

Abstract
RNA interference through non-coding microRNAs (miRNAs) represents a vital component of the innate antiviral immune response in plants and invertebrate animals; however, a role for cellular miRNAs in the defence against viral infection in mammalian organisms has thus far remained elusive1. Here we show that interferon beta (IFNβ) rapidly modulates the expression of numerous cellular miRNAs, and that eight of these IFNβ-induced miRNAs have sequence-predicted targets within the hepatitis C virus (HCV) genomic RNA. The introduction of synthetic miRNA-mimics corresponding to these IFNβ-induced miRNAs reproduces the antiviral effects of IFNβ on HCV replication and infection, whereas neutralization of these antiviral miRNAs with anti-miRNAs reduces the antiviral effects of IFNβ against HCV. In addition, we demonstrate that IFNβ treatment leads to a significant reduction in the expression of the liver-specific miR-122, an miRNA that has been previously shown to be essential for HCV replication2. Therefore, our findings strongly support the notion that mammalian organisms too, through the interferon system, use cellular miRNAs to combat viral infections.
miRNAs are a class of small non-coding RNA molecules that function through post-transcriptional regulation of gene expression by a process termed RNA interference (RNAi). Over 500 miRNA-encoding genes, which seem to be exclusively transcribed by RNA polymerase II, have been identified in mammals. These primary microRNAs are processed by the enzymes Drosha/DGCR8/RNASEN into hairpin-loop-containing pre-miRNAs, which are then subject to nuclear export via exportin 5. Further enzymatic processing of the pre-miRNAs by Dicer leads to a mature miRNA duplex that is loaded into the RNA-induced silencing complex (RISC), in which the miRNA guides RISC to complementary messenger RNAs. On the basis of the degree of homology between the miRNA and the mRNA, RISC can inhibit mRNA function by either promoting its cleavage or by inhibiting its translation3,4. Particularly, the sequence complementarity in the 6–8 base pair ‘seed region’ at the 5′ end of the miRNA—mRNA heteroduplex seems to determine the specificity of miRNA—targetRNA interactions5.
RNAi-mediated targeting of viral RNAs was first recognized in plants as an antiviral defence mechanism, but it is now apparent that invertebrates also use RNAi to combat viral infection1,6,7. However, thus far, no evidence has been presented in support of a protective RNAi-based antiviral response in mammalian cells. Rather, the hypothesis has been proposed that “the extremely potent interferon system has displaced RNAi as the key defence against virus infection in mammalian cells”1. Indeed, several type I interferon (IFNα/β)-regulated gene products such as protein kinase R, the 2′-5′ OA synthase/RNase L system, the adenosine deaminase ADAR1 or the Mx GTPases are important contributors to the antiviral properties of these cytokines8,9. However, the possibility that IFNα/β might induce cellular miRNAs that target viral transcripts and thereby use RNAi as part of their arsenal against invading viruses has been left unexplored. To test whether IFNα/β could alter the expression of cellular miRNAs, we used microarray technology to analyse RNA derived from IFNα/β- or IFNγ-stimulated cells. This initial screening effort identified ~30 miRNAs, the expression levels of which were increased or attenuated in response to IFNα/β or IFNγ. Because we were interested in determining whether these miRNAs could potentially contribute to the antiviral effects of interferons, we performed sequence complementarity analysis of these miRNAs against viral transcripts or viral genomic RNAs with an initial focus on the crucial seed sequence. This approach revealed promising matches among several viruses, most of which harbour an RNA-based genome. Specifically, eight of the IFNβ-induced miRNAs (miR-1, miR-30, miR-128, miR-196, miR-296, miR-351, miR-431 and miR-448) displayed nearly perfect complementarity in their seed sequences with hepatitis C virus (HCV) RNA genomes. This finding was rather intriguing considering that IFNα and IFNβ are the most common treatment regimen for HCV infection10,11. Interestingly, a similar analysis of the hepatitis B virus (a DNA virus) yielded no significant matches.
