was also confirmed by the presence of GCLM (Fig. 9D, lower panel). Importantly, treatment with As3+ resulted in an increase in GCL holoenzyme formation detected with either α-GCLC or α-GCLM antisera, which was associated with a concomitant decrease in monomeric GCLC. Increased GCL holoenzyme formation and activity could result from either de novo synthesis of the GCL subunits or post-translational control of pre-existing GCL protein [27, 28]. To determine whether increased GCL subunit protein expression per se was required for As3+-induced GCL holoenzyme formation and activity, cells were pretreated with the protein synthesis inhibitor cycloheximide (CHX). As shown in Fig. 9E and 9F, pretreatment with CHX abolished As3+-induced GCL activity and GCL holoenzyme formation, respectively. CHX alone had no effect on basal GCL activity (data not shown). In aggregate, these findings are consistent with a model in which As3+-induced GCL activity is mediated by the induction of Gclc and Gclm gene expression, increased GCL subunit protein expression and increased GCL holoenzyme formation.
Discussion
Chronic exposure to low levels of As3+ induces transformation in vitro, while acute exposure to high concentrations induces apoptotic cell death [21]. Interestingly, As3+-transformed cells often exhibit increased resistance to As3+-induced apoptosis and the GSH antioxidant defense system may play a central role in the development of this resistant phenotype. While both acute and chronic As3+ exposure induce an adaptive up-regulation of the GSH antioxidant system, the molecular mechanism(s) mediating these responses are not known. In this study, we have identified distinct transcriptional and post-transcriptional mechanisms and signal transduction pathways that mediate As3+-induced expression of the subunits of GCL, the rate-limiting enzyme in GSH biosynthesis.
As3+ has been reported to induce Gclc gene expression in human keratinocytes [9, 29, 30] and rat lung epithelial cells [31]. Our findings confirm and extend these studies by demonstrating that As3+ coordinately induces both Gclc and Gclm gene expression in the TAMH murine hepatocyte cell line, albeit by quite distinct mechanisms. Transcriptional and post-transcriptional mechanisms contribute to the steady-state levels of Gclc and Gclm mRNA in response to various oxidative stressors [24, 28, 32, 54]. While As3+ increased the rate of transcription of both Gclc and Gclm, As3+ only enhanced the post-transcriptional stabilization of Gclm mRNA in TAMH cells. These findings are in contrast to the post-transcriptional stabilization of both Gclc and Gclm mRNA in response to 4-hydroxynonenal (4-HNE) or diethylmaleate (DEM) [50, 53]. As3+-induced Gclm gene transcription and enhanced Gclm mRNA stability, together with the longer t1/2 of Gclm mRNA compared to Gclc mRNA, likely contribute to the more prolonged increase in steady-state levels of Gclm mRNA relative to Gclc mRNA. While the relative translational efficiency of the Gclc and Gclm mRNAs is not known, this may provide the molecular basis for the greater increase in GCLM protein (4-fold) versus GCLC protein (2-fold) in response to As3+ treatment. As3+ also induced Gcl subunit gene expression in a similar dose-dependent manner. Importantly, while maximal induction occurred at slightly toxic concentrations of As3+ (>10 uM), both subunits were induced at toxicologically and physiologically relevant As3+ concentrations (1–2 uM) in the TAMH cell model.
As3+-induced Gclc and Gclm expression were both dependent on de novo protein synthesis. In contrast, inhibition of de novo protein synthesis selectively prevents 4-HNE-induced Gclm expression and menadione-induced Gclc expression, in rat [50] and human [56] lung epithelial cells, respectively, while having no effect on Gclc or Gclm gene induction in response to ACNU or cigarette smoke condensate [52, 55]. There are also conflicting reports of the involvement of de novo protein synthesis in constitutive Gclc and Gclm expression. Cycloheximide alone had no effect on basal Gclc expression in our cell model, which is consistent with previous reports in HepG2 cells [69] and human bronchial epithelial cells [56], but in contrast to findings in human glioma cells [52]. These findings highlight the differential involvement of de novo protein synthesis in mediating the constitutive and inducible expression of the Gcl subunits in response to As3+ and other types of oxidative stress in various cell types.
Similar to the human Gclc and Gclm gene promoters [33], the proximal region of the murine Gclc and Gclm promoters contain numerous cis-elements that could potentially mediate As3+-inducibility, including ARE, AP1, MTF-1 and NF-kB sites [70–72]. Nrf1 and Nrf2 play important roles in ARE-mediated gene transcription and the constitutive and inducible expression of multiple phase II enzymes, including Gclm and Gclc [28, 32–34, 73, 74]. However, Nrf1 and Nrf2 are distinct in their ability to modulate Gcl subunit gene expression [43, 44, 63]. While paraquat- and DEM-inducible Gclm expression is suppressed in Nrf1-deficient MEFs [43, 63], deletion of Nrf1 had no effect on As3+- or tBHQ-induced Gclc and Gclm mRNA expression. In contrast, tBHQ-induced Gclc and Gclm mRNA expression was abolished in Nrf2-deficient MEFs, which is consistent with previous studies [65]. Surprisingly, deletion of Nrf2 had little effect on As3+-induced Gclm and Gclc expression. The only notable effect was an attenuation and delay in As3+-induced Gclc expression in Nrf2-deficient MEFs. In vitro studies indicate that Nrf1/2-dependent Gcl subunit expression is at least partially mediated by antioxidant response elements (ARE) in the Gclc and Gclm promoters [28, 33, 54]. However, while the murine Gclc and Gclm promoters contain functional ARE sites [70–72] and As3+ increases Nrf2 protein expression and ARE-mediated gene transcription [22, 30, 66, 67, 75], Nrf2 is not required for As3+-induced Gclc and Gclm gene transcription. In contrast, Nrf2 is required for Gclc induction in response to tBHQ ([65] and Fig. 6), DEM [63], and indomethacin [76, 77]. Such findings do not exclude the possibility that Nrf1 and Nrf2 exhibit stimulus-dependent functional redundancy and can compensate for each other in As3+-induced, but not tBHQ-induced Gcl subunit expression. Interestingly, while Nrf1/2 mediate tBHQ-induced rat Gclc expression this occurs via AP-1 and NF-kB binding sites and not ARE sites, which are not present in the rat promoter [65, 78, 79]. Nrf2 also plays an essential role in mediating As3+-induced transcription of certain target genes. For instance, As3+-induced NAD(P)H:quinone oxidoreductase I (NQO1), and heme oxygenase-1 (HO-1) and MSP23 (peroxiredoxin 1) expression is attenuated or abolished in Nrf2-deficient MEFs [66, 80] and macrophages [81], respectively. A dominant-negative Nrf2 mutant also inhibited As3+-induced ARE-mediated transcriptional activity [75] and HO-1 expression in L929 fibroblasts [82] and HepG2 hepatoma cells [83]. Cell type-specific differences have also been reported in the constitutive and inducible expression of Gclc and Gclm [84]. In aggregate, these findings suggest that while As3+-induced expression of some phase II enzymes are Nrf2-dependent in certain cell types, Nrf2 is not required for As3+-induced Gclc or Gclm gene expression in murine hepatocytes even though the murine promoters of both subunits contain functional ARE binding sites [70–72].
