

Abstract
Oxidative damage and inflammation are postulated to be involved in age-related macular degeneration (AMD). However, the molecular signal(s) linking oxidation to inflammation in this late-onset disease is unknown. Here we describe AMD-like lesions in mice after immunization with mouse serum albumin adducted with carboxyethylpyrrole, a unique oxidation fragment of docosahexaenoic acid that has previously been found adducting proteins in drusen from AMD donor eye tissues1 and in plasma samples2 from individuals with AMD. Immunized mice develop antibodies to this hapten, fix complement component-3 in Bruch's membrane, accumulate drusen below the retinal pigment epithelium during aging, and develop lesions in the retinal pigment epithelium mimicking geographic atrophy, the blinding end-stage condition characteristic of the dry form of AMD. We hypothesize that these mice are sensitized to the generation of carboxyethylpyrrole adducts in the outer retina, where docosahexaenoic acid is abundant and conditions for oxidative damage are permissive. This new model provides a platform for dissecting the molecular pathology of oxidative damage in the outer retina and the immune response contributing to AMD.
AMD is the most common cause of legal blindness in elderly individuals of industrialized countries3,4. Clinicians have long recognized that debris (termed drusen) below the retinal pigment epithetlium (RPE) in the macula is a risk factor for AMD. The presence of complement factor proteins in drusen in AMD eyes1,5–7 and genetic variation in several complement factor genes in individuals with AMD8–13 implicate inflammation as an important component in this disease. However, little is known about the signal(s) from the outer retina that initiates the immune system's involvement in AMD.
As a potential initiating signal we evaluated carboxyethylpyrrole (CEP; Fig. 1a), an adduct that forms from an oxidation fragment of docosahexaenoic acid (DHA)2. DHA, the most oxidizable of all long-chain polyunsaturated fatty acids, is abundant in the outer retina, where high oxygen tension and light provide a permissive environment for oxidation14–16. CEP forms when the aldehyde group on a newly formed seven-carbon oxidation fragment of DHA covalently interacts with an ε-lysyl amino group in a tissue protein1,2. AMD donor eyes contain more CEP-modified proteins in the outer retina than are present in age-matched controls1, and CEP-adducted proteins are also more abundant in AMD plasma than in control samples2. CEP autoantibodies are also present in plasma and are more abundant in AMD than in controls2. This oxidation-generated hapten is noteworthy because of the long-recognized association of AMD with oxidative damage17–21.
Figure 1.

CEP antigen and CEP-specific antibody titers in immunized mice. (a) Drawing of CEP adducted to MSA. (b) SDS-PAGE gel (left) and western blot for CEP (right) showing the MSA (lane 1) and two preparations of the CEP-adducted MSA (lanes 2 and 3, respectively) used. The bolus of the MSA protein in lane 1 is between 66 and 97 kDa. Lanes 2 and 3 show a higher mass range after adduction with CEP. Molar ratios of CEP to MSA were 11:1 in lane 2 and 6.6:1 in lane 3. The western blot shows strong CEP immunoreactivity in lanes 2 and 3. Minor CEP immunoreactivity is present in lane 1 (between the brackets). MWM, molecular weight marker. (c) CEP-specific antibody titer at the end of the short-term protocol (3 months) in each of the six treatment groups. (d) CEP-specific antibody titer at the end of the long-term protocol (12–14 months) in mice immunized with CEP-MSA or the indicated control treatments. Paired comparisons between CEP-MSA and all other groups are indicated with P values. NS, not significant.
Because DHA is concentrated in RPE and photoreceptor cells14–16, we reasoned that these tissues are a probable source of CEP adducts during aging. Because serum albumin is one of the major proteins modified with CEP in people with AMD2, we immunized mice with CEP-modified mouse serum albumin (CEP-MSA) in an attempt to raise the level of sensitivity to endogenously generated CEP (Fig. 1b). Our hypothesis was that immunized mice would generate a stronger immune response to CEP adducts, making the outer retina more vulnerable to immune-mediated damage.
