

Abstract
Background
Inherited long QT syndrome (LQTS) is characterized by prolonged QT interval on the EKG, syncope and sudden death due to ventricular arrhythmia. Causative mutations occur mostly in cardiac potassium and sodium channel subunit genes. Confidence in mutation pathogenicity is usually reached through family genotype-phenotype tracking, control population studies, molecular modelling and phylogenetic alignments, however, biophysical testing offers a higher degree of validating evidence.
Methods and Results
By using in-vitro electrophysiological testing of transfected mutant and wild-type LQTS constructs into Chinese Hamster Ovary cells, we investigated the biophysical properties of 9 KCNQ1 missense mutations (A46T, T265I, F269S, A302V, G316E, F339S, R360G, H455Y, and S546L) identified in a New Zealand based LQTS screening programme. We demonstrate through electrophysiology and molecular modeling that seven of the missense mutations have profound pathological dominant negative loss-of-function properties confirming their likely disease-causing nature. This supports the use of these mutations in diagnostic family screening. Two mutations (A46T, T265I) show suggestive evidence of pathogenicity within the experimental limits of biophysical testing, indicating that these variants are disease-causing via delayed or fast activation kinetics. Further investigation of the A46T family has revealed an inconsistent co-segregation of the variant with the clinical phenotype.
Conclusions
Electrophysiological characterisation should be used to validate LQTS pathogenicity of novel missense channelopathies. When such results are inconclusive, great care should be taken with genetic counselling and screening of such families, and alternative disease causing mechanisms should be considered.
Keywords: Long QT, Mutations, Arrhythmia, Ion Channels, Sudden Cardiac Death
Introduction
Long QT syndrome (LQTS) is a potentially fatal arrhythmic syndrome typically associated with a prolonged QT interval on the surface 12-lead electrocardiogram (EKG). It represents a diverse range of disorders associated with prolonged ventricular repolarisation [1]. Current estimates are that LQTS mutation carriers can be present in 1 in 1000 – 5000 [2, 3]. Recent advances in the molecular basis of LQTS has revealed a greater than expected incidence within the population due to the extent of incomplete penetrance within families with causative potassium and sodium cardiac ion-channel genes [4, 5]. More recently, LQTS and the allelic SCN5A disorder, Brugada syndrome, have been strongly associated with a proportion of sudden unexpected death syndrome cases including in infancy [6].
To date, ten genes have proven association with LQTS; LQT1 (KCNQ1) and LQT2 (HERG) encoding α-subunits of the voltage-gated K+ channel, IKs and IKr, respectively; LQT3 (SCN5A) encodes the α-subunit of a voltage-gated Na+ channel; LQT5 (KCNE1) and LQT6 (KCNE2) code for the β-subunit of IKs and IKr, respectively; and LQT4 (Ankyrin-B), a member of a membrane adapter protein family [7]. Recently, the Andersen (KCNJ1) and Timothy (Cav1.2) syndrome genes have been suggested as loci for LQT7 and LQT8 respectively [8, 9]. Voltage-gated K+ channels require four α-subunits with six transmembrane domains (S1-S6), a voltage sensor (S4) and a pore loop containing a conserved K+-selective signature sequence between S5 and S6, and one β-subunit to form a functional ion channel.
Several LQTS genetic screening studies have been published identifying over six hundred mutations which have revealed important pathophysiological mechanisms of arrhythmogensis [3, 10, 11]. Pathological mutations in these channel subunits can reduce the depolarising / repolarising cardiac current through several mechanisms including allelic haploinsufficiency, heterotetrameric dominant-negative mechanisms, nonsense-mediated decay and trafficking defects [12, 13]. Whilst the outcome of mutations which cause frameshift RNA messages and truncated polypeptides are generally unequivocal, the novel and recurrent missense variants are more problematic as observed in many other genetic disorders [14]. It is imperative that genotype pathogenicity is convincing to assign a degree of assurance that family cascade testing is truly informative. This has led to a trend in clinical and scientific best practice where variants are determined to be mutations of pathological significance by using both larger control cohorts with ethnic stratification and electrophysiological testing of missense mutations. In response to this we tested the biophysical properties of 5 novel and 4 literature-recurrent KCNQ1 mutations that were detected in a genetic screening programme [15]. We demonstrate that seven of the missense mutations we assayed and modelled have pathological dominant-negative properties which abolish IKs dynamics. Two novel mutations, that are phylogenetically conserved and excluded from the control population, have alternative explanations where we present suggestive data on the mechanism of abnormal receptor activity.
Materials and Methods
Patients and Cases
The cases were referred to tertiary cardiac arrhythmia clinics for a variety of reasons. Most patients had experienced syncope or resuscitated sudden cardiac death. This study also included eight patients previously diagnosed with epilepsy who had exhibited prolonged or borderline QTc intervals on ECG examination; and four surviving parent-pairs of young children who were victims of sudden cardiac death. Informed consent for genetic testing was obtained in all cases as established in the multi-centre ethical approval protocols covering this project (Auckland Regional Ethics Committee). DNA was extracted from blood samples using standard phenol-chloroform extraction.
Mutation Detection
The mutations have been described previously [15, 16] and were discovered by dHPLC analysis of LQTS genes in index cases. Any sample that displayed a variant chromatogram profile was selected for further analysis by DNA sequencing. Purified DNA fragments were sequenced using Big Dye Terminator kits and an ABI 3100 automated sequencer (Applied Biosystems, Foster City, US) at the Centre for Gene Technology, University of Auckland. Population frequency of single nucleotide polymorphisms (SNP's) or mutations were assessed in 300 control chromosomes as well as data from other screening studies and database sites. Various Genebank databases were aligned with LQTS gene-families and homologues at the point of mutation to assess the degree of evolutionary conservation.