Multiple MAP kinase pathways have also been implicated in oxidative stress-induced Gcl subunit expression [37–40]. In TAMH cells, As3+ treatment induced the dose-dependent activation of JNK, p38 MAPK, and ERK and pharmacological inhibitors of these pathways were employed to determine their involvement in As3+-induced Gclc and Gclm gene expression. While inhibition of the p38 MAPK or ERK pathway alone partially suppressed As3+-induced Gclc expression, inhibition of both pathways was required to abolish Gclc induction. These findings are analogous to the involvement of both p38 MAPK and ERK in PDTC-induced Gclc expression in HepG2 cells [40, 85], while ERK activation alone appears to be required for 4-HNE-induced Gclc expression in rat L2 alveolar epithelial cells [37, 39, 54]. Interestingly, the JNK inhibitor SP600125 dramatically potentiated As3+-induced Gclc expression. However, SP600125 is known to inhibit a number of protein kinases with similar efficacies that could potentially mediate this response [68]. Importantly, the inability of the highly specific peptide JNK inhibitor JNKi to elicit a similar superinduction of Gclc expression indicates that the SP600125 effects are not mediated by JNK inhibition. These findings also suggest that JNK is not involved in As3+-induced Gclc gene expression, which contrasts with previous reports that JNK activation is required for 4-HNE- and glucose-deprivation-induced Gclc expression in HBE1 human bronchial epithelial cells [37] and MCF7 cells [38], respectively. While the cellular and molecular basis for these disparities are not known, there appear to be distinct stimuli-, cell type-, concentration-, and species-dependent differences in the mechanisms regulating both the constitutive and inducible expression of the Gclc and Gclm genes [37, 39, 40, 54, 84].
While the transcriptional effects of SP600125 appear to be JNK-independent, SP600125 clearly potentiated As3+-induced Gclc expression in the TAMH murine hepatocyte cell line. This response was subunit specific as SP600125 had no effect on As3+-induced Gclm expression. SP600125 also had no effect on basal Gclc gene expression, suggesting that this superinduction resulted from inhibition of an As3+-activated signal transduction pathway. While the molecular target mediating this SP600125 response is not known, these findings imply that As3+ is activating an SP600125-sensitive protein kinase that is suppressing Gclc expression in the TAMH cell model. This is reminiscent of p38 MAPK-mediated suppression of ARE-mediated gene transcription and induction of phase II enzymes in human HepG2 and murine Hepa1C1C7 hepatoma cell lines [86]. However, the ability of the p38 MAPK inhibitor SB203580 to abolish SP600125-mediated potentiation of As3+-induced Gclc expression suggests that this response is mediated by activation of the p38 MAPK pathway, which we have shown contributes to As3+-induced Gclc expression (Fig. 8). Interestingly, SP600125 rapidly stimulates p38 MAPK phosphorylation and activity in the MIN6 mouse pancreatic cell line [87]. Additional studies are required to reveal the molecular mechanisms and putative involvement of p38 MAPK in SP600125-mediated potentiation of As3+-induced Gclc expression.
In contrast to As3+-induced Gclc expression, p38 MAPK is the only MAPK involved in As3+-induced Gclm expression and it only accounts for ~30% of steady-state Gclm mRNA levels, suggesting the involvement of additional signaling pathways. Furthermore, the effects of p38 MAPK could result from either enhanced Gclm mRNA stabilization or activation of Gclm gene transcription, both of which contribute to steady-state Gclm mRNA levels. In fact, p38 MAPK has been implicated in both As3+-induced gene transcription and post-transcriptional stabilization of various mRNA species [88]. While our findings indicate that As3+ did not increase steady-state murine Gclc mRNA levels via mRNA stabilization, Song et al. have shown that p38 MAPK activation enhances human Gclc mRNA stability in response to several prooxidants, including tBHQ and menadione [51]. An AUUUA HuR recognition sequence in the 3′-UTR of human Gclc was found to be responsible for HuR-mediated regulation of Gclc mRNA stability [51]. It remains to be determined whether the 3′-UTR of murine Gclc and/or human or murine Gclm contain functional AU-rich sequences capable of binding mRNA stabilizing factors such as HuR.
NAC-mediated inhibition of As3+-induced Gclc induction and attenuation of Gclm induction suggests that redox-sensitive mechanisms are involved in the induction of both subunits. Furthermore, the ability of NAC to only partially suppress As3+-induced Gclm expression (~30–50%) is consistent with the selective inhibition of either gene transcription or post-transcriptional Gclm mRNA stabilization. Interestingly, the relative level of NAC-mediated inhibition of As3+-induced Gclm expression correlated quantitatively with the inhibition by the p38 MAPK inhibitor SB203580 (Fig. 7; 30–40%). Given that NAC abolished As3+-induced p38 MAPK, JNK, and ERK in TAMH cells (data not shown) it is intriguing to speculate that the partial suppression of As3+-induced Gclm expression is due to NAC-mediated inhibition of p38 MAPK activity. Correspondingly, NAC-mediated inhibition of As3+-induced Gclc expression could result from inhibition of As3+-induced activation of p38 MAPK and ERK.