We immunized mice with CEP-MSA or MSA controls in complete Freund's adjuvant (CFA) at day 0 and followed this with a challenge at day 10 in incomplete Freund's adjuvant (IFA). To generate a strong immunological response and confirm that CEP-MSA is immunogenic, a group of ‘short-term’ experiments were performed in which animals received three immunizations during a 2–3–month period, with the final immunization given 10 d before the mice were killed. To assess the effect of aging on the CEP-MSA immune response, a second group of ‘long-term’ experiments were performed in which mice were treated with only the first immunization described above and maintained for 12–14 months before they were killed. (Mice used are listed in Supplementary Table 1 online.)
Titers of antibody to CEP in the short-term recovery mice were six to eight times higher in the CEP-MSA–immunized mice compared to naive and control mice immunized with MSA or CFA (Fig. 1c). CEP-specific antibody was not detected in Rag–deficient mice that lack mature T and B cells. Antibody titers were similar in the long-term recovery mice, with CEP-specific antibody titers higher in the CEP-MSA–immunized mice than in the control mice (Fig. 1d). These data show that CEP-MSA can induce an antibody-mediated immune response.
Histology of the CEP-MSA eyes revealed multiple RPE changes, including vesiculation and swelling of individual or multiple adjacent cells (Fig. 2a–c); cell lysis (Fig. 2b,c); pyknosis, as evidenced by intense toluidine blue staining (Fig. 2b); and the presence of monocytes in the interphotoreceptor matrix (Fig. 2d,e). Some of the invading cells expressed the macrophage marker F4/80 (Fig. 2e). RPE expanses with up to 14 contiguous swollen cells that appeared to be undergoing lysis were common (Fig. 2f). In some eyes, the RPE was missing in focal areas and the overlying photoreceptors were greatly swollen (Fig. 2g and Supplementary Figs. 1-4).
Figure 2.
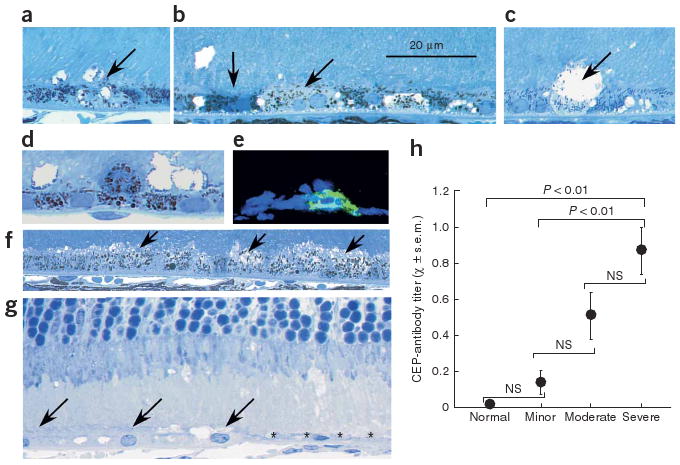
Relationship between pathology and CEP-specific antibody titer in the short-term immunization mice. (a–g) Histology of CEP-MSA eyes. The RPE is located across the lower one-third to one-half of each image, and vesiculation of RPE cells is evident (oblique arrows in a–c and f). Darkly staining RPE cells are present, suggesting pyknosis (vertical arrow, lower left of b). RPE lysis is shown in b and c. A monocyte is located above the RPE layer in d. In e, frozen section stained with the macrophage marker F4/80 is shown. In f, several adjacent RPE cells (arrows) show vesiculation similar to that in a and b. In g, a portion of the RPE layer in a BALB/c mouse is absent (* *) but present on the left (arrows), and the photoreceptors above the RPE-free area are swollen. (h) Comparison of RPE pathology and CEP-specific antibody titer. Only the antibody titers of the severe group were different (at the indicated P values) when tested against the antibody titers in the normal and minor pathology groups.