Site-directed KCNQ1 mutagenesis and transient transfection in Chinese Hamster Ovary (CHO) cells
The human KCNQ1 DNA was originally provided by Dr. Mark Keating (currently at Novartis Institute of Biomedical Research, Cambridge, MA). Individual KCNQ1 mutation constructs were made using QuickChange™ XL site-directed mutagenesis kit and manufacturer instructions (Stratagene Inc., La Jolla, US). The human IKs channel auxiliary subunit KCNE1-IRES-pEGFP construct was a gift from Dr. Al George at Vanderbilt. All inserts were sequenced to ensure that only the desired mutation was obtained. Wild-type or mutated KCNQ1 constructs and KCNE1 (at 1:1 μg ratio) were transiently co-transfected into cultured Chinese hamster ovary (CHO) cells with FuGENE6 transfection reagent (Roche Applied Science, Indianapolis, US). A plasmid encoding the enhanced green fluorescent protein (pEGFP) linked to KCNE1 was used to identify transfected cells for the voltage clamp studies. Cells were grown for 48 hours after transfection before study.
Whole-cell voltage clamp studies and solutions
Whole-cell voltage clamp was performed at room temperature with 3-5MΩ patch microelectrodes and an Axopatch 200A amplifier (Axon Instruments Inc., USA). The cell chamber (extracellular) solution contained (in mmol/L) NaCl 145, KCl 4.0, MgCl2 1.0, CaCl2 1.8, glucose 10, and HEPES 10; the pH was 7.4, adjusted with NaOH. The pipette (intracellular) solution contained (in mmol/L) KCl 110, MgCl2 1.0, ATP-K2 5.0, BAPTA-K4 5.0 and HEPES 10; the pH was 7.2, adjusted with KOH. Data acquisitions were done by use of pClamp9.2 (Axon Instruments Inc), sampled at 1 kHz, and low-pass filtered at 5 kHz. Activating current was elicited with 5-sec depolarizing pulses to + 60 mV from a holding potential of -80 mV at a 10 mV increments, and tail currents was recorded upon return to -40 mV. Pulses were delivered every 30 sec. To mimic a “physiological” action potential duration, a single 400-msec pulse to +20 mV and back to -40 mV for another 100-msec from the holding potential of -80 mV was used to compare the initial IKs magnitudes in wild-type and several particular KCNQ1 mutant channels in some experiments. To determine the membrane potential of the channels activated, I-V relationships were established by fitting data to the Boltzmann equation: I=Imax/{1+exp[(Vt-V0.5)/k]}, where Imax is the maximal current, Vt is the testing potential, V0.5 is the membrane potential at which 50% of the channels are activated, and k is the slope factor. Current densities (pA/pF) were obtained after normalization to cell surface area calculated by Membrane Test (OUT 0) in pClamp9.2. The steady-state activating current at the end of a 5-sec depolarizing pulse to +60 mV and the peak deactivating tail current at -40 mV were measured for comparisons of WT with mutated IKs densities. The time constants (Tau, msec) for activating IKs currents in WT and any mutations with obvious channel gating changes were obtained by using Chebyshev method to fit individual activating current traces to the mono-exponential function: A1*exp{-(t-k)/tau}+C.
Statistical analysis
Data are expressed as mean±SEM. For comparisons among means of several groups, ANOVA is used, with post hoc pair-wise comparisons by Duncan's test if significant differences among means are detected. If only two groups are being compared, Student's t-test is used. A p-value <0.05 is considered statistically significant.
Structural modelling
The locations of the newly identified KCNQ1 mutations were investigated using the recently published homology model of the transmembrane regions of the open and closed KCNQ1 channel in tetrameric form [17]. All models were visualized using the molecular graphics program Chimera (http://www.cgl.ucsf.edu/chimera/).
Results
The LQTS Cases
All nine patients with 5 novel and 4 literature-recurrent KCNQ1 mutations selected for functional analysis had Romano-Ward syndrome, an autosomal dominant hereditary disorder which is characterized by a prolonged QT interval on the electrocardiogram (ECG), syncope and sudden death. The heart rate-corrected QT interval (QTc) ranged from 480 ms to 660 ms (average: 534±20 ms). Eight patients were of European descent and one was of Maori descent and the presenting clinical features are indicated in Table 1. Three cases were identified from follow-up of sudden unexpected death in a 1st degree family member.
Table 1. KCNQ1 gene variants characterised in this study.
Representation of novel/recurrent KCNQ1 variants and phenotypic presentation of the index patients (refer to reference [15]): The patients presented with syncope, resuscitated sudden cardiac death (RSCD), or surviving parents of sudden cardiac death (SCD) in young children. All were subsequently identified as Romano-Ward Syndrome (RWS) patients by presentation of QT interval elongation and mutations in KCNQ1 (LQT1).
| Sequence Variants and Amino Acid Changes | Protein Position | Age (yrs) |
Gender | Ethnicity | Syncope | SCD of 1° Relative | RSCD | Identified trigger/s | QTc (ms) |
|---|---|---|---|---|---|---|---|---|---|
| 136G>A : A46T | N-terminal | 57 | F | European | Y | Y | Stress | 620 | |
| 794C>T : T265I | S5 | 40 | F | European | Y | Y | Rest / Sibutramine | 660 | |
| 887T>C : F296S | S5/pore | 14 | F | European | 490 | ||||
| 905C>T : A302V | Pore | 12 | F | Maori | Y | Exercise/stress | 490 | ||
| 947G>A : G316E | Pore | 8 | M | European | Y | Y* | Exercise (water) | 560 | |
| 1016T>C : F339S | S6 | 9 | M | European | Y | Y | Exercise | 480 | |
| 1078A>G : R360G | C-terminal | 15 | F | European | Y | Exercise (water) | 600 | ||
| 1363C>T : H445Y | C-terminal | 38 | F | European | Y | 470 | |||
| 1637C>T : S546L | C-terminal | 9 | M | European | Y | Exercise (water) | 480 |
Y* = DC cardioversion required
Novel KCNQ1 Mutations in LQTS
Between 2001 and 2005, 48 gene-positive probands were identified in an LQTS screening program in New Zealand [15]: 25 had KCNQ1 mutations, nine of which (in 2004) had not been reported in the literature (Table 1). Subsequently, four of the variants (A46T, A302V, G316E and S546L) have been reported by other LQTS screening programs, but without any supporting electrophysiological data; whilst there are alternative amino acid substitutions or frameshifts recorded at positions 265 (T265fs+22X), at 302 (A302T), and at 360 (R360T) [10, 18]. Therefore, all nine variants described here are novel observations based upon receptor properties recorded by in-vitro functional electrophysiology. Sequence analysis revealed that these mutations are located in a variety of domain regions in KCNQ1 (Figure 1): the N-terminus (A46T), S5 (T265I), the S5 and P-loop linker (F296S), the P-loop (A302V and G316E), S6 (F339S) and the C-terminus (R360G, H445Y, and S546L). Population studies confirmed that these sequence variations were not represented in unrelated controls, and phylogenetic alignment of KCNQ1 homologues confirmed the mutations have occurred at conserved sites. Mutation constructs of KCNQ1 were prepared using a QuickChange™ XL site-directed mutagenesis kit and submitted for electrophysiological characterisation.