HCV is the sole member of the hepacivirus genus of the Flaviviridiae family, and is represented by six major genotypes. The virion contains a 9.6 kb single-stranded RNA genome of positive polarity, with highly invariant 5′ and 3′ untranslated regions12,13. After virus entry into the host cell, the viral genome is uncoated and serves as a template for the translation of a single polyprotein, which is subsequently processed by host and viral proteases. The non-structural viral proteins then initiate the synthesis of a negative-strand RNA, which serves as a replication template for the generation of new positive-strand viral genomes12,13. Sequence alignment of the six HCV genotypes illustrated that the putative miRNA target sites for the IFNβ-induced miRNAs are located in areas strictly conserved among all HCV genotypes (see examples in Supplementary Fig. 1a) as well as in regions that differ between the genotypes such that high seed-sequence complementarity occurs only with selected HCV genotypes (see examples in Supplementary Fig. 1b).
To verify our microarray studies we analysed miRNA induction in response to IFNβ in the human hepatoma cell line Huh7, as well as in freshly isolated primary murine hepatocytes by quantitative PCR (qPCR). As shown in Fig. 1a, b, IFNβ treatment resulted in a similar induction of the prospective antiviral miRNAs in both cell types to that observed in ISG54, a well-characterized IFNα/β-regulated gene14. Two miRNAs (miR-125 and miR-142) that were found IFNβ-unresponsive in the microarray analysis were included as negative controls. We next performed kinetic, as well as dose—response, analyses of the induction of miRNAs by IFNβ. Time course analysis revealed that induction of miR-1 and miR-196 occurs very rapidly, with peak concentrations within 30 min, and thus even precedes the upregulation of ISG54 (Fig. 1c). In a similar manner to ISG54 induction, upregulation of miR-1 and miR-196 followed a classical dose—response curve between 1 and 1,000 U ml−1 IFNβ (Fig. 1d), and could be blocked by actinomycin D (data not shown), indicating that the increased levels of miRNAs are the result of transcriptional induction.
Figure 1. Regulation of miRNA expression by IFNβ in Huh7 cells and primary hepatocytes.

a, b, Huh7 cells (a) or primary hepatocytes (b) were stimulated with 100 U ml−1 IFNβ for 2 h, and the indicated miRNAs were quantified by qPCR. ISG54 induction is shown for comparison. c, Time course of miRNA induction by IFNβ: Huh7 cells were stimulated with 100 U ml−1 IFNβ for the indicated times, and miR-1, miR-196 or ISG54 expression was quantified by qPCR. d, Dose—response analysis of miRNA induction by IFNβ: Huh7 cells were stimulated with the indicated doses of IFNβ for 2 h, and miR-1, miR-196 or ISG54 expression was quantified by qPCR. e, Time course and dose—response analysis of miR-122 downregulation by IFNβ: Huh7 cells were stimulated as described in c and d, and miR-122 was quantified by qPCR. Error bars, means ± s.d. of at least four independent experiments.
miR-122 is specifically expressed in the liver, and previous studies using anti-miRNAs elegantly demonstrated that miR-122, and consequently Dicer15, are essential for HCV replication2. We therefore tested whether miR-122 was also subject to regulation by IFNβ. As shown in Fig. 1e, IFNβ-stimulation of Huh7 cells resulted in a transient, but pronounced (~80%) down-modulation of miR-122. In a similar manner to miRNA induction, 100 U ml−1 IFNβ induced maximal attenuation of miR-122 expression, and no additional effect was observed with increased IFNβ concentrations (Fig. 1e).
