

Abstract
This study uses microarray analyses to examine gene expression profiles for Mycobacterium tuberculosis (Mtb) induced by exposure in vitro to bovine lung surfactant preparations that vary in apoprotein content: (i) whole lung surfactant (WLS) containing the complete mix of endogenous lipids and surfactant proteins (SP)-A, -B, -C, and -D; (ii) extracted lung surfactant (CLSE) containing lipids plus SP-B and -C; (iii) column-purified surfactant lipids (PPL) containing no apoproteins, and (iv) purified human SP-A. Exposure to WLS evoked a multitude of transcriptional responses in Mtb, with 52 genes up-regulated and 23 genes down-regulated at 30 min exposure, plus 146 genes up-regulated and 27 genes down-regulated at 2 h. Notably, WLS rapidly induced several membrane-associated lipases that presumptively act on surfactant lipids as substrates, and a large number of genes involved in the synthesis of phthiocerol dimycocerosate (PDIM), a cell wall component known to be important in macrophage interactions and Mtb virulence. Exposure of Mtb to CLSE, PPL, or purified SP-A caused a substantially weaker transcriptional response (≤20 genes were induced) suggesting that interactions among multiple lipid-protein components of WLS may contribute to its effects on Mtb transcription.
Keywords: Mycobacterium tuberculosis, microarray, transcriptional responses, surfactant, surfactant proteins
1. Introduction
Infection with Mycobacterium tuberculosis (Mtb) is widespread throughout the world, and tuberculosis is one of the leading causes of death in developing countries [1]. Mtb is a facultative intracellular pathogen that survives and replicates within macrophages [2]. Interactions between Mtb and pulmonary macrophages are crucial in the disease process caused by this microorganism. One factor that may be important in moderating the interactions of Mtb with alveolar macrophages is pulmonary surfactant, a complex mixture of lipids and specific proteins that is synthesized, packaged, stored, and secreted by type II epithelial cells in the alveolar lining [3–5]. Because surfactant is present throughout the alveolar lining layer (hypophase), it is likely that respired microorganisms such as Mtb will frequently encounter and interact with this physiologically essential material. Interactions between Mtb and components of surfactant at the alveolar air-liquid interface or in the hypophase may significantly influence the subsequent behavior and interactions of the mycobacteria with macrophages [6–9].
In addition to its essential biophysical actions in lowering surface tension and stabilizing alveolar inflation-deflation, surfactant is known to play a role in host defense against infection through the biological activity of the two hydrophilic surfactant proteins A and D (SP-A and SP-D; [10–13]. These oligomeric carbohydrate binding proteins are members of the collectin family, and have been shown to influence the opsonization, phagocytosis, and agglutination of microorganisms within the respiratory tract [10–13]. The localization of SP-A/D in the alveolar hypophase ideally positions these proteins (and other components of the surfactant system) to act as “first responders” to inhaled pathogens. Lung surfactant also contains two hydrophobic apoproteins (SP-B and SP-C) plus a substantial lipid composition (~90% by weight) that have the potential to influence the early interactions of Mtb with the host airways.
We and others have recently shown that peripheral cell wall lipids isolated from Mtb can directly impair the biophysical function of lung surfactant [14,15]. In the present study, we examined the transcriptional responses of Mtb to surfactant and subfractions of its components. Particular focus was on the induction of genes relevant for Mtb-macrophage interactions, and on genes coding for bacterial lipases that might utilize surfactant phospholipids as substrate. Whole genome microarrays have been used previously to determine gene expression patterns of tubercle bacilli in response to various environmental conditions [16–21]. However, microarray studies on specific gene expression in Mtb in response to surfactant exposure have not been done. Experiments here used surfactant preparations that varied systematically in apoprotein content to assess the roles and importance of these components in the transcriptional responses of Mtb in vitro. Preparations studied are (i) whole lung surfactant (WLS) from calf lung lavage, (ii) a chloroform/methanol-extract of WLS (calf lung surfactant extract (CLSE)), and (iii) column-purified surfactant lipids (PPL) isolated from CLSE. These surfactant preparations are identical in lipid content, but differ in protein content as follows: WLS contains all four surfactant apoproteins (SP-A, -B, -C, and -D); CLSE contains only the hydrophobic surfactant proteins (SP-B and -C), and PPL contains no proteins. In addition, purified SP-A was also examined for its effects on Mtb transcription in vitro.