Figure 1.
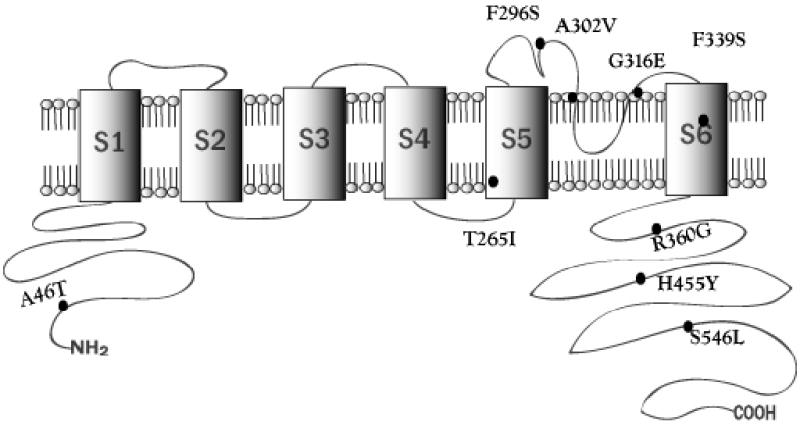
Positioning of the KCNQ1 variants on a 2-D representation of the KCNQ1 channel protein subunit domains.
Seven KCNQ1 mutations are loss-of-function channels
To compare the biophysical properties of normal IKs with those of the KCNQ1 subunit mutations, we used single 5-sec pulsing to +60 mV from a holding potential of -80 mV to elicit the IKs current. The tail current was recorded upon return to -40 mV. Wild type (WT) KCNQ1 and the auxiliary subunit KCNE1 were first co-expressed to generate an outward potassium current typical to cardiac IKs current (Figure 2A). Then, individual KCNQ1 mutations were studied with KCNE1 co-expression for comparisons. As illustrated in Figure 2 and 3A/B, seven of the nine KCNQ1 mutations generated a reduced IKs current when co-expressed with KCNE1. The IKs magnitudes by other two mutations (A46T and T265I) were similar to normal IKs, except that A46T-IKs showed a fast activation (Figure 2B): the time constants for activation at +60 mV were 1717.3±192.4 ms (WT) and 977.6±68.9 ms (A46T-IKs, p<0.01, n=6 cells for each group), respectively. The half-maximal voltage (V0.5) of IKs activation was not different among WT, A46T, T265I, G316E F339S and H455Y (p>0.05) (Figure 3C). However, the V0.5 for the mutation F296S was ∼10 mV more negative than WT-IKs (p<0.05), while the V0.5 for S546L was significantly shifted toward more positive direction (+20 mV greater than WT-IKs, p<0.01). Because of very small IKs magnitude from A302V and R360G, the current-voltage curves for the two mutations were not established to generate the V0.5 values.
Figure 2.

IKs for WT (A) and mutated KCNQ1 constructs (B-J) in the presence of KCNE1 in CHO cells. Raw IKs traces were obtained by using the voltage clamp protocol shown at room temperature. Cells were clamped to a holding potential of -80 mV. Activating IKs was elicited with a 5-sec pulse from -80 to +60 mV and deactivating tail current was recorded upon repolarisation to -40 mV for another 5 sec.
Figure 3.

Summarized data for WT and mutated IKs currents. A. Steady-state IKs current levels for WT and mutations. The currents were measured at the end of a 5-sec depolarizing pulse. B. The peak IKs tail current for WT and mutations. The peak tails were measured after a 5-sec repolarising pulse to -40 mV. C. The half-maximal voltage of IKs activation (V0.5, mV) for WT and mutations. After the current-voltage relations were obtained, the V0.5 values were obtained after fitting individual data points to the Boltzmann equation: I=Imax/{1+exp[(Vt-V0.5)/k]}. Two mutations (A302V and R360G) expressed too tiny tail currents to generate a V0.5 value.
Enhanced Initial IKs Currents and Loss of the Delay Phase of the IKs in Particular KCNQ1 Mutations
In our experiments with a 5-sec pulse protocol, we found that two KCNQ1 mutations (A46T and H445Y) activated more rapidly without an initial delay at the very beginning phase of the IKs (Figure 2B and 2I). In addition, T265I seems to have a longer delay before the current activation (Figure 2C). Accordingly, we applied a single 500-msec pulse protocol which mimics a “physiological” action potential duration. In this protocol, a 400-msec depolarization pulse to +20 mV with a 100-msec repolarization to -40 mV was used. The current magnitudes at 100th- and 400th-msec points were measured for comparisons. As illustrated in figure 4A, WT-IKs showed an initial delay at first ∼50-msec before activation (rising phase in dotted box). The mutations A46T-IKs and H445Y-IKs lost this initial current delay with a rapid activation/rising phase (Figure 4B/E). However, the mutation T265I-IKs displayed a longer delay (∼150 msec) before activation (Figure 4D). The summarized data on the current magnitudes at 100th- and 400th-msec points were presented in Figure 4C/F.