To evaluate whether the eight IFNβ-induced miRNAs with sequence matches in the HCV genome are indeed capable of inhibiting HCV replication, we transfected synthetic miRNA-mimics corresponding to these miRNAs into Huh-7 cells that harbour an autonomously replicating, dicistronic full-length HCV replicon16,17. An anti-miRNA against miR-122 served as a positive control, because it had previously been shown to significantly reduce the abundance of replicon RNA in this system2. As expected, non-specific control miRNAs (individually or combined) or anti-miRNA oligos did not alter the amounts of HCV replicon RNA, whereas introduction of anti-miR-122 resulted in a ~70% reduction in viral RNA levels, as previously reported (Fig. 2a). Transfection of the eight candidate miRNAs individually revealed that miRNAs miR-196, miR-296, miR-351, miR-431 and miR-448 were indeed able to substantially attenuate viral replication, whereas miRNAs miR-1, miR-30 and miR-128 were without effect (Fig. 2a). Transfection of a mixture of the five functional miRNA-mimics yielded a >80% reduction in viral RNA load. As IFNβ induces miRNAs miR-196, miR-296, miR-351, miR-431 and miR-448 but downregulates miR-122, we decided to imitate this cellular response by transfecting the mix of miRNA-mimics in combination with anti-miR-122. Indeed, a small, but reproducible additive inhibitory effect on HCV replication was observed that was similar in efficacy to treatment of the cells with IFNβ (Fig. 2a). Virtually identical results were obtained when Huh7 cells were infected with live JFH1 HCV and either viral RNA levels (Fig. 2b) or viral foci formation (Supplementary Fig. 2) were analysed, further corroborating the fact that the IFNβ-induced alterations in the miRNA expression profile endow the cells with a pronounced antiviral state.
Figure 2. IFNβ-induced miRNAs display anti-viral activity against HCV.

a, JFH1-replicon-containing Huh7 cells were transfected with single non-specific miRNA-mimics (miR-ctrl), a pool of control miRNAs (miR-ctrl(mix5)) or anti-miRNAs (anti-miR-ctrl), or specific miRNA-mimics corresponding to the eight IFNβ-induced miRNAs, or specific anti-miRNAs. In addition, a combination of the five miRNAs that displayed anti-viral activity individually was used (miR-mix5) with or without anti-miR-122, as indicated, and HCV RNA was quantified by qPCR after 48 h. b, Same as a, except Huh7 cells were infected with live JFH-1 virus for 48 h (error bars, means ± s.d. of at least four independent experiments; P values are from paired Student’s t-tests).
To investigate whether these anti-viral miRNAs are targeting the HCV genome rather than inducing a non-specific antiviral state through alteration of cellular gene expression, we took advantage of the fact that the J6CF HCV molecular clone has single-nucleotide variations in the predicted target seed sequences for miR-196 and miR-448 as opposed to JFH1 (Fig. 3a). As J6CF is non-infectious in vitro, we employed an infection-competent J6CF/JFH1 chimaeric virus that carried the ‘mutant’ miR-448 and miR-196 target sites (chimaera J6/JFH) (Fig. 3a). As anticipated, miR-196 was effective against JFH1, but not against J6/JFH, which contained the ‘mutant’ target site. Similarly, miR-448 only inhibited the replication of JFH1 containing the ‘correct’ target site, but was ineffective against J6/JFH (Fig. 3b). Introduction of a compensatory single nucleotide change into miR-196 and miR-448 (designated miR-196* and miR-448*) to match their seed sequence to J6/JFH yielded a reversed efficacy profile compared to the wild-type miRNAs when they were tested against the two viruses. As such, both miR-196* and miR-448* were unable to subdue replication of JFH1, but clearly inhibited replication of J6/JFH (Fig. 3b). Together, these results suggest that at least miR-196 and miR-448 are directly targeting the HCV genomic RNA.
Figure 3. IFNβ-induced miRNAs directly target viral genomic RNA.