While transcriptional induction of the Gcl subunit mRNAs invariably leads to increased GCL protein translation, it was important to demonstrate functionality with regard to GCL enzymatic activity. As expected, As3+-induced Gcl subunit gene expression correlated with increased GCL subunit protein expression and GCL enzymatic activity. Direct quantitation and functional studies indicate that GCLM is limiting for GCL holoenzyme formation and GCL activity both in vivo and in cultured cell systems [48, 89–91]. While we did not directly measure the molar amounts and ratios of the GCL subunits, analysis of GCL holoenzyme composition by native gel electrophoresis confirmed that GCLM was limiting in TAMH cells. This was based on the presence of both monomeric GCLC and heterodimeric holo-GCL bands upon immunoblotting with α-GCLC antisera and the detection of only a single band comigrating with holo-GCL when immunoblotting with α-GCLM antisera. Importantly, As3+ treatment enhanced GCL holoenzyme formation as judged by the shift of monomeric GCLC protein to heterodimeric GCL holoenzyme and the increased amount of GCLM protein present in the holo-GCL complex (Fig. 8D). The greater relative induction of the GCLM subunit (~4 fold for GCLM vs. ~2-fold for GCLC) and the fact that GCLM is limiting suggest that increased expression of GCLM likely contributes more to As3+-induced GCL enzymatic activity via increased GCL holoenzyme formation than increased GCLC expression per se. Preferential induction of the GCLM subunit is also observed in response to 4-HNE in rat lung epithelial cells [39], cysteine deprivation in HepG2 cells and the rat liver in vivo [91], oxidized LDL in murine macrophages [71], and thyroid hormone in rat astrocytes [92]. Irrespective of the relative contribution of GCLC or GCLM protein, the inhibitory effects of cycloheximide suggest that As3+-induced GCL holoenzyme formation and activity result from increased de novo synthesis of the GCL subunits and not post-translational modification of pre-existing GCL protein.
In aggregate, this study identified multiple molecular mechanisms that contribute to increased GCL subunit expression and activity in response to acute As3+ exposure. While As3+ coordinately induced Gclc and Gclm gene expression, distinct signal transduction pathways and transcriptional and post-transcriptional regulatory mechanisms mediated these responses. Importantly, these transcriptional events were functionally relevant, leading to increased GCL subunit protein expression, GCL holoenzyme formation, and GCL activity. In light of the importance of intracellular GSH levels in dictating cellular sensitivity to As3+-induced apoptosis, induction of GCL expression and activity is likely a cytoprotective adaptive response to As3+ exposure. In this regard, increased GSH biosynthetic capacity may play a fundamental role in As3+ adaptation and apoptosis evasion during As3+-induced hepatocarcinogenesis and could also have clinical implications with regard to the use of arsenic trioxide as a chemotherapeutic in the treatment of acute promyelocytic leukemia. However, it remains to be determined whether up-regulation of cellular GSH biosynthetic capacity per se affects As3+-induced apoptosis and/or transformation.
Acknowledgments
This work was supported by NIH grants CA75316, CA90473, and ES07033 (to CCF), CA23226 and CA74131 (to NF), and ES04696, ES10849, AG01751, and ES07033 (to TJK). James A. Thompson is a Mary Gates Scholar. The authors thank Zahra Afsharinejad for performing the real-time RT-PCR analyses and Donald S. Backos for assistance with statistical analyses.
List of Abbreviations
- As3+
arsenite
- GSH
glutathione
- GSSG
glutathione disulfide
- GCL
glutamate-cysteine ligase
- GCLC
catalytic subunit of glutamate-cysteine ligase
- GCLM
modifier subunit of glutamate-cysteine ligase
- GST
glutathione S-transferase
- Nrf1/2
NF-E2-related factor 1/2
- ROS
reactive oxygen species
- NAC
N-acetylcysteine
- CHX
cycloheximide
- ActD
actinomycin D
- BSO
buthionine sulfoximine
- tBHQ
tert-butylhydroquinone
- 4-HNE
4-hydroxynonenal
- GAPDH
glyceraldehyde-3-phosphate dehydrogenase
- ARE
antioxidant response element
- UTR
untranslated region
- MEF
mouse embryo fibroblast
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Yoshida T, Yamauchi H, Fan Sun G. Chronic health effects in people exposed to arsenic via the drinking water: dose-response relationships in review. Toxicol Appl Pharmacol. 2004;198:243–252. doi: 10.1016/j.taap.2003.10.022. [DOI] [PubMed] [Google Scholar]