Figure 4.

500-msec short pulse-elicited currents and responses to isoproterenol (ISO, 1 μM). In several particular KCNQ1 mutations, a voltage-clamp pulsing protocol to +20 mV for 500 msec (corresponding to the plateau membrane potential and duration of a cardiac action potential, inset) was used to further compare very beginning (initial) currents. The responses of WT and mutation A46T to the β-adrenergic stimulation by ISO were also compared.
Given the fact that the IKs channel is highly up-regulated by β-adrenergic stimulation and that the mutation A46T was located in the N-terminus of the KCNQ1 channel, we further compared the effects of a β-receptor agonist isoproterenol (ISO, 1 μM) on WT-IKs and this mutant. The experimental results in Figure 4G/H/I showed that the β-stimulation increased the current in both WT and A46T to similar extent, implying that the A46T mutation did not influence the β-stimulation mediated phosphorylation of the KCNQ1 channel. Previously, we and others have already demonstrated that several amino-acid residues in the N-terminus (S27) and C-terminus (S468 and T470) are critical phosphorylation sites of the KCNQ1 channel [19, 20].
Two Mutations have Suggestive Outcomes
There is inconclusive but suggestive evidence for functional consequences of 2 variants in this study (A46T and T265I) representing challenges for the clinical interpretation of the genotype. The mutation C794T (with resulting T265I) was found in a 40-year old woman who presented following resuscitated sudden death during sleep (Figure 5A). She had been taking Sibutramine, a medication to aid with weight loss, for approximately 3 weeks prior to her presentation and her case has been previously reported [16]. Her QTc was 600-660msec and she went on to have an intracardiac cardioverter defibrillator (ICD) inserted. A previous electrocardiogram (EKG) was also abnormal with prolonged QTc of 500ms. Subsequent EKGs have had QTc values in the upper normal range. She has four children who all have prolonged QTc intervals, and one of whom has a history of syncope on exertion. Cascade testing in three younger children has identified the same mutation as the proband whilst the oldest child has declined genetic testing. The proband's mother and three siblings have all had EKGs with normal QTc intervals. Her father has a prolonged QTc of 0.48 seconds, and is gene-positive.
Figure 5.


Pedigrees and Clinical Data of the gene-positive families with uncertain, ambiguous outcomes. A. Family with KCNQ1 A46T and associated clinical presentation and incidences; B. Family with T265I and associated clinical presentation and incidences. Refer to Figure for the key guide.
The mutation G136A (with resulting A46T) has previously been reported by Napolitano et al (2005) [10]. In our cohort, it was detected in a 57-year old woman following presentation with acute syncope, whilst taking Cisapride (Figure 5B). She had a prior history of syncope at times of stress. Her initial QTc interval was 620msec, while subsequent EKGs have revealed QTc intervals of 400-470msec. Her presenting episode of collapse occurred in the days following the unexpected death of her son, who had died suddenly, during gentle activity, aged 37 years. He also had a history of syncope, but there were no prior EKGs. The post-mortem examination revealed no cause, but no DNA was available for posthumous genetic screening. Despite the proband's presentation, the acutely prolonged QTc interval and family history of young sudden death, further investigation of the family to date lends some doubt as to the malignancy of this mutation. The proband has three other children who all have a history of syncope and have undergone cascade genetic screening. One son (QTc 380-420msec) and one daughter (QTc 420-480msec) have tested positive for this mutation, the other daughter (QTc 420-440msec) has had negative cascade testing. The proband's two living siblings have not presented for assessment, but are reportedly asymptomatic. Another sibling died in an accident at age 20 years. With regard to the proband's parents, her father died at age 71 years and her mother's genetic screening was negative. As a result of the poor co-segregation of the mutation with clinical findings in this family, together with the in-vitro electrophysiological testing result, we are not using cascade screening as a definitive test for the presence or absence of disease in this family.
Molecular Modelling of the KCNQ1 Mutations
The tertiary positions of mutations T265I, F296S, A302V, G316E and F339S were examined in relation to the recently published homology model of the transmembrane regions of the protein [17]. The N-terminal A46T mutation, and the C-terminal R360G, H455Y and S546L fell outside the current coverage of published models and crystal structures. A crystal structure for the C-terminal region of human KCNQ1 has recently been elucidated [21], but this is restricted to residues 584-621 of the C-terminus.
As shown in Figure 6A, in the closed state, T265 and F339 are closely located and likely to interact by means of intra-chain hydrophobic interactions between S5 and S6. F296 and A302 are also closely located, but on the extra-membrane portion of the P loop. Comparison of the closed and open states of the channel (Figure 6A and B) reinforces the importance of the mutated positions. In the open state, the interactions between the residues of each pairing are less intimate, suggesting that engagement and loss of specific hydrophobic interactions is central to the conformational changes in the transmembrane and loop regions required for activity of the channel. We speculate that mutations involving these residues disrupt the fine architecture of the channel and preclude normal opening and closure. G316 is located on the P loop, squarely at the entry to the pore (Figure 6C). The G316E mutation directly restricts the aperture of the pore by introducing a much larger and negatively charged glutamic acid residue to this critical gating position (Figure 6D). It is also possible that the mutation may bring about mis-folding of the protein, which may subsequently affect its trafficking or tetramerisation, though homology modelling of the E316 variant (data not shown) suggests that this is unlikely.
Figure 6.