2. Results
2.1. Verification of the presence of SP-A and SP-D in WLS
A large number of studies have demonstrated that isolates of whole surfactant from animal lung lavage contain both of the hydrophilic apoproteins SP-A and SP-D. Immunoblot analysis indicated that the WLS preparation used here contained both SP-A and SP-D, and that CLSE did not contain either of these proteins (Fig. 1).
Fig. 1. Immunoblots showing the presence of SP-A and SP-D in whole lung surfactant (WLS) but not in calf lung surfactant extract (CLSE).
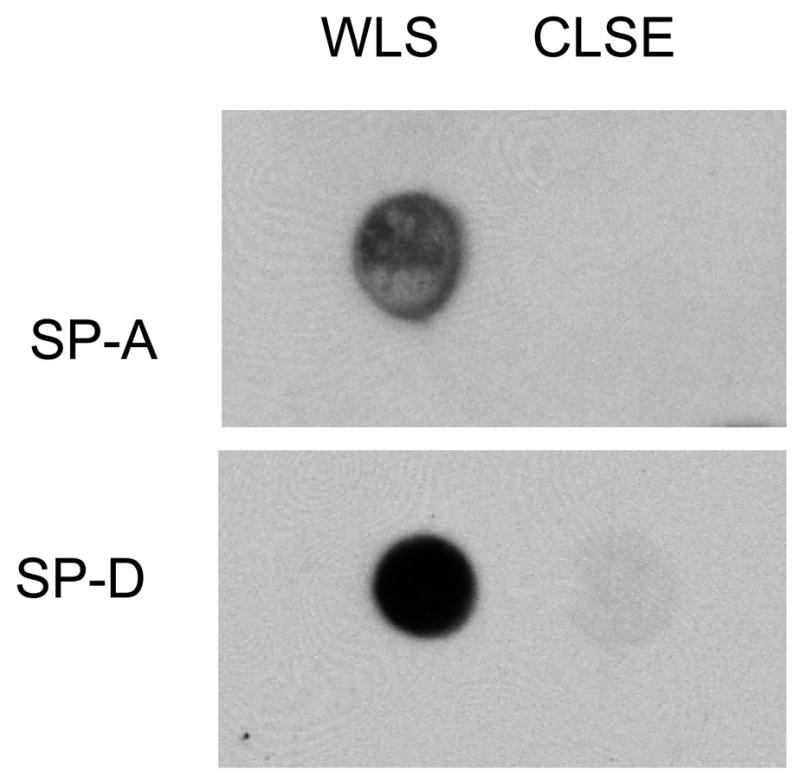
SP-A and SP-D were assessed in WLS and CLSE by immunoblotting as detailed in Materials and methods. Both surfactant preparations are also known to contain the hydrophobic lung surfactant apoproteins SP-B and SP-C (not tested for in the figure). Data shown are representative blots from 3 independent experiments on each surfactant preparation.