- 2.Centeno JA, Mullick FG, Martinez L, Page NP, Gibb H, Longfellow D, Thompson C, Ladich ER. Pathology related to chronic arsenic exposure. Environ Health Perspect. 2002;110(Suppl 5):883–886. doi: 10.1289/ehp.02110s5883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Liu J, Waalkes MP. Liver is a target of arsenic carcinogenesis. Toxicol Sci. 2008;105(1):24–32. doi: 10.1093/toxsci/kfn120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dong Z. The molecular mechanisms of arsenic-induced cell transformation and apoptosis. Environ Health Perspect. 2002;110(Suppl 5):757–759. doi: 10.1289/ehp.02110s5757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhao CQ, Young MR, Diwan BA, Coogan TP, Waalkes MP. Association of arsenic-induced malignant transformation with DNA hypomethylation and aberrant gene expression. Proc Natl Acad Sci U S A. 1997;94:10907–10912. doi: 10.1073/pnas.94.20.10907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sens DA, Park S, Gurel V, Sens MA, Garrett SH, Somji S. Inorganic cadmium- and arsenite-induced malignant transformation of human bladder urothelial cells. Toxicol Sci. 2004;79:56–63. doi: 10.1093/toxsci/kfh086. [DOI] [PubMed] [Google Scholar]
- 7.Achanzar WE, Brambila EM, Diwan BA, Webber MM, Waalkes MP. Inorganic arsenite-induced malignant transformation of human prostate epithelial cells. J Natl Cancer Inst. 2002;94:1888–1891. doi: 10.1093/jnci/94.24.1888. [DOI] [PubMed] [Google Scholar]
- 8.Chien CW, Chiang MC, Ho IC, Lee TC. Association of chromosomal alterations with arsenite-induced tumorigenicity of human HaCaT keratinocytes in nude mice. Environ Health Perspect. 2004;112:1704–1710. doi: 10.1289/ehp.7224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pi J, Diwan BA, Sun Y, Liu J, Qu W, He Y, Styblo M, Waalkes MP. Arsenic-induced malignant transformation of human keratinocytes: Involvement of Nrf2. Free Radic Biol Med. 2008;45:651–658. doi: 10.1016/j.freeradbiomed.2008.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Huang C, Ma WY, Li J, Goranson A, Dong Z. Requirement of Erk, but not JNK, for arsenite-induced cell transformation. J Biol Chem. 1999;274:14595–14601. doi: 10.1074/jbc.274.21.14595. [DOI] [PubMed] [Google Scholar]
- 11.Miller WH, Jr, Schipper HM, Lee JS, Singer J, Waxman S. Mechanisms of action of arsenic trioxide. Cancer Res. 2002;62:3893–3903. [PubMed] [Google Scholar]
- 12.Liu SX, Athar M, Lippai I, Waldren C, Hei TK. Induction of oxyradicals by arsenic: implication for mechanism of genotoxicity. Proc Natl Acad Sci U S A. 2001;98:1643–1648. doi: 10.1073/pnas.031482998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kitchin KT, Ahmad S. Oxidative stress as a possible mode of action for arsenic carcinogenesis. Toxicol Lett. 2003;137:3–13. doi: 10.1016/s0378-4274(02)00376-4. [DOI] [PubMed] [Google Scholar]
- 14.Romach EH, Zhao CQ, Del Razo LM, Cebrian ME, Waalkes MP. Studies on the mechanisms of arsenic-induced self tolerance developed in liver epithelial cells through continuous low-level arsenite exposure. Toxicol Sci. 2000;54:500–508. doi: 10.1093/toxsci/54.2.500. [DOI] [PubMed] [Google Scholar]
- 15.Brambila EM, Achanzar WE, Qu W, Webber MM, Waalkes MP. Chronic arsenic-exposed human prostate epithelial cells exhibit stable arsenic tolerance: mechanistic implications of altered cellular glutathione and glutathione S-transferase. Toxicol Appl Pharmacol. 2002;183:99–107. [PubMed] [Google Scholar]
- 16.Qu W, Bortner CD, Sakurai T, Hobson MJ, Waalkes MP. Acquisition of apoptotic resistance in arsenic-induced malignant transformation: role of the JNK signal transduction pathway. Carcinogenesis. 2002;23:151–159. doi: 10.1093/carcin/23.1.151. [DOI] [PubMed] [Google Scholar]
- 17.Somji S, Zhou XD, Garrett SH, Sens MA, Sens DA. Urothelial cells malignantly transformed by exposure to cadmium (Cd(+2)) and arsenite (As(+3)) have increased resistance to Cd(+2) and As(+3)-induced cell death. Toxicol Sci. 2006;94:293–301. doi: 10.1093/toxsci/kfl108. [DOI] [PubMed] [Google Scholar]
- 18.Pi J, He Y, Bortner C, Huang J, Liu J, Zhou T, Qu W, North SL, Kasprzak KS, Diwan BA, Chignell CF, Waalkes MP. Low level, long-term inorganic arsenite exposure causes generalized resistance to apoptosis in cultured human keratinocytes: potential role in skin co-carcinogenesis. Int J Cancer. 2005;116:20–26. doi: 10.1002/ijc.20990. [DOI] [PubMed] [Google Scholar]
- 19.Coppin JF, Qu W, Waalkes MP. Interplay between cellular methyl metabolism and adaptive efflux during oncogenic transformation from chronic arsenic exposure in human cells. J Biol Chem. 2008 doi: 10.1074/jbc.M802942200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Qu W, Kasprzak KS, Kadiiska M, Liu J, Chen H, Maciag A, Mason RP, Waalkes MP. Mechanisms of arsenic-induced cross-tolerance to nickel cytotoxicity, genotoxicity, and apoptosis in rat liver epithelial cells. Toxicol Sci. 2001;63:189–195. doi: 10.1093/toxsci/63.2.189. [DOI] [PubMed] [Google Scholar]
- 21.Bode AM, Dong Z. The paradox of arsenic: molecular mechanisms of cell transformation and chemotherapeutic effects. Crit Rev Oncol Hematol. 2002;42:5–24. doi: 10.1016/s1040-8428(01)00215-3. [DOI] [PubMed] [Google Scholar]
- 22.Kann S, Estes C, Reichard JF, Huang MY, Sartor MA, Schwemberger S, Chen Y, Dalton TP, Shertzer HG, Xia Y, Puga A. Butylhydroquinone protects cells genetically deficient in glutathione biosynthesis from arsenite-induced apoptosis without significantly changing their prooxidant status. Toxicol Sci. 2005 doi: 10.1093/toxsci/kfi253. [DOI] [PubMed] [Google Scholar]