The degree of pathology in the short-term tissues was evaluated in three or four sections per eye from the dorsal to the ventral retinal margin. Each sample was placed in one of the following categories based on the average number of lesions present per section: normal, minor pathology, moderate pathology or severe pathology. Two of the six naive mice studied had one and three lysed RPE cells, respectively, and one of these also contained a single pyknotic RPE cell. Because these were naive mice that had not been immunized, these lesions were considered normal background variability (Supplementary Fig. 1). When four or fewer lesions were found, the tissue was scored as normal. Samples in the minor pathology category contained five to ten separate areas of RPE or outer retinal pathology in each section studied (Supplementary Fig. 2). Samples in the moderate pathology category contained 11–15 separate sites of RPE or outer retinal pathology (Supplementary Fig. 3). Mice in the severe pathology category had 16 or more sites of RPE pathology in each section. Four eyes in this group had lesions in the RPE that were 20–70 μm long (Supplementary Fig. 4). We then plotted the degree of pathology against the mean antibody titer of the mice in these groupings (Fig. 2h). These data show a close relationship between the CEP-specific antibody titer and the severity of outer retina pathology.
C3d is a degradation product of C3b, a key complement protein required for the generation of the C3 and C5 convertases in the classical, lectin and alternate pathways22. We evaluated short-term tissues for C3d immunolocalization (Fig. 3a–h). C3d was observed in Bruch's membrane below the RPE in the mice receiving CEP-MSA immunization (Fig. 3b). Some of the CEP-MSA tissues contained lysed RPE cells that could be labeled with an antibody to C3d (Fig. 3c), suggesting that complement may be fundamentally involved in the lysis of these cells. Although the C3d immunofluorescence was somewhat patchy in eyes from CEP-MSA–immunized mice, it was not observed in control mice (Fig. 3d,e), except for minor fluorescence occasionally seen in the MSA–treated mice (Fig. 3d). CEP-MSA–immunized mice showed ten times more immunofluorescence in Bruch's membrane than did control mice (Fig. 3f–h).
Figure 3.

C3d localization in the outer eye wall of the short-term immunization mice. (a) Frozen section showing the four tissue lamina in a differential interference contrast image. (b–e) Confocal images of Bruch's membrane in mice. (b) Confocal image of the outer eye wall from a CEP-MSA–immunized mouse showing the bright green fluorescence of the C3d-FITC probe along Bruch's membrane (arrow). (c) Image from another CEP-MSA–immunized mouse in an area where Bruch's membrane (arrow) labeling is not pronounced but intense C3d labeling is associated with two lysed RPE cells (on upper left of image) with profiles similar to those illustrated in the histology presented in Figure 2b–d. (d) Minor C3d labeling of Bruch's membrane (arrow) is present in a mouse immunized with non-adducted MSA. Differences in intensity and distribution of FITC fluorescence are present in b, c and d. (e) No C3d localization is evident in Bruch's membrane (arrow) in a naive mouse. Red fluorescence is the nuclear stain propidium iodide. Fluorescent intensity in an MSA immunized mouse (f) and a CEP-MSA-immunized mouse (g) was measured in the overlays shown. The long axis on the overlay represents 150 μm. Images in a–g are at identical magnification. (h) C3d fluorescence intensity from the groups specified. C3d immunofluorescence in the CEP-MSA group is five to six times higher than in the other groups.
Examination of the fundus from the long-term protocol mice revealed patchy, reticular changes not present in controls (Supplementary Fig. 5 online). Histology of 15 eyes from the long-term protocol revealed an accumulation of sub-RPE deposits in the CEP-MSA-immunized mice (compare Fig. 4a to Fig. 4b and Fig. 4c to Fig. 4d). These deposits stained less intensely than did the RPE cytoplasm, forming a near continuous band of variable thickness (2–4 μm) below the RPE (Supplementary Fig. 6 online). Choroidal neovascularization was not observed. Sub-RPE material did not develop in controls, although increased thickening of RPE basal infoldings with aging was evident.