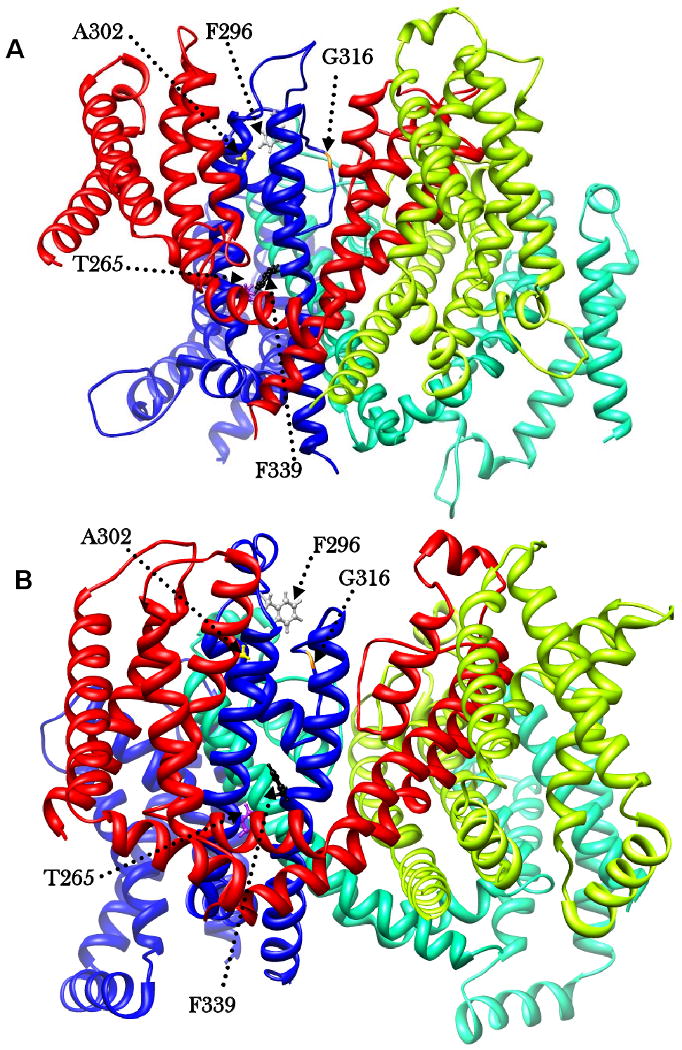
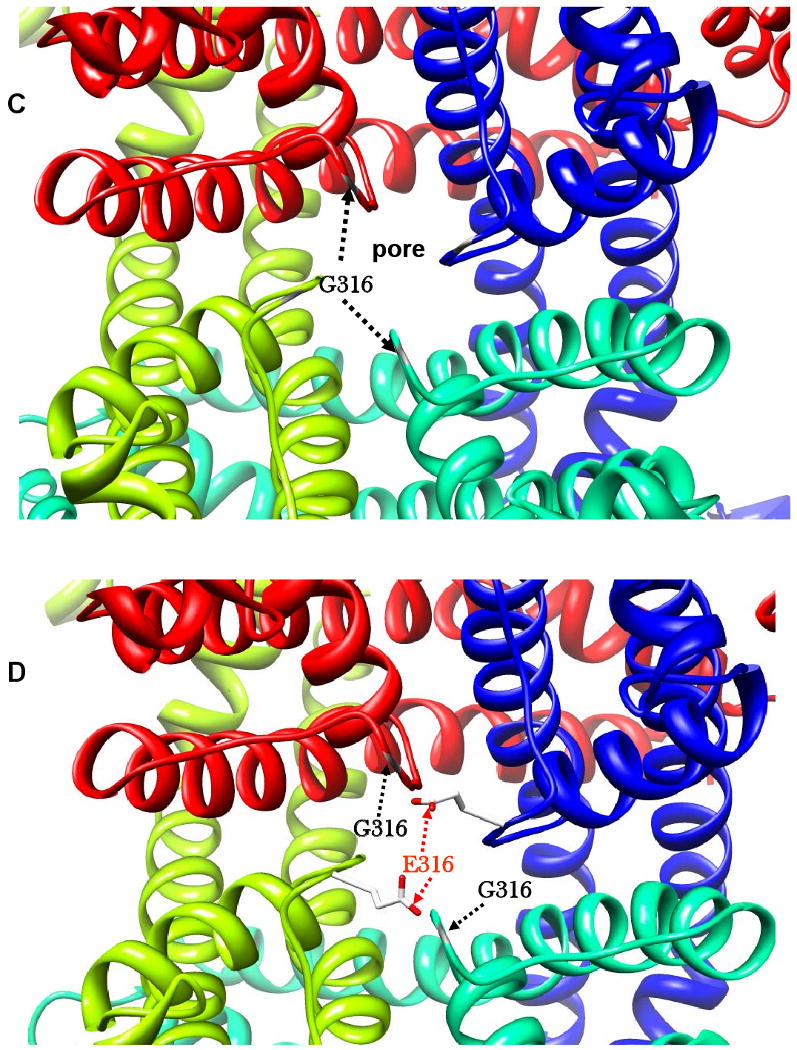
The chains of the tetramer shown in blue (a), green (b), lime (c), and red (d). Residues involved in mutations shown in ball and stick. A) Closed - the two pairings, F296 (grey) and A302 (yellow); T265 (purple) and F339 (black) in close proximity. G316 (orange) on P loop, projected outward into pore of channel. B) Open - the two pairings are drawn apart, and G316 (orange) on P loop, withdrawn from the pore of the channel. C) KCNQ1 in open conformation showing the pore region. The G316 wild type, with positions of Glycine residues, which possess no side chain, shown in grey, leaving the channel unobstructed. D) KCNQ1 in open conformation showing restriction of the pore by the G316E mutation. E316 on P loop shown in orange for chains a and c, projected outward into the pore. For illustration, wild type G316 shown in yellow on P loop of chains b and d, leaving the channel unobstructed. All models were visualized using the molecular graphics program Chimera (http://www.cgl.ucsf.edu/chimera/).
Figure 6: Structural modelling of location of mutations in the closed channel (A), and the open channel (B), showing the changes in positions of mutated residues.