2.2. Global expression profile of M. tuberculosis (Mtb) after exposure to WLS
We initially examined exposure of Mtb to WLS containing the complete mix of endogenous lipids and surfactant proteins, since this mimics the in vivo situation where Mtb presumptively contacts alveolar surfactant prior to interacting with macrophages. Among the 4,295 open reading frames (ORFs) represented on the arrays, 52 genes were up-regulated >1.5-fold (p<0.05) when Mtb was exposed to WLS for 30 min compared to “no treatment” control (Fig. 2A; Supplemental Table 1). Eleven genes (efpA, modA, lipX, Rv1057, rpfE, Rv2897c, Rv2949c, ethR, ethA, MT1321, and MT1627) that were induced after 30 min exposure to WLS remained induced at the 2 h exposure time. A total of 146 genes were up-regulated after WLS treatment for 2h (Fig. 2B; Supplemental Table 2). Most of these genes were induced at ≤3-fold, but some showed considerably higher levels of induction (e.g., Rv3481c, MT0915.1 and MT1321). Sixty-eight of the Mtb genes induced by WLS after a 2 h exposure encoded products with proposed or known functions, while the remaining 78 genes had no recognizable features or current annotations by which to predict function. Although the majority of Mtb responses to WLS involved gene up-regulation, transcript levels of 35 genes were down-regulated at 30 min of exposure (Supplemental Table 1) and 13 genes were down-regulated at 2 h of exposure (Supplemental Table 2). Seven genes remained repressed for 2h (Rv0124, fadA2, Rv0516c, Rv1216c, Rv3614c–Rv3616c). Interestingly, among the 30 min-repressed genes are several genes belonging to the 24-membered Mtb Mce (mammalian cell entry) protein family (Supplemental Table 1).
Fig. 2. Scatter plots of Mtb gene expression after (A, B) WLS and (C, D) CLSE treatment for 30min and 2h.

Vertical axis depicts expression ratios in surfactant-treated relative to no treatment control. Note the few genes induced by CLSE when compared to WLS.
2.3 Global expression profile of Mtb after exposure to CLSE
When Mtb was exposed to CLSE, which does not contain the collectins SP-A and SP-D, a significantly lower gene induction response was seen compared to WLS (Fig. 2C and D; Supplemental Table 3). Among the 14 genes that were significantly induced (>1.5 fold) by CLSE after a 2 h exposure, the majority of them (11 genes) matched those induced by WLS at 2 h (Fig. 3B). Three genes were unique to CLSE, i.e. Rv3041c (probable conserved ATP binding protein), Rv0862c (conserved hypothetical protein), and rmlD (involved in dTDP-L-rhamnose biosynthesis). When exposure time to CLSE was reduced to 30 min, only 4 genes (desA3, Rv0081, Rv2897c, and MT1118.2) were significantly induced in Mtb. With the exception of MT1118.2, the remaining three genes were also induced by WLS at 30 min exposure (Fig. 3A). Again, fewer genes were down-regulated by CLSE treatment compared to WLS. At both 30 min and 2 h of exposure to CLSE, different sets of genes in Mtb were down-regulated. Among the six genes being repressed by CLSE at 30 min (Supplemental Table 3) two genes (Rv0124 and cyp138) were also repressed by WLS. After a 2 h exposure to CLSE, the transcription levels of six genes were reduced, and three of these (Rv0292, Rv1036c, and Rv2280) were also repressed by WLS at 2h (Supplemental Table 3).
Fig. 3. Comparison of genes induced by WLS, CLSE, and PPL treatment.


Venn diagram showing overlap of genes induced by WLS, CLSE, and PPL after 30 min (A) and 2 h (B) treatment. Genes induced >1.5-fold (P<0.05) in at least one condition and whose array signals passed quality filters in all three conditions were included.
2.4. Global expression profile of Mtb after exposure to PPL
To test the effect of surfactant phospholipids alone on Mtb, we analyzed responses to PPL, which contained all the lung surfactant lipids but no surfactant proteins. Twenty genes in Mtb were up-regulated (> 1.5-fold) following a 30 min exposure to PPL, and thirteen genes were up-regulated at 2 h exposure (Supplemental Table 4). These responses represented an increase over CLSE at 30 min (20 genes vs. 4 genes) and a similar number of induced genes relative to CLSE at 2 h (13 genes vs. 14 genes). Comparison of the gene expression profiles revealed that there was no overlap of the PPL profile with CLSE at 30 min. There was only one gene of the PPL profile that was also induced after WLS treatment (Fig. 3A). When the 30 min PPL profile was compared with the 2h profile of WLS six genes were also induced by WLS (Supplemental Tables 2 and 4). At 2h five genes of the PPL profile were also significantly induced by WLS and three of these genes were also induced by CLSE (Fig. 3B). No down-regulation of genes was found after PPL treatment at either exposure time.