Figure 4.

Light and electron microscopy of the RPE–Bruch's membrane interface with quantitative comparisons. (a–d) Light microscopy images. Images in a and c are from the 1-year naive control mouse, and images in b and d are from the long-term CEP-MSA mice. The basal side of the RPE in a is thinner than in b. In c and d this sub-RPE area is delineated by two lines (shown in red). Images in a–d are at identical magnification. (e,f) Area comparisons of basal RPE infoldings in short-term (e) and long-term (f) mice. (g,h) Electron micrographs of the interface between RPE and Bruch's membrane from a 1-year naive control mouse (g) and a 1-year CEP-MSA–immunized mouse (h). The RPE basal lamina is aligned along the horizontal double arrow. The basal lamina of the choriocapillaris is thicker than that of the RPE and is indicated in both images by the oblique double arrow. Bruch's membrane in h is thicker than in g, shown by the length of the vertical line across Bruch's membrane on the left in g and right in h. The height of the basal infoldings is indicated by the vertical line along the right margin of g. In g, basal infoldings are evident as parallel membranes, but in h they surround a flocculent, fluffy material that appears to be extracellular. The vertical bar length along the left side of this debris zone in h is greater than the length of the vertical bar on the right side of the basal infoldings in g.
Sub-RPE deposits were measured with an image-processing algorithm that recognizes Bruch's membrane and the basal infoldings and sub-RPE deposits in digitized images (Figs. 4c,d). We measured the area occupied by basal infoldings in short-term recovery mice (Fig. 4e) and the area of sub-RPE deposits plus basal infoldings in long-term recovery mice (Fig. 4f). Short-term recovery tissues had similar basal infolding dimensions (Fig. 4e). All eyes in the long-term protocol showed greater thickening below the RPE than was measured in the short-term tissues, except for the Rag-deficient mice. This probably reflects age-related changes. However, the CEP-MSA–immunized mice showed greater accumulation of basal deposits than with any other treatment (Fig. 4f).
In electron micrographs, the Bruch's membrane of long-term naive mice measured 0.54 ± 0.06 μm (mean ± s.d.) thick, and RPE basal infoldings had a mean height of 1.19 ± 0.12 μm (Fig. 4g). In contrast, Bruch's membrane in CEP-MSA tissues measured 1.71 ± 0.05 μm. Basal infoldings were evident in CEP-MSA samples, but these profiles surrounded extracellular flocculent material that expanded this sub-RPE compartment to a thickness of 4.21 ± 1.14 μm (Fig. 4h). Similar sub-RPE debris is reported in mice subjected to dietary, lighting and/or genetic manipulations23–26.
Our studies demonstrate that mice mount an antibody-mediated response to CEP-MSA. The immune system responds by depositing complement below the RPE, as evidenced by the localization of C3d in Bruch's membrane and the development of lytic changes in RPE cells. An intact immune system is required for this process, because CEP-MSA–immunized Rag-deficient mice, which are missing mature T cells and B cells27, showed none of the changes observed in normal mice.
Although macrophages were found near some RPE lesions, it is unlikely that they initiate the pathology observed, as many lesions occurred in the absence of these cells. Macrophage movement into this compartment may be due to the release of cytokines from lysed cells. Indeed, melanin-containing macrophages were observed, suggesting that there is debris removal after RPE lysis (Fig. 2d). Aging mice deficient in the macrophage chemokine Ccl2 or its receptor Ccr2 also show features similar to those in AMD23, suggesting a role for macrophages in maintaining the outer retina.