Discussion
Many KCNQ1 mutations and rare variants have now been identified in a variety of KCNQ1 domains and regions (http://www.fsm.it/cardmoc/), some of which are recurrent in populations whilst some remain private to one index case or extended family. This represents a dilemma in the clinical setting because it is difficult to assign pathogenicity to novel variants without several levels of validating evidence, the best of which is the demonstration of biophysical deficits by ‘in-vitro’ electrophysiological experiments. This is a highly-specialised and challenging technology platform which is unlikely to cross over to the diagnostic domain, nevertheless, it can provide definitive evidence and clinical confidence that a gene variant can translate into a functional mutation.
For this reason we describe in this study the biophysical characterisation of 9 KCNQ1 variants in patients with Romano Ward syndrome [15, 16]. The gene-positive patients had unequivocally prolonged QTc intervals and all but one had typical presentation scenarios such as syncope during exercise, a history of sudden death in the family or resuscitated cardiac arrest. Within the KCNQ1 subunit structure, these mutations are located in different regional domains: the N-terminus (A46T), S5 (T265I), the S5 and P-loop linker (F296S), P-loop (A302V and G316E), S6 (F339S) and the C-terminus (R360G, H445Y, and S546L). Four variants (A46T, A302V, G316E and S546L) have subsequently been detected by other LQTS screening programs, but without any supporting electrophysiological data; whilst there are alternative amino acid substitutions or frameshifts recorded at positions 265 (T265fs+22X), at 302 (A302T), and at 360 (R360T) [10, 18]. Whatever the status and origin of the KCNQ1 variants, the biophysical investigation of their pathogenicity has not been investigated.
The value of discovering the physiological properties of channel variants has contributed much to the understanding of the pathophysiological mechanisms of LQTS [12-14]. Cardiac slowly-activating delayed rectifier K+ current (IKs) is generated by co-assembly of the α-subunit KCNQ1 (KvLQT1) and its auxiliary subunit KCNE1 (minK). The IKs current does not play a major role in regulation of cardiac repolarisation under normal physiological conditions. However, mutations in the KCNQ1 gene could cause inherited prolongation of the QT interval by reducing the quality of IKs current, thereby affecting cardiac repolarisation. Expression studies have demonstrated that LQTS mutations of genes related with K+ currents can produce loss of channel function by either a net current reduction by altering the channel gating and kinetic properties, prevention of assembly of functional channel protein, or an abnormal intracellular protein trafficking.
We examined the electrophysiological properties of the nine KCNQ1 mutations in the presence KCNE1 by heterologous expression in CHO cells and by using a whole-cell voltage clamp. We found seven KCNQ1 mutations causing a reduced IKs current density with co-expression of KCNE1, indicating the loss-of-function in the heterotetrametric state channel. The electrophysiological data for the seven KCNQ1 mutations with decrease the IKs density, suggest several possible mechanisms: (1) a positive shift of the channel activation voltage could lead to a reduction of the current (this effect is more obvious in the mutation S546L); (2) the dominant negative effect, and (3) failure of the channel protein expression within the cell membrane. The studies relating to the latter two effects are separate projects that are being pursued, but irrespective of the specific mechanism involved, it is now possible to label these seven LQTS genotypes as definitive pathological mutations.
Two other mutations (A46T and T265I) displayed no alterations in the current density, although there is evidence that T265I-IKs displayed a longer delay (∼150 msec) before activation, and A46T-IKs (as well as H445Y-IKs) lost initial current delay with a rapid activation/rising phase. In the mutation T265I, a longer initial 100-msec current delay and small current magnitude would be responsible for prolonged QT intervals. Studies have demonstrated that the initial delay phase of IKs channel activation is caused by its β-subunit KCNE1 via moving the channel through multiple closed states before opening during a depolarization [22, 23], thereby decreasing the initial current activation. In the cases of A46T-IKs and H445Y-IKs, whether the interaction between the mutation and KCNE1 is affected to some degree remains to be studied further. The mutation-caused loss of the delay of the initial IKs activation could be caused by channel accumulation in open states between depolarization pulses. The mutation caused loss of the delay of the initial IKs activation could be associated with more channel accumulations in Zone 1 of closed states that are near open states in Markove model of the IKs channel kinetics. The channel accumulation in Zone 1 of closed states between cardiac action potentials provides an “available reserve” [24] which represents an important mechanism for IKs participation in repolarisation and its dependence on rate. During the action potential repolarisation, especially at fast heart rates, these readily available channels can very quickly open on demand to cause rapid IKs activation and rise, an effect to shorten cardiac action potential duration. Again, the current magnitude at the 1st 100-msec of IKs activation was larger in the mutation A46T than in WT current (Figure 4C). Therefore, these alterations in IKs properties could explain failure to observe a prolonged QT interval in the A46T patient.
Structural modelling helps us to conceptualize the different functional effects of particular mutations. Mutation of T265 and F339 affect interactions between S5 and S6. This is consistent with the T265I mutation displaying a longer delay (∼150 msec) before activation (Figure 4D) as the normal movements of the S5 helix required for efficient opening and closure are impeded by the substitution of threonine by the larger more hydrophobic isoleucine. F339S, on the other hand, involves substitution by a smaller more polar residue, which is less physically impeding, but nevertheless remains pathogenic as indicated by the functional assays (Figure 2G).
Though the A302V substitution involves residues of similar size, the observed functional effects may be due to the external aqueous environment being less forgiving of changes in hydrophobic interactions. Similarly, changes in polarity of exposed extramembrane residues upon mutation affect V0.5 values, resulting accordingly in more negative (F296S, change to polar) or more positive S546L (change to hydrophobic) values. The G316E mutation appears to directly obstruct the normal aperture of the pore by introducing a much larger and negatively charged glutamic acid side-chain to one of the most sensitive gating positions. Modelling and comparison of the heteromeric mutation of two of the four chains (Figure 6D) with the wild type (Figure 6C) reveals the extent to which the pore is likely to be obstructed. It is interesting to note that the mutation also disrupts the second glycine residue in a combinatorial sequence pattern [S/T]××[S/T]×G[F/Y]G that has been identified in 90% of 134 potassium channel re-entrant loops analysed [25], which suggests that this glycine residue may also be important for the stable insertion of the P loop within the membrane.