- 23.Habib GM, Shi ZZ, Lieberman MW. Glutathione protects cells against arsenite-induced toxicity. Free Radic Biol Med. 2007;42:191–201. doi: 10.1016/j.freeradbiomed.2006.10.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Griffith OW, Mulcahy RT. The enzymes of glutathione synthesis: gamma-glutamylcysteine synthetase. Adv Enzymol Relat Areas Mol Biol. 1999;73:209–267. doi: 10.1002/9780470123195.ch7. [DOI] [PubMed] [Google Scholar]
- 25.Hayes JD, McLellan LI. Glutathione and glutathione-dependent enzymes represent a coordinately regulated defence against oxidative stress. Free Radic Res. 1999;31:273–300. doi: 10.1080/10715769900300851. [DOI] [PubMed] [Google Scholar]
- 26.Ballatori N, Hammond CL, Cunningham JB, Krance SM, Marchan R. Molecular mechanisms of reduced glutathione transport: role of the MRP/CFTR/ABCC and OATP/SLC21A families of membrane proteins. Toxicol Appl Pharmacol. 2005;204:238–255. doi: 10.1016/j.taap.2004.09.008. [DOI] [PubMed] [Google Scholar]
- 27.Franklin CC, Backos DS, Mohar I, White CC, Forman HJ, Kavanagh TJ. Structure, Function, and Post-Transcriptional Regulation of the Catalytic and Modifier Subunits of Glutamate Cysteine Ligase. Mol Aspects Med. 2008 doi: 10.1016/j.mam.2008.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lu SC. Regulation of glutathione synthesis. Mol Aspects Med. 2008 doi: 10.1016/j.mam.2008.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schuliga M, Chouchane S, Snow ET. Upregulation of glutathione-related genes and enzyme activities in cultured human cells by sublethal concentrations of inorganic arsenic. Toxicol Sci. 2002;70:183–192. doi: 10.1093/toxsci/70.2.183. [DOI] [PubMed] [Google Scholar]
- 30.Pi J, Qu W, Reece JM, Kumagai Y, Waalkes MP. Transcription factor Nrf2 activation by inorganic arsenic in cultured keratinocytes: involvement of hydrogen peroxide. Exp Cell Res. 2003;290:234–245. doi: 10.1016/s0014-4827(03)00341-0. [DOI] [PubMed] [Google Scholar]
- 31.Li M, Cai JF, Chiu JF. Arsenic induces oxidative stress and activates stress gene expressions in cultured lung epithelial cells. J Cell Biochem. 2002;87:29–38. doi: 10.1002/jcb.10269. [DOI] [PubMed] [Google Scholar]
- 32.Wild AC, Mulcahy RT. Regulation of gamma-glutamylcysteine synthetase subunit gene expression: insights into transcriptional control of antioxidant defenses. Free Radic Res. 2000;32:281–301. doi: 10.1080/10715760000300291. [DOI] [PubMed] [Google Scholar]
- 33.Dickinson DA, Levonen AL, Moellering DR, Arnold EK, Zhang H, Darley-Usmar VM, Forman HJ. Human glutamate cysteine ligase gene regulation through the electrophile response element. Free Radic Biol Med. 2004;37:1152–1159. doi: 10.1016/j.freeradbiomed.2004.06.011. [DOI] [PubMed] [Google Scholar]
- 34.Zhang DD. Mechanistic studies of the Nrf2-Keap1 signaling pathway. Drug Metab Rev. 2006;38:769–789. doi: 10.1080/03602530600971974. [DOI] [PubMed] [Google Scholar]
- 35.Nguyen T, Sherratt PJ, Pickett CB. Regulatory mechanisms controlling gene expression mediated by the antioxidant response element. Annu Rev Pharmacol Toxicol. 2003;43:233–260. doi: 10.1146/annurev.pharmtox.43.100901.140229. [DOI] [PubMed] [Google Scholar]
- 36.Myhrstad MC, Husberg C, Murphy P, Nordstrom O, Blomhoff R, Moskaug JO, Kolsto AB. TCF11/Nrf1 overexpression increases the intracellular glutathione level and can transactivate the gamma-glutamylcysteine synthetase (GCS) heavy subunit promoter. Biochim Biophys Acta. 2001;1517:212–219. doi: 10.1016/s0167-4781(00)00276-1. [DOI] [PubMed] [Google Scholar]
- 37.Dickinson DA, Iles KE, Watanabe N, Iwamoto T, Zhang H, Krzywanski DM, Forman HJ. 4-hydroxynonenal induces glutamate cysteine ligase through JNK in HBE1 cells. Free Radic Biol Med. 2002;33:974. doi: 10.1016/s0891-5849(02)00991-7. [DOI] [PubMed] [Google Scholar]
- 38.Lee YJ, Galoforo SS, Sim JE, Ridnour LA, Choi J, Forman HJ, Corry PM, Spitz DR. Dominant-negative Jun N-terminal protein kinase (JNK-1) inhibits metabolic oxidative stress during glucose deprivation in a human breast carcinoma cell line. Free Radic Biol Med. 2000;28:575–584. doi: 10.1016/s0891-5849(99)00267-1. [DOI] [PubMed] [Google Scholar]
- 39.Liu RM, Borok Z, Forman HJ. 4-Hydroxy-2-nonenal increases gamma-glutamylcysteine synthetase gene expression in alveolar epithelial cells. Am J Respir Cell Mol Biol. 2001;24:499–505. doi: 10.1165/ajrcmb.24.4.4307. [DOI] [PubMed] [Google Scholar]
- 40.Zipper LM, Mulcahy RT. Inhibition of ERK and p38 MAP kinases inhibits binding of Nrf2 and induction of GCS genes. Biochem Biophys Res Commun. 2000;278:484–492. doi: 10.1006/bbrc.2000.3830. [DOI] [PubMed] [Google Scholar]
- 41.Wang XJ, Sun Z, Chen W, Li Y, Villeneuve NF, Zhang DD. Activation of Nrf2 by arsenite and monomethylarsonous acid is independent of Keap1-C151: enhanced Keap1-Cul3 interaction. Toxicol Appl Pharmacol. 2008;230:383–389. doi: 10.1016/j.taap.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wu JC, Merlino G, Cveklova K, Mosinger B, Fausto N. Autonomous growth in serum-free medium and production of hepatocellular carcinomas by differentiated hepatocyte lines that overexpress transforming growth factor alpha 1. Cancer Res. 1994;54:5964–5973. [PubMed] [Google Scholar]