To our knowledge, this is the first study showing that immunization with a hapten generated by oxidative damage to the DHA28 present in the drusen1 and plasma from AMD-affected individuals2 is sufficient to produce AMD-like lesions in mice. This model provides a new resource for understanding the early changes in the outer retina in AMD, as well as the disease progression in mice with mutations or polymorphisms in complement pathway genes that are linked to AMD in humans.
Methods
Mice
We used C57BL/6 (Jackson Labs) and BALB/c (Taconic Labs) mice of both sexes at 2–3 months of age. The Rag-deficient mice were on a C57BL/6 background. Protocols used here were approved by the Institutional Animal Care and Use Committee at the Cleveland Clinic.
Antigens
We prepared the CEP-MSA and CEP-BSA from commercially available mouse serum albumin and bovine serum albumin (Sigma), which we converted to CEP-modified MSA and BSA following published procedures28.
Immunization
We used standard mouse immunization protocols29. We anesthetized mice with ketamine-xylazine in PBS (80–90 mg/kg ketamine, 2–10 mg/ml xylazine). We used 200 μg of CEP-MSA in CFA or IFA (Difco Labs) for initial and all booster doses. We used the following control mice for comparisons with CEP-MSA immunizations: mice immunized with non-adducted MSA (200 μg per inoculum), mice injected with CFA and subsequently boosted with IFA without any antigen, and age-matched naive mice. We also used Rag-deficient mice for identical immunization protocols.
Carboxyethylpyrrole-specific antibody assay
We performed direct ELISA for the detection of antibody to CEP in 96-well plates coated with CEP-BSA (100 μl/well) at a 1:1,000 dilution in PBS and incubated at 37 °C for 1 h, using 1% BSA solution (Sigma) as a blank control. We used CEP-BSA as the coating agent (Supplementary Fig. 7 online). We washed the plate with PBS three times (300 μl/well) and blocked with 1% ovalbumin in PBS (Sigma) and then incubated at 37 °C for 1 h. We then washed the plate with 0.1% ovalbumin and 0.05% Tween-20 before loading the standards and mouse serum samples (at 1:10, 1:100 and 1:1,000 dilutions) and incubating them at 22 °C on a shaker for 1 h. We applied the alkaline phosphatase–conjugated secondary antibody to mouse IgG (Sigma) at a 1:2,000 dilution at 22 °C for 1 h, then rinsed and incubated the plate with Fast p-nitrophenyl phosphate tablets (Sigma). We measured absorbance (at 405 nm, using 655 nm as a reference) on a Spectra Max Plus (Molecular Devices) and defined the titer as the ratio of serum binding to antigen versus serum binding to BSA. The antibody used on this western blot was described previously2.
Fundus photography
We examined the mouse fundus at various ages and post-immunization times. We anesthetized and dilated the mice as described above. We used a handheld Kowa Genesis-D digital retinal camera (Kowa Optimed) and a 90-mm intermediate lens.
Histology
We prepared one eye from each mouse for histopathology and electron microscopy. We fixed the eye in 2% glutaraldehyde and 2% formaldehyde (freshly prepared from paraformaldehyde) in 0.1 M cacodylate buffer, pH 7.2 at 4 °C overnight, then post-fixed it in 1% osmium tetroxide, dehydrated it in graded ethanol, and then in propylene oxide, and then transferred it to a plastic resin mixture containing Polybed 812 (Polysciences) and Araldite 502 (Polysciences) with polymerizer. After polymerization, we cut five to ten 1-μm-thick sections, mounted them on microscope slides and stained them with toluidine blue.
Image analysis
We photographed retinal sections with a Zeiss Axiophot microscope equipped with a Hamamatsu digital camera using a 63× oil-immersion lens. For quantitative analysis of Bruch's membrane and sub-RPE boundaries, we took retinal images from three sections in the fundus of each of three eyes per treatment group, and we then batch-processed them with customized macros and algorithms generated for Image-Pro Plus 6.1 (Media Cybernetics).