In summary, the biophysical characterisation of these 9 KCNQ1 missense mutations has provided unequivocal heterogeneous proof of pathogenicity in 8 variants to support the various clinical and genetics studies. We remain uncertain as to the pathogenicity of A46T given the suggestive biophysical evidence and co-segregation studies are inconclusive in part due to resistance to wider family screening. In this family it is possible that the genetic basis of the QT prolongation lies elsewhere and further molecular screening of the index cases must remain an option. Whilst cellular electrophysiological testing is unlikely to move easily into the clinical diagnostic area, its clinical value is very important and not in question given the high proportion of novel genetic variants being discovered presenting a challenge to the clinical and genetic counselling teams. For the time-being a collaborative multi-disciplinary approach is needed between clinical and research domains to permit relative and informative proof for cardiologists and genetic counselling teams.
Acknowledgments
Sources of Funding: This work was supported by Cure Kids for New Zealand (Child Health Research Foundation of New Zealand – JS, MIR, S-KC), The Royal Society (MIR), and The U.S. National Institutes of Health grants HL-46681 (DMR) and HL-49989 (DMR and TY) as well as The American Heart Association grant 0565306B (TY). DMR is the holder of the William Stokes chair in Experimental Therapeutics, a gift of the Dai-ichi Corporation.
Footnotes
Long QT syndrome (LQTS) is characterized by prolonged QT interval on the EKG, syncope and sudden death due to ventricular arrhythmia. Inherited LQTS is caused by mutations in cardiac ion channel genes and mutation screening in LQTS is now a well-established part of genetic service programs around the world. Identification of mutations that are known to cause arrhythmias is useful. The significance of novel mutations is often unclear. Is such a mutation a rare natural variation in the human genetic code or a pathophysiological entity predisposing to sudden death? This study supports the use of biophysical testing to provide evidence for functional pathogenicity of such mutations. The protein coded for by the mutated gene can be expressed in-vitro to allow electrophysiologic characterization of the resulting ion channels, providing an indication of the mechanisms by which the clinical phenotype is produced. Electrophysiology analysis identified dominant negative / loss-of-function properties in seven of 9 novel LQTS mutations evaluated in this study, supporting the disease-causing nature of these mutations. These findings support the use of electrophysiological characterisation to potentially validate LQTS pathogenicity of novel missense mutations in ion channels. This approach may be helpful in assessing risk and providing genetic counselling and screening of families with novel mutations.
Subject Code: [106] Electrophysiology, [132] Arrhythmias-basic studies
Disclosures: All authors: None
References
- 1.Ackerman MJ. The long QT syndrome: Ion channel diseases of the heart. Mayo Clinic Proceedings. 1998;73:250–269. doi: 10.4065/73.3.250. [DOI] [PubMed] [Google Scholar]
- 2.Yang P, Kanki H, Drolet B, Yang T, Wei J, Viswanathan PC, Hohnloser SH, Shimizu W, Schwartz PJ, Stanton M, Murray KT, Norris K, George ALJ, Roden DM. Allelic variants in long-QT disease genes in patients with drug-associated torsades de pointes. Circulation. 2002;105:1943–1948. doi: 10.1161/01.cir.0000014448.19052.4c. [DOI] [PubMed] [Google Scholar]
- 3.Tester DJ, Will ML, Haglund CM, Ackerman MJ. Compendium of cardiac channel mutations in 541 consecutive unrelated patients referred for long QT syndrome genetic testing. Heart Rhythm. 2005;2:507–517. doi: 10.1016/j.hrthm.2005.01.020. [DOI] [PubMed] [Google Scholar]
- 4.Gouas L, Bellocq C, Berthet M, Potet F, Demolombe S, Forhan A, Lescasse R, Simon F, Balkau B, Denjoy I, Hainque B, Baro I, Guicheney P. New KCNQ1 mutations leading to haploinsufficiency in a general population; Defective trafficking of a KvLQT1 mutant. Cardiovasc Res. 2004;63:60–68. doi: 10.1016/j.cardiores.2004.02.011. [DOI] [PubMed] [Google Scholar]
- 5.Wehrens XHT, Vos MA, Doevendans PA, Wellens HJJ. Novel Insights in the Congenital Long QT Syndrome. Ann Intern Med. 2002;137:981–992. doi: 10.7326/0003-4819-137-12-200212170-00012. [DOI] [PubMed] [Google Scholar]
- 6.Vincent GM. The Long QT and Brugada syndromes: causes of unexpected syncope and sudden cardiac death in children and young adults. Semin Pediatr Neurol. 2005;12:15–24. doi: 10.1016/j.spen.2004.11.008. [DOI] [PubMed] [Google Scholar]
- 7.Mohler PJ, Schott JJ, Gramolini AO, Dilly KW, Guatimosim S, duBell WH, Song LS, Haurogne K, Kyndt F, Ali ME, Rogers TB, Lederer WJ, Escande D, Le Marec H, Bennett V. Ankyrin-B mutation causes thype 4 long-QT cardiac arrhythmia and sudden cardiac death. Nature. 2003;421:634–639. doi: 10.1038/nature01335. [DOI] [PubMed] [Google Scholar]