2.5 Global expression profile of Mtb after exposure to purified SP-A
Since SP-A is the most abundant apoprotein in native lung surfactant, we carried out further testing on the expression profile of Mtb following exposure to purified human SP-A for 2 h. SPA exposure was accomplished by coating the apoprotein onto the bottom surface of a cell culture flask as done with WLS, CLSE and PPL, and also by a second method where SP-A was directly incubated with Mtb. Transcriptional responses to SP-A varied with the method of exposure, but were both different from those observed following exposure to WLS. Eighteen genes in Mtb were up-regulated following a 2 h exposure to pre-coated SP-A (Supplemental Table 5), and all of these were distinct from genes induced by exposure to WLS at both exposure times (Supplemental Tables 1 and 2). No genes in Mtb were down-regulated by pre-coated SP-A at the cut-off level of >1.5 fold, in contrast to 13 genes down-regulated by WLS at 2 h (Supplemental Table 2). When Mtb was incubated with SP-A as opposed to being pre-coated on the culture flask, the resulting induction profile was weaker and different (Supplemental Table 6). Nine genes were induced in the Mtb response to SP-A after 2 h of incubation and two of them (pknF and cyp141) were also induced by WLS at 2h and one gene (atsF) by WLS at 30 min. Surprisingly, eighty-eight genes were repressed and none of these genes was repressed by WLS. Only one gene (Rv0111) was also repressed by CLSE treatment (Supplemental Tables 3 and 6). There was no overlap of genes between these two different exposure methods of SP-A, indicating that exposure method (SP-A in solution versus SP-A coated to a plastic surface) had a significant effect on Mtb gene expression.
2.6. Induction of lipase genes and genes relevant for Mtb/macrophage interactions
One group of genes of particular interest was related to mycobacterial responses potentially involved in metabolizing surfactant lipids. Exposure of Mtb to WLS for 2 h induced two known membrane-located mycobacterial lipases: Rv3097 (lipY) encoding PE30 [22] and Rv1169c (lipX) encoding PE11 [23] (Supplemental Table 2). The following genes related to PDIM metabolism [24,25] were also significantly up-regulated (>1.5 fold, P<0.05): ppsA- ppsD (type-I modular polyketide synthase responsible for the synthesis of phthiocerol), drrB (daunorubicin DIM transport protein), lppX (lipoprotein), pks1 (polyketide synthase), fadD22 (acyl-CoA synthase), Rv2949c (hypothetical protein), and fadD29 (acyl-CoA synthase) (Supplemental Table 2). Four other PDIM cluster genes (tesA, thioesterase; fadD26, acyl-adenylate synthase; drrC, ABC-transporter; and pks15, polyketide synthase) were also induced at the >1.5-fold level by WLS at 2 h, but were not statistically significant for the number of biological replicates studied (P>0.05). Although below the 1.5 fold cutoff the genes ppsE, papA5 (acyltransferase that is involved in production of PDIM esters), and mmpl7 (transporter of the RND permease superfamily), all belonging to the PDIM cluster, were also modestly induced. Our observation that additional genes in this region putatively involved in the synthesis of cell wall components (Rv2954c, hypothetical protein; Rv2958c, possible glycosyltransferase; Rv2959c, possible methyltransferase; and Rv2962c, possible glycosyltransferase) were significantly induced (Supplemental Table 2) further highlights the impact of WLS on Mtb cell wall remodeling. Analyzing overall expression profiles of the PDIM genes revealed that all four stimuli tested (WLS, CLSE, PPL and SP-A) triggered the expression of this gene set to some degree (Fig. 4). Finally, gene MT3290.1 encoding for the putative transcriptional regulator WhiB7 that has been related to macrophage/bacterial interactions was also significantly induced at > 1.5 fold in Mtb following a 2 h exposure to WLS.
Fig. 4. Effect of different surfactant treatments on the expression of the PDIM gene cluster in Mtb.