a, b, An infectious chimaeric virus was constructed from JFH1 and J6CF as shown in a; the numbers in brackets indicate the location in the virion (nucleotides) that the miRNA binds. The Huh7-cell subclone Huh7.5.1c2 was transfected with either miR-196 or miR-448, or with the mutant miR-196* and miR-448* harbouring a compensatory mutation in the seed sequence as outlined in a. Transfected Huh7.5.1c2 cells were infected with JFH1(D183) (ref. 23) or chimaeric J6/JFH, and HCV RNA was quantified by qPCR after 24 or 60 h post infection, respectively, during the phase of exponential viral RNA amplification, to accommodate the difference in replication kinetics between the two viruses (error bars, means ± s.e.m. of 8 independent experiments; P values are from paired Student’s t-tests).
Although the above described experiments demonstrate that IFNβ-induced miRNAs, in conjunction with the downregulation of miR-122, are sufficient to induce an antiviral state, the question remained whether this modulation of cellular miRNA levels was necessary for IFNβ to prevent HCV replication. To this end, we decided to counteract the IFNβ-elicited changes in miRNA expression by transfecting anti-miRNAs against miRNAs miR-196, miR-296, miR-351, miR-431 and miR-448, with and without the inclusion of an miR-122 mimic. IFNβ treatment leads to a >90% reduction in the amount of viral HCV replicon RNA and this inhibition is unaffected by transfected non-specific control anti-miRNAs. As shown in Fig. 4, introduction of the anti-miRNA mix, or of the miR-122 mimic separately, attenuated the IFNβ effect to ~75% inhibition. Co-transfection of the anti-miRNA mix and the miR-122 mimic further reduced the efficacy of IFNβ to ~50%, indicating that modulation of the expression levels of the identified miRNAs has an important, albeit not exclusive, role in the antiviral effects of IFNβ against HCV. It remains to be determined whether alterations in the expression levels of the identified cellular miRNAs account for only a part of the antiviral response, or if additional unidentified miRNAs that have not been ‘neutralized’ by anti-miRNAs mediate the remaining antiviral IFNβ effects.
Figure 4. IFNβ-induced miRNAs mediate anti-viral IFNβ responses against HCV.
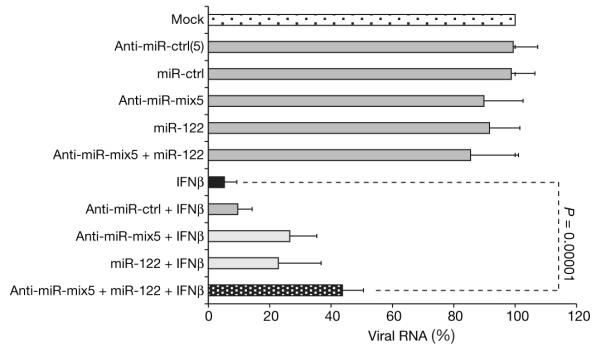
JFH1-replicon-containing Huh7 cells were transfected with non-specific miRNA-mimics (miR-ctrl), or non-specific anti-miRNAs (anti-miR-ctrl), or a pool of anti-miRNAs (anti-miR-ctrl(5)), or a combination of anti-miRNA complementary to the five IFNβ-induced miRNAs and with potent anti-viral effect (anti-miRNA-mix5), and/or with specific miRNAs and anti-miRNAs, as indicated, before stimulation with IFNβ for 48 h. HCV RNA was quantified by qPCR (means ± s.d. of at least four independent experiments; P values are from paired Student’s t-tests).