Immunocytochemistry
We froze one eye from each mouse in optimal cutting temperature medium for immunocytochemistry. We prepared 7-μm cryosections through the center of each eye on a cryostat HM 505E (Microm). For C3d localization, we used a polyclonal rabbit antibody to human C3d (Dako) followed by the secondary antibody (FITC-conjugated swine antibody to rabbit immunoglobulin, Dako). We mounted the samples onto slides with Vectashield and examined them for fluorescence. For F4/80 localization, we used rat antibody to mouse F4/80 antigen (Serotec), followed by secondary antibody (Alexa Fluor 488–conjugated goat antibody to rabbit IgG, Molecular Probes). We mounted the sections with Vectashield and evaluated them with a Zeiss Axiophot microscope equipped with epifluorescence illumination. The mounting medium contained either propidium iodide or DAPI for nuclear counterstaining. We performed the final imaging on a Leica TCS-SP (Exton) confocal microscope.
To compare fluorescence intensities in the C3d immunocytochemistry, we acquired a series of 1.0-μm sections from the full retinal expanse. In the Leica confocal software, we saved the pinhole channel gain and offset as a macro and used it to collect all the images. We performed quantitative analysis using the stack profile function. We generated an area template with a dimension of 4,512 μm2 (similar to that shown in Fig. 3f,g). We moved the template onto each image and positioned it so that fluorescence levels in equal areas along Bruch's membrane could be measured. We generated a graphic with the mean fluorescence intensity in each section of the stack. We defined ten areas from each section and measured sections from three or four eyes per treatment.
Electron microscopy
We thin-sectioned selected blocks on an RMC MT-XL ultramicrotome, placed the sections on nickel grids, stained them with uranyl acetate and lead citrate and viewed them with a Tecnai 20 200-kV digital electron microscope equipped with a Gatan image filter. We evaluated at least three tissue blocks from each treatment and captured over 50 images from each block at a 3,500× magnification, and we measured the dimensions of the indicated compartments directly from prints.
Statistical analysis
We analyzed antibody titer and RPE basal deposit thickness data by ANOVA. Where the ANOVA was significant, we made pairwise comparisons using Dunnett's test.
Supplementary Material
Acknowledgments
Supported by the State of Ohio Biomedical Research and Technology Transfer Program, a Research Center Grant from the Foundation Fighting Blindness, and a Challenge Grant from Research to Prevent Blindness. The project was also supported by grants R56EY10240, R01EY014240, R24EY015638 (J.G.H.), R21EY017153 (V.L.B.), R01GM21249 (R.G.S.) and K08EY014912 (V.L.P.) from the US National Institutes of Health. The content is solely the responsibility of the authors and does not necessarily represent the official views of the US National Eye Institute of the National Institutes of Health. We thank J.W. Crabb for valuable discussions, B. Anand-Apte for critical comments on the manuscript, N.S. Peachey for help with the statistical comparisons, R.L. Fairchild for providing the Rag-deficient mice, Y. Li for help with the histology, K. Sayanagi and T. Yakamoto for the fundus photography and A. Vasanji for developing the image-analysis algorithm used to define the areas of sub-RPE deposits.
Footnotes
Note: Supplementary information is available on the Nature Medicine website.
Author Contributions J.G.H. and V.L.P. designed and initiated the experiments. L.L. and R.G.S. prepared the CEP-MSA and CEP-BSA. R.L.U., V.L.P., K.G.S. and X.Y. immunized the mice. K.G.S. and X.Y. performed the protein chemistry and ELISA assays, as well as managing the day-to-day maintenance of the mice. M.E.R. and J.G.H. performed all of the histological and electron microscopic analysis. V.L.B. performed the confocal microscopy. J.G.H. analyzed all of the data and wrote the manuscript. R.G.S., R.L.U. and V.L.P. made critical comments and suggestions for revisions of the manuscript in response to the reviewers.
Competing Interests Statement The authors declare competing financial interests: details accompany the full-text HTML version of the paper at www.nature.com/naturemedicine/.