- 43.Kwong M, Kan YW, Chan JY. The CNC basic leucine zipper factor, Nrf1, is essential for cell survival in response to oxidative stress-inducing agents. Role for Nrf1 in gamma-gcs(l) and gss expression in mouse fibroblasts. J Biol Chem. 1999;274:37491–37498. doi: 10.1074/jbc.274.52.37491. [DOI] [PubMed] [Google Scholar]
- 44.Chan JY, Kwong M. Impaired expression of glutathione synthetic enzyme genes in mice with targeted deletion of the Nrf2 basic-leucine zipper protein. Biochim Biophys Acta. 2000;1517:19–26. doi: 10.1016/s0167-4781(00)00238-4. [DOI] [PubMed] [Google Scholar]
- 45.Franklin CC, Rosenfeld-Franklin ME, White C, Kavanagh TJ, Fausto N. TGFbeta1-induced suppression of glutathione antioxidant defenses in hepatocytes: caspase-dependent posttranslational and caspase-independent transcriptional regulatory mechanisms. Faseb J. 2003;17:1535–1537. doi: 10.1096/fj.02-0867fje. [DOI] [PubMed] [Google Scholar]
- 46.Botta D, Shi S, White CC, Dabrowski MJ, Keener CL, Srinouanprachanh SL, Farin FM, Ware CB, Ladiges WC, Pierce RH, Fausto N, Kavanagh TJ. Acetaminophen-induced liver injury is attenuated in male glutamate-cysteine ligase transgenic mice. J Biol Chem. 2006;281:28865–28875. doi: 10.1074/jbc.M605143200. [DOI] [PubMed] [Google Scholar]
- 47.Franklin CC, Krejsa CM, Pierce RH, White CC, Fausto N, Kavanagh TJ. Caspase-3-Dependent Cleavage of the Glutamate-L-Cysteine Ligase Catalytic Subunit during Apoptotic Cell Death. Am J Pathol. 2002;160:1887–1894. doi: 10.1016/S0002-9440(10)61135-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Krzywanski DM, Dickinson DA, Iles KE, Wigley AF, Franklin CC, Liu RM, Kavanagh TJ, Forman HJ. Variable regulation of glutamate cysteine ligase subunit proteins affects glutathione biosynthesis in response to oxidative stress. Arch Biochem Biophys. 2004;423:116–125. doi: 10.1016/j.abb.2003.11.004. [DOI] [PubMed] [Google Scholar]
- 49.Franklin CC, Srikanth S, Kraft AS. Conditional expression of mitogen-activated protein kinase phosphatase- 1, MKP-1, is cytoprotective against UV-induced apoptosis. Proc Natl Acad Sci U S A. 1998;95:3014–3019. doi: 10.1073/pnas.95.6.3014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Liu RM, Gao L, Choi J, Forman HJ. γ-glutamylcysteine synthetase: mRNA stabilization and independent subunit transcription by 4-hydroxy-2-nonenal. Am J Physiol. 1998;275:L861–869. doi: 10.1152/ajplung.1998.275.5.L861. [DOI] [PubMed] [Google Scholar]
- 51.Song IS, Tatebe S, Dai W, Kuo MT. Delayed mechanism for induction of gamma-glutamylcysteine synthetase heavy subunit mRNA stability by oxidative stress involving p38 mitogen-activated protein kinase signaling. J Biol Chem. 2005;280:28230–28240. doi: 10.1074/jbc.M413103200. [DOI] [PubMed] [Google Scholar]
- 52.Gomi A, Masuzawa T, Ishikawa T, Kuo MT. Posttranscriptional regulation of MRP/GS-X pump and gamma- glutamylcysteine synthetase expression by 1-(4-amino-2-methyl-5- pyrimidinyl) methyl-3-(2-chloroethyl)-3-nitrosourea and by cycloheximide in human glioma cells. Biochem Biophys Res Commun. 1997;239:51–56. doi: 10.1006/bbrc.1997.7423. [DOI] [PubMed] [Google Scholar]
- 53.Sekhar KR, Long M, Long J, Xu ZQ, Summar ML, Freeman ML. Alteration of transcriptional and post-transcriptional expression of gamma-glutamylcysteine synthetase by diethyl maleate. Radiat Res. 1997;147:592–597. [PubMed] [Google Scholar]
- 54.Iles KE, Liu RM. Mechanisms of glutamate cysteine ligase (GCL) induction by 4-hydroxynonenal. Free Radic Biol Med. 2005;38:547–556. doi: 10.1016/j.freeradbiomed.2004.11.012. [DOI] [PubMed] [Google Scholar]
- 55.Rahman I, Smith CA, Lawson MF, Harrison DJ, MacNee W. Induction of gamma-glutamylcysteine synthetase by cigarette smoke is associated with AP-1 in human alveolar epithelial cells. FEBS Lett. 1996;396:21–25. doi: 10.1016/0014-5793(96)01027-7. [DOI] [PubMed] [Google Scholar]
- 56.Ray S, Watkins DN, Misso NL, Thompson PJ. Oxidant stress induces gamma-glutamylcysteine synthetase and glutathione synthesis in human bronchial epithelial NCI-H292 cells. Clin Exp Allergy. 2002;32:571–577. doi: 10.1046/j.0954-7894.2002.01294.x. [DOI] [PubMed] [Google Scholar]
- 57.Shi MM, Iwamoto T, Forman HJ. gamma-Glutamylcysteine synthetase and GSH increase in quinone-induced oxidative stress in BPAEC. Am J Physiol. 1994;267:L414–421. doi: 10.1152/ajplung.1994.267.4.L414. [DOI] [PubMed] [Google Scholar]
- 58.Barchowsky A, Roussel RR, Klei LR, James PE, Ganju N, Smith KR, Dudek EJ. Low levels of arsenic trioxide stimulate proliferative signals in primary vascular cells without activating stress effector pathways. Toxicol Appl Pharmacol. 1999;159:65–75. doi: 10.1006/taap.1999.8723. [DOI] [PubMed] [Google Scholar]