In summary, our results demonstrate that IFNα/β upregulates several cellular miRNAs that are capable of inhibiting HCV replication and infection. In addition, downregulation of miR-122 in response to IFNβ further contributes to the antiviral effects of this cytokine. These findings not only offer a new model of the host defence mechanisms that exist in mammalian cells, but also add a new component to the antiviral arsenal employed by interferons. Furthermore, although ample documentation is present in the literature for the developmentally regulated or tissue-specific expression of miRNAs and their dysregulation in malignant cells18,19, little evidence existed for a direct and immediate transcriptional regulation of miRNAs by an endogenous ligand such as a cytokine or growth factor. Our results provide some of the first evidence for rapid changes in cellular miRNA levels in response to ligand stimulation. During the preparation of this manuscript, it was reported that IFNβ induced upregulation of miR-155 (ref. 20), a miRNA with, as of yet, no identified targets. The delayed induction kinetics observed in their studies supports the model that miR-155 upregulation is mediated by TNFα as part of an autocrine response elicited by IFNβ, rather than by IFNβ itself20. Nevertheless, this study and our present report provide the first examples of rapid modulation of cellular miRNAs as components of the mammalian innate immune response.
METHODS SUMMARY
Cell culture
The human hepatoma-derived cell lines Huh7 and their JFH-1-replicon-containing subclone were maintained in DMEM with 10% FCS/10mM Hepes, penicillin/streptomycin and 2 mM L-glutamine. The JFH-1 full-length genomic replicon construct pFGR-JFH-1 was obtained from T. Wakita21. A Huh7 cell clone that stably replicates the full-length genomic HCV RNA was selected and used in these experiments, as described previously22. The Huh7.5.1c2 cell line was derived from curing replicon-containing Huh-7.5.1 cells by interferon treatment. Primary murine hepatocytes (BalbC) were collected after collagenase perfusion of the livers for 30 min at 37 °C.
miRNA array analysis
Microarray analysis of total RNA of interferon-stimulated lymphocytes was performed at the Ohio State University Comprehensive Cancer Center Microarray Shared Resource, as described23.
RNA extraction
Total RNA was isolated using TriZol according to the manufacturer’s instructions.
Real-time PCR
Quantification of HCV genomic RNA was performed using HCV- and β-actin- or GAPDH-specific primers as described16. Real-time PCR-based quantification of miRNAs was performed using miRNA analysis kits specific for each individual miRNA (Applied Biosystems), according to the manufacturers instructions.
METHODS
Transfection of miRNA mimics and anti-miRNAs into Huh7 or Huh7 replicon cells
When JFH1-replicon-containing Huh7 cells (70% confluent) were used for transfection experiments, constant total amounts of miRNA mimics or anti-miRNAs (Dharmacon and Applied Biosystems, respectively) were combined with Mirus transfection reagent (Mirus Bio/Fisher Scientific) according to the manufacturers instructions and added to the cells (transfection efficiency >90%). Where indicated, IFNβ (DBL Biomedical Laboratories) was added to the cultures after 8 h, and cells were harvested 48 h post transfection. Viral replication was determined by real-time PCR analysis of viral genomic RNA. Similarly, for experiments using infectious HCV, Huh7 cells were transfected with miRNAs and anti-miRNAs, as described above. HCV infection (multiplicity of infection = 0.05) was performed 8 h post transfection, and IFNβ was added where indicated to the cultures at the same time. Cells were harvested at the indicated time points during the phase of exponential viral RNA amplification, and viral replication was determined by real-time PCR analysis of viral genomic RNA, or by determination of the number and size of viral foci after 72 h.