Published online at http://www.nature.com/naturemedicine
Reprints and permissions information is available online at http://npg.nature.com/reprintsandpermissions
References
- 1.Crabb JW, et al. Drusen proteome analysis: an approach to the etiology of age-related macular degeneration. Proc Natl Acad Sci USA. 2002;99:14682–14687. doi: 10.1073/pnas.222551899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gu X, et al. Carboxyethylpyrrole protein adducts and autoantibodies, biomarkers for age-related macular degeneration. J Biol Chem. 2003;278:42027–42035. doi: 10.1074/jbc.M305460200. [DOI] [PubMed] [Google Scholar]
- 3.Javitt JC, Zhou Z, Maguire MG, Fine SL, Willke RJ. Incidence of exudative age-related macular degeneration among elderly Americans. Ophthalmology. 2003;110:1534–1539. doi: 10.1016/S0161-6420(03)00495-0. [DOI] [PubMed] [Google Scholar]
- 4.Augood CA, et al. Prevalence of age-related maculopathy in older Europeans: the European Eye Study (EUREYE) Arch Ophthalmol. 2006;124:529–535. doi: 10.1001/archopht.124.4.529. [DOI] [PubMed] [Google Scholar]
- 5.Hageman GS, et al. An integrated hypothesis that considers drusen as biomarkers of immune-mediated processes at the RPE-Bruch's membrane interface in aging and age-related macular degeneration. Prog Retin Eye Res. 2001;20:705–732. doi: 10.1016/s1350-9462(01)00010-6. [DOI] [PubMed] [Google Scholar]
- 6.Anderson DH, Mullins RF, Hageman GS, Johnson LV. A role for local inflammation in the formation of drusen in the aging eye. Am J Ophthalmol. 2002;134:411–431. doi: 10.1016/s0002-9394(02)01624-0. [DOI] [PubMed] [Google Scholar]
- 7.Johnson LV, Ozaki S, Staples MK, Erickson PA, Anderson DH. A potential role for immune complex pathogenesis in drusen formation. Exp Eye Res. 2000;70:441–449. doi: 10.1006/exer.1999.0798. [DOI] [PubMed] [Google Scholar]
- 8.Edwards AO, et al. Complement factor H polymorphism and age-related macular degeneration. Science. 2005;308:421–424. doi: 10.1126/science.1110189. [DOI] [PubMed] [Google Scholar]
- 9.Hageman GS, et al. A common haplotype in the complement regulatory gene factor H (HF1/CFH) predisposes individuals to age-related macular degeneration. Proc Natl Acad Sci USA. 2005;102:7227–7232. doi: 10.1073/pnas.0501536102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Haines JL, et al. Complement factor H variant increases the risk of age-related macular degeneration. Science. 2005;308:419–421. doi: 10.1126/science.1110359. [DOI] [PubMed] [Google Scholar]
- 11.Klein RJ, et al. Complement factor H polymorphism in age-related macular degeneration. Science. 2005;308:385–389. doi: 10.1126/science.1109557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gold B, et al. Variation in factor B (BF) and complement component 2 (C2) genes is associated with age-related macular degeneration. Nat Genet. 2006;38:458–462. doi: 10.1038/ng1750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yates JR, et al. Complement C3 variant and the risk of age-related macular degeneration. N Engl J Med. 2007;357:553–561. doi: 10.1056/NEJMoa072618. [DOI] [PubMed] [Google Scholar]
- 14.Anderson RE, Lissandrello PM, Maude MB, Matthes MT. Lipids of bovine retinal pigment epithelium. Exp Eye Res. 1976;23:149–157. doi: 10.1016/0014-4835(76)90198-6. [DOI] [PubMed] [Google Scholar]