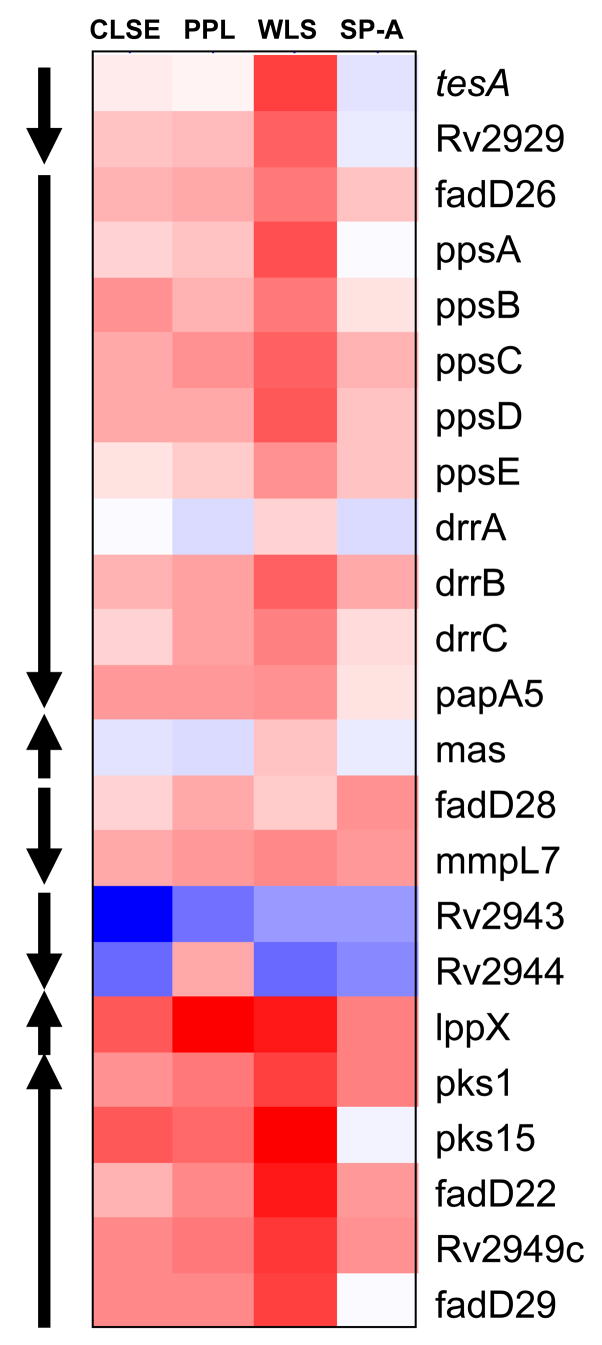
Heat map showing differentially regulated PDIM genes in each of the 2 h-surfactant treatments (i.e. WLS, CLSE, PPL, and SP-A). Genes are organized according to genomic location. Red indicates up-regulation, blue indicates down-regulation and white indicates no change. Arrows indicate predicted operons within the PDIM locus.
2.7. Confirmation of a subset of genes differentially expressed in Mtb by real-time RT-PCR
To validate array data, eight differentially expressed genes in Mtb treated for 30 min or 2h with WLS (Table 1, Supplemental Table 2) were analyzed further by real-time RT-PCR. To confirm a subset of up-regulated genes, we quantified the expression of ppsA, ppsC, fadD22, and pks1, all of which belong to the PDIM gene cluster (located on two different operons), and rpfE, which encodes a resuscitation-promoting factor and plays a role in growth and/or reactivation from dormancy [26]. Real time RT-PCR showed that all genes were consistently up-regulated in Mtb by WLS treatment (Fig. 5). Similarly, we used real-time RT-PCR to validate a set of genes down-regulated by WLS identified by microarray analysis. The genes chosen included Rv0168, which encodes a hypothetical protein and part of the mce1 operon and member of the YrbE family; the gene Rv3616c, part of the putative operon Rv3614c – Rv3616c encoding critical components of SNM secretion [27] and the gene gltA1, involved in carbon metabolism. Again, real-time RT-PCR analysis of these genes confirmed down-regulation (Fig. 5) as determined by microarray analysis. The discrepancy in the magnitude of induction of rpfE as measured by the two methods is likely due to the limited dynamic range of microarrays for accurate quantification of highly expressed genes.