J6CF—JFH1 chimaeric HCV genomes
Recombinant PCR was used to replace the J6CF NS5B-3′ UTR region in pCV-J6CF24 with the corresponding sequences from JFH-1 present in pUC-vJFH25. First, using primers XbaJFH (GATTACGCCAAGCTTGCATGCCTGCAG), and NS5Bup (CTCCATGTCATACTCCTGGACCGGGGCTC) and primers NS5Blo (GAGCCCCGGTCCAGGAGTATGACATGGAG) and XhoJ6CF (AGGTTCCATCTCTTCCATGCCCCCCCTCG) the NS5B-3′ UTR region of JFH-1 and an NS5A-NS5B fragment containing a unique XhoI restriction site from J6CF were amplified, respectively. The two PCR products were mutually extended and amplified using the primers XhoJ6CF and XbaJFH, and the resultant PCR product was cloned into pGEM-Teasy (Invitrogen) yielding pTe-J6/JFH, and the insert was verified by DNA sequencing. Finally, the XhoI to XbaI fragment in pCV-J6CF was replaced by the 2.1-kb XhoI/XbaI fragment excised from pTe-J6/JFH, yielding pCV-J6CF/JFHNS5B3′UTR containing the J6/JFH chimaeric HCV genome with the NS5B-3′ UTR of J6CF replaced by the corresponding sequences from JFH-1. A similar recombinant PCR approach was used to replicate the J6CF—JFH-1 chimaeric HCV genome Jc126 by replacing the corresponding JFH-1 core-NS2 region in pUC-vJFH with the corresponding sequences from J6CF to yield the plasmid pUC-vJc1. Infectious JFH-1 and chimaeric viruses were produced by transfection of in vitro synthesized genomic HCV RNA into Huh-7.5.1 cells and virus stocks containing 104–105 focus forming units per ml (f.f.u. ml−1) were prepared as described previously27.
Supplementary Material
Acknowledgements
We thank G. Calin (OSU) for help with the microarray studies, D. Burton (TSRI) for E2-antibodies, and J. Zhong (TSRI) for many discussions. This work was supported by the NIH (I.M.P.), a private donation from C. Evans to F.V.C, and the NIH and UCSD Academic Senate Funding (M.D.).
Footnotes
Full Methods and any associated references are available in the online version of the paper at www.nature.com/nature.
Supplementary Information is linked to the online version of the paper at www.nature.com/nature.
Author Information Reprints and permissions information is available at www.nature.com/reprints.
References
- 1.Cullen BR. Is RNA interference involved in intrinsic antiviral immunity in mammals? Nature Immunol. 2006;7:563–567. doi: 10.1038/ni1352. [DOI] [PubMed] [Google Scholar]
- 2.Jopling CL, Yi M, Lancaster AM, Lemon SM, Sarnow P. Modulation of hepatitis C virus RNA abundance by a liver-specific microRNA. Science. 2005;309:1577–1581. doi: 10.1126/science.1113329. [DOI] [PubMed] [Google Scholar]
- 3.Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–297. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- 4.Ambros V. The functions of animal microRNAs. Nature. 2004;431:350–355. doi: 10.1038/nature02871. [DOI] [PubMed] [Google Scholar]
- 5.Lewis BP, Burge CB, Bartel DP. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell. 2005;120:15–20. doi: 10.1016/j.cell.2004.12.035. [DOI] [PubMed] [Google Scholar]
- 6.Zamore PD. Plant RNAi: How a viral silencing suppressor inactivates siRNA. Curr. Biol. 2004;14:R198–R200. doi: 10.1016/j.cub.2004.02.021. [DOI] [PubMed] [Google Scholar]
- 7.Baulcombe D. RNA silencing in plants. Nature. 2004;431:356–363. doi: 10.1038/nature02874. [DOI] [PubMed] [Google Scholar]
- 8.Katze MG, He Y, Gale M., Jr. Viruses and interferon: a fight for supremacy. Nature Rev. Immunol. 2002;2:675–687. doi: 10.1038/nri888. [DOI] [PubMed] [Google Scholar]
- 9.Samuel CE. Antiviral actions of interferons. Clin. Microbiol. Rev. 2001;14:778–809. doi: 10.1128/CMR.14.4.778-809.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liang TJ, Rehermann B, Seeff LB, Hoofnagle JH. Pathogenesis, natural history, treatment, and prevention of hepatitis C. Ann. Intern. Med. 2000;132:296–305. doi: 10.7326/0003-4819-132-4-200002150-00008. [DOI] [PubMed] [Google Scholar]