- 59.Barchowsky A, Klei LR, Dudek EJ, Swartz HM, James PE. Stimulation of reactive oxygen, but not reactive nitrogen species, in vascular endothelial cells exposed to low levels of arsenite. Free Radic Biol Med. 1999;27:1405–1412. doi: 10.1016/s0891-5849(99)00186-0. [DOI] [PubMed] [Google Scholar]
- 60.Huang HS, Chang WC, Chen CJ. Involvement of reactive oxygen species in arsenite-induced downregulation of phospholipid hydroperoxide glutathione peroxidase in human epidermoid carcinoma A431 cells. Free Radic Biol Med. 2002;33:864–873. doi: 10.1016/s0891-5849(02)00983-8. [DOI] [PubMed] [Google Scholar]
- 61.Chan K, Han XD, Kan YW. An important function of Nrf2 in combating oxidative stress: detoxification of acetaminophen. Proc Natl Acad Sci U S A. 2001;98:4611–4616. doi: 10.1073/pnas.081082098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.McMahon M, Itoh K, Yamamoto M, Chanas SA, Henderson CJ, McLellan LI, Wolf CR, Cavin C, Hayes JD. The Cap‘n’Collar basic leucine zipper transcription factor Nrf2 (NF-E2 p45-related factor 2) controls both constitutive and inducible expression of intestinal detoxification and glutathione biosynthetic enzymes. Cancer Res. 2001;61:3299–3307. [PubMed] [Google Scholar]
- 63.Leung L, Kwong M, Hou S, Lee C, Chan JY. Deficiency of the Nrf1 and Nrf2 transcription factors results in early embryonic lethality and severe oxidative stress. J Biol Chem. 2003;278:48021–48029. doi: 10.1074/jbc.M308439200. [DOI] [PubMed] [Google Scholar]
- 64.Chen L, Kwong M, Lu R, Ginzinger D, Lee C, Leung L, Chan JY. Nrf1 is critical for redox balance and survival of liver cells during development. Mol Cell Biol. 2003;23:4673–4686. doi: 10.1128/MCB.23.13.4673-4686.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Yang H, Magilnick N, Lee C, Kalmaz D, Ou X, Chan JY, Lu SC. Nrf1 and Nrf2 Regulate Rat Glutamate-Cysteine Ligase Catalytic Subunit Transcription Indirectly via NF-{kappa}B and AP-1. Mol Cell Biol. 2005;25:5933–5946. doi: 10.1128/MCB.25.14.5933-5946.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.He X, Chen MG, Lin GX, Ma Q. Arsenic induces NAD(P)H:quinone oxidoreductase I by disrupting the NRF2/KEAP1/CUL3 complex and recruiting NRF2/MAF to are enhancer. J Biol Chem. 2006 doi: 10.1074/jbc.M604120200. [DOI] [PubMed] [Google Scholar]
- 67.Aono J, Yanagawa T, Itoh K, Li B, Yoshida H, Kumagai Y, Yamamoto M, Ishii T. Activation of Nrf2 and accumulation of ubiquitinated A170 by arsenic in osteoblasts. Biochem Biophys Res Commun. 2003;305:271–277. doi: 10.1016/s0006-291x(03)00728-9. [DOI] [PubMed] [Google Scholar]
- 68.Bain J, McLauchlan H, Elliott M, Cohen P. The specificities of protein kinase inhibitors: an update. Biochem J. 2003;371:199–204. doi: 10.1042/BJ20021535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wild AC, Moinova HR, Mulcahy RT. Regulation of gamma-glutamylcysteine synthetase subunit gene expression by the transcription factor Nrf2. J Biol Chem. 1999;274:33627–33636. doi: 10.1074/jbc.274.47.33627. [DOI] [PubMed] [Google Scholar]
- 70.Hudson FN, Kavanagh TJ. Cloning and characterization of the proximal promoter region of the mouse glutamate-L-cysteine ligase regulatory subunit gene. Biochim Biophys Acta. 2000;1492:447–451. doi: 10.1016/s0167-4781(00)00128-7. [DOI] [PubMed] [Google Scholar]
- 71.Bea F, Hudson FN, Chait A, Kavanagh TJ, Rosenfeld ME. Induction of glutathione synthesis in macrophages by oxidized low-density lipoproteins is mediated by consensus antioxidant response elements. Circ Res. 2003;92:386–393. doi: 10.1161/01.RES.0000059561.65545.16. [DOI] [PubMed] [Google Scholar]
- 72.Solis WA, Dalton TP, Dieter MZ, Freshwater S, Harrer JM, He L, Shertzer HG, Nebert DW. Glutamate-cysteine ligase modifier subunit: mouse Gclm gene structure and regulation by agents that cause oxidative stress. Biochem Pharmacol. 2002;63:1739–1754. doi: 10.1016/s0006-2952(02)00897-3. [DOI] [PubMed] [Google Scholar]
- 73.Chanas SA, Jiang Q, McMahon M, McWalter GK, McLellan LI, Elcombe CR, Henderson CJ, Wolf CR, Moffat GJ, Itoh K, Yamamoto M, Hayes JD. Loss of the Nrf2 transcription factor causes a marked reduction in constitutive and inducible expression of the glutathione S-transferase Gsta1, Gsta2, Gstm1, Gstm2, Gstm3 and Gstm4 genes in the livers of male and female mice. Biochem J. 2002;365:405–416. doi: 10.1042/BJ20020320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.McWalter GK, Higgins LG, McLellan LI, Henderson CJ, Song L, Thornalley PJ, Itoh K, Yamamoto M, Hayes JD. Transcription factor Nrf2 is essential for induction of NAD(P)H:quinone oxidoreductase 1, glutathione S-transferases, and glutamate cysteine ligase by broccoli seeds and isothiocyanates. J Nutr. 2004;134:3499S–3506S. doi: 10.1093/jn/134.12.3499S. [DOI] [PubMed] [Google Scholar]
- 75.Gong P, Stewart D, Hu B, Vinson C, Alam J. Multiple basic-leucine zipper proteins regulate induction of the mouse heme oxygenase-1 gene by arsenite. Arch Biochem Biophys. 2002;405:265–274. doi: 10.1016/s0003-9861(02)00404-6. [DOI] [PubMed] [Google Scholar]