- 8.Tristani-Firouzi M, Jensen JL, Donaldson MR, Sansone V, Meola G, Hahn A, Bendahhou S, Kwiecinski H, Fidzianska A, Plaster N, Fu YH, Ptacek LJ, Tawil R. Functional and clinical characterization of KCNJ2 mutations associated with LQT7 (Andersen syndrome) J Clin Invest. 2002;110:381–388. doi: 10.1172/JCI15183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Splawski I, Timothy KW, Sharpe LM, Decher N, Kumar P, Bloise R, Napolitano C, Schwartz PJ, Joseph RM, Condouris K, Tager-Flusberg H, Priori SG, Sanguinetti MC, Keating MT. Ca(V)1.2 calcium channel dysfunction causes a multisystem disorder including arrhythmia and autism. Cell. 2004;119:19–31. doi: 10.1016/j.cell.2004.09.011. [DOI] [PubMed] [Google Scholar]
- 10.Napolitano C, Priori SG, Schwartz PJ, Bloise R, Ronchetti E, Nastoli J, Bottelli G, Cerrone M, Leonardi S. Genetic testing in the long QT syndrome: development and validation of an efficient approach to genotyping in clinical practice. Jama. 2005;294:2975–2980. doi: 10.1001/jama.294.23.2975. [DOI] [PubMed] [Google Scholar]
- 11.Splawski I, Shen J, Timothy KW, Lehmann MH, Priori S, Robinson JL, Moss AJ, Schwartz PJ, Towbin JA, Vincent GM, Keating MT. Spectrum of mutations in long QT syndrome genes. Circulation. 2000;102:1178–1185. doi: 10.1161/01.cir.102.10.1178. [DOI] [PubMed] [Google Scholar]
- 12.Huang L, Bitner-Glindzicz M, Tranebaerg L, Tinker A. A spectrum of functional effects for disease causing mutations in the Jervell and Lange-Nielsen syndrome. Cardiovascular Research. 2001;51:670–680. doi: 10.1016/s0008-6363(01)00350-9. [DOI] [PubMed] [Google Scholar]
- 13.Anderson CL, Delisle BP, Anson BD, Kilby JA, Will ML, Tester DJ, Gong Q, Zhou Z, Ackerman MJ, January CT. Most LQT2 mutations reduce Kv11.1 (hERG) current by a class 2 (trafficking-deficient) mechanism. Circulation. 2006;113:365–373. doi: 10.1161/CIRCULATIONAHA.105.570200. [DOI] [PubMed] [Google Scholar]
- 14.Wilson AJ, Quinn KV, Graves FM, Bitner-Glindzicz M, Tinker A. Abnormal KCNQ1 trafficking influences disease pathogenesis in hereditary long QT syndromes (LQT1) Cardiovasc Res. 2005;67:476–486. doi: 10.1016/j.cardiores.2005.04.036. [DOI] [PubMed] [Google Scholar]
- 15.Chung SK, MacCormick JM, McCulley CH, Crawford J, Eddy CA, Mitchell EA, Shelling AN, French JK, Skinner JR, Rees MI. Long QT and Brugada syndrome gene mutations in New Zealand. Heart Rhythm. 2007;4:1306–1314. doi: 10.1016/j.hrthm.2007.06.022. [DOI] [PubMed] [Google Scholar]
- 16.Harrison-Woolrych M, Clark DW, Hill GR, Rees MI, Skinner JR. QT interval prolongation associated with sibutramine treatment. Br J Clin Pharmacol. 2006;61:464–469. doi: 10.1111/j.1365-2125.2006.02574.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Smith JA, Vanoye CG, George AL, Jr, Meiler J, Sanders CR. Structural models for the KCNQ1 voltage-gated potassium channel. Biochemistry. 2007;46:14141–14152. doi: 10.1021/bi701597s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Choi G, Kopplin LJ, Tester DJ, Will ML, Haglund CM, Ackerman MJ. Spectrum and Frequency of Cardiac Channel Defects in Swimming-Triggered Arrhythmia Syndromes. Circulation. 2004;110:2119–2124. doi: 10.1161/01.CIR.0000144471.98080.CA. [DOI] [PubMed] [Google Scholar]
- 19.Marx SO, Kurokawa J, Reiken S, Motoike HK, D'Armiento J, Marks AR, Kass RS. Requirement if a macromolecular signaling complex for β adrenergic receptor modulation of the KCNQ1-KCNE1 potassium channel. Science. 2002;295:496–499. doi: 10.1126/science.1066843. [DOI] [PubMed] [Google Scholar]
- 20.Yang T, Kanki H, Roden DM. Phosphorylation of the IKs Channel Complex Inhibits Drug Block: Novel Mechanism Underlying Variable Antiarrhythmic Drug Actions. Circulation. 2003;108:132–134. doi: 10.1161/01.CIR.0000082708.86266.B8. [DOI] [PubMed] [Google Scholar]
- 21.Wiener R, Haitin Y, Shamgar L, Fernandez-Alonso MC, Martos A, Chomsky-Hecht O, Rivas G, Attali B, Hirsch JA. The KCNQ1 (Kv7.1) COOH terminus, a multitiered scaffold for subunit assembly and protein interaction. J Biol Chem. 2008;283:5815–5830. doi: 10.1074/jbc.M707541200. [DOI] [PubMed] [Google Scholar]
- 22.Balser JR, Bennett PB, Roden DM. Time-dependent outward current in guinea pig ventricular myocytes. Gating kinetics of the delayed rectifier. J Gen Physiol. 1990;96:835–863. doi: 10.1085/jgp.96.4.835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Silva J, Rudy Y. Subunit interaction determines IKs participation in cardiac repolarization and repolarization reserve. Circulation. 2005;112:1384–1391. doi: 10.1161/CIRCULATIONAHA.105.543306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rudy Y, Silva JR. Computational biology in the study of cardiac ion channels and cell electrophysiology. Q Rev Biophys. 2006;39:57–116. doi: 10.1017/S0033583506004227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lasso G, Antoniw JF, Mullins JG. A combinatorial pattern discovery approach for the prediction of membrane dipping (re-entrant) loops. Bioinformatics. 2006;22:e290–297. doi: 10.1093/bioinformatics/btl209. [DOI] [PubMed] [Google Scholar]