- 15.Anderson RE. Lipids of the ocular tissues. IV. A comparison of the phospholipids from the retina of six mammalian species. Exp Eye Res. 1970;10:339–344. doi: 10.1016/s0014-4835(70)80046-x. [DOI] [PubMed] [Google Scholar]
- 16.Fliesler SJ, Anderson RE. Chemistry and metabolism of lipids in the vertebrate retina. Prog Lipid Res. 1983;22:79–131. doi: 10.1016/0163-7827(83)90004-8. [DOI] [PubMed] [Google Scholar]
- 17.Seddon JM, Willett WC, Speizer FE, Hankinson SE. A prospective study of cigarette smoking and age-related macular degeneration in women. J Am Med Assoc. 1996;276:1141–1146. [PubMed] [Google Scholar]
- 18.Klein R, Klein BE, Cruickshanks KJ. The prevalence of age-related maculopathy by geographic region and ethnicity. Prog Retin Eye Res. 1999;18:371–389. doi: 10.1016/s1350-9462(98)00025-1. [DOI] [PubMed] [Google Scholar]
- 19.Snow KK, Seddon JM. Do age-related macular degeneration and cardiovascular disease share common antecedents? Ophthalmic Epidemiol. 1999;6:125–143. doi: 10.1076/opep.6.2.125.1558. [DOI] [PubMed] [Google Scholar]
- 20.Christen WG, Glynn RJ, Manson JE, Ajani UA, Buring JE. A prospective study of cigarette smoking and risk of age-related macular degeneration in men. J Am Med Assoc. 1996;276:1147–1151. [PubMed] [Google Scholar]
- 21.Solberg Y, Rosner M, Belkin M. The association between cigarette smoking and ocular diseases. Surv Ophthalmol. 1998;42:535–547. doi: 10.1016/s0039-6257(98)00002-2. [DOI] [PubMed] [Google Scholar]
- 22.Peakman M, Senaldi G, Vergan D. Review: assessment of complement activation in clinical immunology laboratories: time for reappraisal? J Clin Pathol. 1989;42:1018–1025. doi: 10.1136/jcp.42.10.1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ambati J, et al. An animal model of age-related macular degeneration in senescent Ccl-2– or Ccr-2–deficient mice. Nat Med. 2003;9:1390–1397. doi: 10.1038/nm950. [DOI] [PubMed] [Google Scholar]
- 24.Gottsch JD, Bynoe LA, Harlan JB, Rencs EV, Green WR. Light-induced deposits in Bruch's membrane of protoporphyric mice. Arch Ophthalmol. 1993;111:126–129. doi: 10.1001/archopht.1993.01090010130039. [DOI] [PubMed] [Google Scholar]
- 25.Cousins SW, et al. The role of aging, high-fat diet and blue light exposure in an experimental mouse model for basal laminar deposit formation. Exp Eye Res. 2002;75:543–553. doi: 10.1006/exer.2002.2047. [DOI] [PubMed] [Google Scholar]
- 26.Malek G, et al. Apolipoprotein E allele–dependent pathogenesis: a model for age-related retinal degeneration. Proc Natl Acad Sci USA. 2005;102:11900–11905. doi: 10.1073/pnas.0503015102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mombaerts P, et al. RAG-1–deficient mice have no mature B and T lymphocytes. Cell. 1992;68:869–877. doi: 10.1016/0092-8674(92)90030-g. [DOI] [PubMed] [Google Scholar]
- 28.Gu X, Sun M, Hazen S, Crabb JW, Salomon RG. Oxidatively truncated docosahexaenoate phosphoplipids: total synthesis, generation and peptide adduction chemistry. J Org Chem. 2003;68:3749–3761. doi: 10.1021/jo026721t. [DOI] [PubMed] [Google Scholar]
- 29.Percopo CM, Hooks JJ, Shinohara T, Caspi R, Detrick B. Cytokine-mediated activation of a neuronal retinal resident cell provokes antigen presentation. J Immunol. 1990;145:4101–4107. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.