Table 1.
Primers used in this study for real-time RT-PCR.
| Gene name | Sense primer (5′ – 3′) | Antisense primer (5′ – 3′) |
|---|---|---|
| sigA | AAGACCACGAAGACCTCGAA | GCCGATCTGTTTGAGGTAGG |
| ppsA | CGAGCAGATCGATGAGTTGA | GCGTAGGTGGTGGAGATGAT |
| ppsC | ATCATCTCCACCACCTACGC | TGCCGATGCTCAGATAGTTG |
| fadD22 | AAATGACAGTCACGCTGTGC | CCAGCTCTTCGACCTTGTTC |
| pks1 | TCATCGAAGAGTTGGTGCTG | CCGTCCGAGATATCCACACT |
| rpfE | CTGCACCCGTCGATACTCC | TGTTGATCGACCAGTTTCCA |
| Rv0168 | ATGGGCATCAAGTCGATCTC | ACGATCGCAGTGATGATGAG |
| Rv3616c | TGATCAACGCCACTCAACTC | GACCACCCTCGAGAGAACAG |
| gltA1 | AGCGATGGCTGGACATCTAC | CAGCCGGTGATCCTACTCAT |
Fig. 5.

Real-time RT-PCR analysis of selected up- and down-regulated genes: verification of microarray results. Several genes that were significantly affected by WLS treatment at 30 min or 2h according to microarray results were chosen for confirmation with real-time RT-PCR. Real-time RT-PCR was performed on the same aRNA samples as were used for the microarray analysis. Real-time RT-PCR (n=2; n=3 for gltA1) and microarray data (n=3; n=5 for gene Rv0168) are presented as the mean fold change ± standard deviation for each gene.
3. Discussion
Mtb interacts with different microenvironments in the tissue of infected hosts, and the outcomes of these host-pathogen interactions reflect selective gene expression by the bacteria as well as by the host at different phases of infection. To aid in identifying specific Mtb gene expression related to interactions with pulmonary surfactant, we used microarray and RT-PCR to measure changes in Mtb CDC1551 transcription following in vitro exposure to WLS (surfactant lipids plus SP-A, -B, -C, and -D), CLSE (surfactant lipids plus SP-B and -C), PPL (surfactant lipids alone), and purified SP-A. Acute exposure times of 30 min and 2 h were studied, yielding results most relevant for early stages of infection when Mtb first enters the alveoli and encounters surfactant at the air-liquid interface and in the hypophase before being phagocytosed by macrophages. A substantial percentage of mycobacterial particles reaching the alveolar level will experience such exposure in vivo, since the surfactant-rich hypophase is known to cover an appreciable fraction of the respiratory surface ([28] for review). Our results show that exposure to WLS in particular induced a multitude of transcriptional responses in Mtb, including the induction of membrane-located lipase genes and the induction of PDIM cluster genes relevant for interactions with alveolar macrophages [29]. Since surfactant has physicochemical similarities to detergent-like materials, it has the potential to produce a stress response in analogy to substances such as SDS [17]. However, when we analyzed changes in the global expression profile of Mtb after 30 min of exposure to 0.05% SDS or 0.05% NP-40 (data not shown), there was little to no correlation with the response to WLS suggesting that surfactant is not recognized by Mtb as a detergent stress signal.