- 11.Lauer GM, Walker BD. Hepatitis C virus infection. N. Engl. J. Med. 2001;345:41–52. doi: 10.1056/NEJM200107053450107. [DOI] [PubMed] [Google Scholar]
- 12.Wieland SF, Chisari FV. Stealth and cunning: hepatitis B and hepatitis C viruses. J. Virol. 2005;79:9369–9380. doi: 10.1128/JVI.79.15.9369-9380.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rehermann B, Nascimbeni M. Immunology of hepatitis B virus and hepatitis C virus infection. Nature Rev. Immunol. 2005;5:215–229. doi: 10.1038/nri1573. [DOI] [PubMed] [Google Scholar]
- 14.Larner AC, et al. Transcriptional induction of two genes in human cells by beta interferon. Proc. Natl Acad. Sci. USA. 1984;81:6733–6737. doi: 10.1073/pnas.81.21.6733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Randall G, et al. Cellular cofactors affecting hepatitis C virus infection and replication. Proc. Natl Acad. Sci. USA. 2007;104:12884–12889. doi: 10.1073/pnas.0704894104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhong J, et al. Robust hepatitis C virus infection in vitro. Proc. Natl Acad. Sci. USA. 2005;102:9294–9299. doi: 10.1073/pnas.0503596102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Moradpour D, et al. Insertion of green fluorescent protein into nonstructural protein 5A allows direct visualization of functional hepatitis C virus replication complexes. J. Virol. 2004;78:7400–7409. doi: 10.1128/JVI.78.14.7400-7409.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Calin GA, Croce CM. MicroRNA—cancer connection: the beginning of a new tale. Cancer Res. 2006;66:7390–7394. doi: 10.1158/0008-5472.CAN-06-0800. [DOI] [PubMed] [Google Scholar]
- 19.Calin GA, Croce CM. MicroRNA signatures in human cancers. Nature Rev. Cancer. 2006;6:857–866. doi: 10.1038/nrc1997. [DOI] [PubMed] [Google Scholar]
- 20.O’Connell RM, Taganov KD, Boldin MP, Cheng G, Baltimore D. MicroRNA-155 is induced during the macrophage inflammatory response. Proc. Natl Acad. Sci. USA. 2007;104:1604–1609. doi: 10.1073/pnas.0610731104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kato T, et al. Efficient replication of the genotype 2a hepatitis C virus subgenomic replicon. Gastroenterology. 2003;125:1808–1817. doi: 10.1053/j.gastro.2003.09.023. [DOI] [PubMed] [Google Scholar]
- 22.Cheng G, Zhong J, Chisari FV. Inhibition of dsRNA-induced signaling in hepatitis C virus-infected cells by NS3 protease-dependent and -independent mechanisms. Proc. Natl Acad. Sci. USA. 2006;103:8499–8504. doi: 10.1073/pnas.0602957103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu CG, et al. An oligonucleotide microchip for genome-wide microRNA profiling in human and mouse tissues. Proc. Natl Acad. Sci. USA. 2004;101:9740–9744. doi: 10.1073/pnas.0403293101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yanagi M, Purcell RH, Emerson SU, Bukh J. Hepatitis C virus: an infectious molecular clone of a second major genotype (2a) and lack of viability of intertypic 1a and 2a chimeras. Virology. 1999;262:250–263. doi: 10.1006/viro.1999.9889. [DOI] [PubMed] [Google Scholar]
- 25.Wakita T, et al. Production of infectious hepatitis C virus in tissue culture from a cloned viral genome. Nature Med. 2005;11:791–796. doi: 10.1038/nm1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pietschmann T, et al. Construction and characterization of infectious intragenotypic and intergenotypic hepatitis C virus chimeras. Proc. Natl Acad. Sci. USA. 2006;103:7408–7413. doi: 10.1073/pnas.0504877103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhong J, et al. Robust hepatitis C virus infection in vitro. Proc. Natl Acad. Sci. USA. 2005;102:9294–9299. doi: 10.1073/pnas.0503596102. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.