- 76.Sekhar KR, Spitz DR, Harris S, Nguyen TT, Meredith MJ, Holt JT, Gius D, Marnett LJ, Summar ML, Freeman ML, Guis D. Redox-sensitive interaction between KIAA0132 and Nrf2 mediates indomethacin-induced expression of gamma-glutamylcysteine synthetase. Free Radic Biol Med. 2002;32:650–662. doi: 10.1016/s0891-5849(02)00755-4. [DOI] [PubMed] [Google Scholar]
- 77.Sekhar KR, Crooks PA, Sonar VN, Friedman DB, Chan JY, Meredith MJ, Starnes JH, Kelton KR, Summar SR, Sasi S, Freeman ML. NADPH oxidase activity is essential for Keap1/Nrf2-mediated induction of GCLC in response to 2-indol-3-yl-methylenequinuclidin-3-ols. Cancer Res. 2003;63:5636–5645. [PubMed] [Google Scholar]
- 78.Yang H, Zeng Y, Lee TD, Yang Y, Ou X, Chen L, Haque M, Rippe R, Lu SC. Role of AP-1 in the coordinate induction of rat glutamate-cysteine ligase and glutathione synthetase by tert-butylhydroquinone. J Biol Chem. 2002;277:35232–35239. doi: 10.1074/jbc.M203812200. [DOI] [PubMed] [Google Scholar]
- 79.Yang H, Wang J, Huang ZZ, Ou X, Lu SC. Cloning and characterization of the 5′-flanking region of the rat glutamate-cysteine ligase catalytic subunit. Biochem J. 2001;357:447–455. doi: 10.1042/0264-6021:3570447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Harada H, Sugimoto R, Watanabe A, Taketani S, Okada K, Warabi E, Siow R, Itoh K, Yamamoto M, Ishii T. Differential roles for Nrf2 and AP-1 in upregulation of HO-1 expression by arsenite in murine embryonic fibroblasts. Free Radic Res. 2008;42:297–304. doi: 10.1080/10715760801975735. [DOI] [PubMed] [Google Scholar]
- 81.Ishii T, Itoh K, Takahashi S, Sato H, Yanagawa T, Katoh Y, Bannai S, Yamamoto M. Transcription factor Nrf2 coordinately regulates a group of oxidative stress-inducible genes in macrophages. J Biol Chem. 2000;275:16023–16029. doi: 10.1074/jbc.275.21.16023. [DOI] [PubMed] [Google Scholar]
- 82.Alam J, Stewart D, Touchard C, Boinapally S, Choi AM, Cook JL. Nrf2, a Cap‘n’Collar transcription factor, regulates induction of the heme oxygenase-1 gene. J Biol Chem. 1999;274:26071–26078. doi: 10.1074/jbc.274.37.26071. [DOI] [PubMed] [Google Scholar]
- 83.Yu R, Chen C, Mo YY, Hebbar V, Owuor ED, Tan TH, Kong AN. Activation of mitogen-activated protein kinase pathways induces antioxidant response element-mediated gene expression via a Nrf2- dependent mechanism. J Biol Chem. 2000;275:39907–39913. doi: 10.1074/jbc.M004037200. [DOI] [PubMed] [Google Scholar]
- 84.Dahl EL, Mulcahy RT. Cell-type specific differences in glutamate cysteine ligase transcriptional regulation demonstrate independent subunit control. Toxicol Sci. 2001;61:265–272. doi: 10.1093/toxsci/61.2.265. [DOI] [PubMed] [Google Scholar]
- 85.Zipper LM, Mulcahy RT. Erk activation is required for Nrf2 nuclear localization during pyrrolidine dithiocarbamate induction of glutamate cysteine ligase modulatory gene expression in HepG2 cells. Toxicol Sci. 2003;73:124–134. doi: 10.1093/toxsci/kfg083. [DOI] [PubMed] [Google Scholar]
- 86.Yu R, Mandlekar S, Lei W, Fahl WE, Tan TH, Kong AN. p38 mitogen-activated protein kinase negatively regulates the induction of phase II drug-metabolizing enzymes that detoxify carcinogens. J Biol Chem. 2000;275:2322–2327. doi: 10.1074/jbc.275.4.2322. [DOI] [PubMed] [Google Scholar]
- 87.Vaishnav D, Jambal P, Reusch JE, Pugazhenthi S. SP600125, an inhibitor of c-jun N-terminal kinase, activates CREB by a p38 MAPK-mediated pathway. Biochem Biophys Res Commun. 2003;307:855–860. doi: 10.1016/s0006-291x(03)01287-7. [DOI] [PubMed] [Google Scholar]
- 88.Dean JL, Sully G, Clark AR, Saklatvala J. The involvement of AU-rich element-binding proteins in p38 mitogen-activated protein kinase pathway-mediated mRNA stabilisation. Cell Signal. 2004;16:1113–1121. doi: 10.1016/j.cellsig.2004.04.006. [DOI] [PubMed] [Google Scholar]
- 89.Yang Y, Dieter MZ, Chen Y, Shertzer HG, Nebert DW, Dalton TP. Initial characterization of the glutamate-cysteine ligase modifier subunit Gclm(-/-) knockout mouse. Novel model system for a severely compromised oxidative stress response. J Biol Chem. 2002;277:49446–49452. doi: 10.1074/jbc.M209372200. [DOI] [PubMed] [Google Scholar]
- 90.Chen Y, Shertzer HG, Schneider SN, Nebert DW, Dalton TP. Glutamate Cysteine Ligase Catalysis: DEPENDENCE ON ATP AND MODIFIER SUBUNIT FOR REGULATION OF TISSUE GLUTATHIONE LEVELS. J Biol Chem. 2005;280:33766–33774. doi: 10.1074/jbc.M504604200. [DOI] [PubMed] [Google Scholar]
- 91.Lee JI, Kang J, Stipanuk MH. Differential regulation of glutamate-cysteine ligase subunit expression and increased holoenzyme formation in response to cysteine deprivation. Biochem J. 2006;393:181–190. doi: 10.1042/BJ20051111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Dasgupta A, Das S, Sarkar PK. Thyroid hormone promotes glutathione synthesis in astrocytes by up regulation of glutamate cysteine ligase through differential stimulation of its catalytic and modulator subunit mRNAs. Free Radic Biol Med. 2007;42:617–626. doi: 10.1016/j.freeradbiomed.2006.11.030. [DOI] [PubMed] [Google Scholar]