The induction of genes for bacterial lipases in Mtb following exposure to whole surfactant is consistent with mycobacteria viewing the predominant lipid content of this material as a potential carbon source. Mtb has more than 150 genes encoding enzymes involved in fatty acid degradation, including 21 genes annotated as putative lipase/esterase genes [30], suggesting that the bacterium uses host lipids during in vivo growth. There is strong evidence that fatty acids may be the energy source for Mtb during persistence [31]. In principle, Mtb in vivo could utilize surfactant lipids as a nutrient source not only within the alveolar lumen, but also following Mtb phagocytosis by macrophages. It has previously been shown that alveolar macrophages internalize surfactant lipids [32,33,34], which would thus be potentially accessible for metabolic use by intracellular Mtb. Although Mtb bacilli have their own internal lipid stores, the opportunistic use of surfactant lipids for metabolism would reduce the need for de novo synthesis by these organisms. We found two lipases (Rv3097 (lipY) encoding PE30 [22] and Rv1169c (lipX) encoding PE11 [23]) in Mtb to be significantly induced by WLS. Since both lipX and lipY have been shown to localize into the mycobacterial cell wall [23], it is tempting to speculate that these lipases may be candidates for hydrolyzing host lipids (in this case surfactant lipids) that are contacted by the organism. Precedent for this type of surfactant interaction is provided by the respiratory pathogen Pseudomonas aeruginosa, which has been shown to grow in surfactant [35], and to impair its biophysical properties by degrading surfactant proteins [36].
A major focus of our analysis was on surfactant-related Mtb transcriptional responses relevant for interactions with alveolar macrophages, which are known to be present in vivo in substantial numbers of ~50 per alveolus [37]. WLS induced a cluster of Mtb genes responsible for the synthesis of PDIM cell wall components found only in pathogenic mycobacteria. PDIM has been shown to be an important virulence determinant required for efficient replication in the lung during short-term infection of mice [38]. PDIM is composed of a mixture of long-chain beta-diols that are esterified by multimethyl-branched fatty acids called mycocerosic acids. More than 30 genes are required for the formation and translocation of PDIM. These genes are clustered in a 73-kbp region of the chromosome of Mtb [24,39]. We found that WLS exposure for 2 h resulted in the significant up-regulation (>1.5-fold increase, P<0.05) of 10 members of the PDIM gene cluster (ppsA-ppsD, drrB, pks1, fadD22, lppX, Rv2949c, fadD29) and additional genes below cut-off. Rousseau et al. [40] have shown that the presence of PDIM in the cell envelope contributes to the protection of Mtb against the bactericidal activity of reactive nitrogen intermediates released by macrophages. Surfactant constituents in WLS could thus be recognized by Mtb as a signal to prepare for upcoming interactions with macrophages. This possibility is strengthened by the observation that WLS also significantly induced the gene MT3290.1, which encodes for WhiB7. This protein is a member of the WhiB family of putative transcriptional regulators that are unique to actinomyctes [41], and are among the earliest and most highly induced genes during interactions with macrophages [16,42]. Interestingly, whib7 is also inducible by fatty acids [43].
In terms of Mtb gene induction related to different surfactant apoproteins, the significantly greater transcriptional responses found for WLS compared to CLSE indicate a functional importance for SP-A and/or SP-D in this phenomenon. These collectins are present in WLS, but are removed by chloroform-methanol extraction in CLSE. SP-A and SP-D are broadly distributed throughout the respiratory division of the lungs by virtue of their presence in secreted alveolar surfactant and also in the bronchiolar lining. They are thus ideally positioned to participate in a first line of defense against inhaled microbes that reach the pulmonary periphery [11,44]. SP-A and SP-D differ significantly in size and tertiary structure, and their recognition of microorganisms is not identical [11,44]. SP-A preferentially associates with lipid membranes, and plays a biophysical role in increasing phospholipid aggregation including tubular myelin formation [4,45,46]. SP-A is also more abundant than SP-D, and is tightly associated with surfactant phospholipids both in the interior of the hypophase and in the surface film [4,45,47]. In contrast, the alveolar localization of SP-D is primarily thought to be in the aqueous compartment as opposed to the active surface film [45].
Differences in the location and activity of SP-A and SP-D suggest that they differ to some extent in their roles in the innate immune response of the host against Mtb [8]. SP-A acts as an opsonin and regulator of cell receptor activity in enhancing the phagocytosis of Mtb by macrophages [6,48,49]. The Mtb cell-surface Apa (alanine-and proline-rich antigenic) glycoprotein has been identified as a potential adhesin for the human pulmonary SP-A [50], and SP-A has recently been shown to regulate toll-like receptor expression and activity in human macrophages [51]. SP-A has also been shown to down-regulate nitric oxide production in Mtb-infected murine alveolar macrophages [52]. In contrast to SP-A, agglutination of virulent Mtb associated with SP-D inhibits phagocytosis of the bacilli by alveolar macrophages [9]. Prior work has also shown that SP-D limits the intracellular growth of bacilli in human macrophages by increasing phagosome/lysosome fusion without generating a respiratory burst [7]. Interestingly, both SP-A and SP-D increase the membrane permeability of Gram-negative bacteria and inhibit their growth [10,53]. We did not test the effects of pure SP-D on Mtb gene expression in the present study, but our data indicated that exposure to purified SP-A did lead to alterations in transcription. The non-overlapping Mtb gene expression results found for two different SP-A exposure methods (SP-A in aqueous solution and pre-coated on a plastic culture dish, Supplemental Tables 5 and 6) suggest that environment-induced physicochemical changes in protein conformation might be important in Mtb responses, although this requires further study.
It is not currently known to what extent the overall host defense activities of SP-A and SP-D are modified depending on whether they are present alone, together, or combined with surfactant lipids. However, our results for Mtb indicate that by far the most striking transcriptional responses were caused by exposure to whole surfactant, i.e., WLS containing all of the lipids and proteins of the surfactant system combined together. Responses of Mtb were much less pronounced following exposure to purified SP-A. Isolated SP-A suspended in culture medium may function quite differently than when the apoprotein is present in the complex lipoprotein milieu of surfactant as a whole. As noted above, SP-A has strong biophysical associations with surfactant lipids [54] and also has cooperative interactions with SP-B in forming tubular myelin [46]. Molecular interactions between SP-A and surfactant lipids/apoproteins can similarly be expected to be relevant for its host defense activities. Our studies on isolated SP-A used the human apoprotein, in contrast to WLS that was derived from bovine lungs. However, the high degree of amino acid sequence homology between SP-A from different mammalian species [45,55–58] suggests that species differences do not account for the relative lack of Mtb response to purified human SP-A measured here. It is possible that gene induction related to SP-D in WLS contributed to the differences found between Mtb responses to whole surfactant compared to purified SP-A. However, it is very likely that these differences also reflect at least in part complex interactions between SP-A and multiple lipoprotein components in whole surfactant as noted above. Additional studies will be necessary to clarify further the mechanisms contributing to specific aspects of Mtb gene expression induced by pulmonary surfactant.
In summary, our data indicate that pulmonary surfactant directly affects Mtb gene transcription in ways that suggest a pre-conditioning of the mycobacterium for interactions with macrophages and providing lipids for its growth. Gene induction in Mtb was much more prominent for WLS containing the complete mix of surfactant components compared to chloroform-methanol extracted CLSE or column purified lipids. Differences between WLS and CLSE indicated a functional importance for the collectins SP-A and/or SP-D in modifying Mtb gene expression. Purified SP-A alone had reduced and altered transcriptional activity compared to WLS, suggesting that interactions with other surfactant apoproteins or with surfactant lipids are likely important in this phenomenon. Future analyses of Mtb gene expression and cellular interactions in more complex model systems, such as exposure to alveolar type II epithelial cells producing surfactant in vitro, may provide further insights into the early phase of tuberculosis infection.
4. Materials and Methods
4.1. Mtb growth conditions
CDC1551, a clinical isolate of M. tuberculosis, was grown in Middlebrook 7H9 broth (Difco, Detroit, Mich.) supplemented with OADC enrichment (Difco), Tween 80 (Sigma, St. Louis, Mo.), and glycerol at 37°C to an A600 ~0.7 under static conditions. Bacteria were harvested by centrifugation (10 min, 3,000 rpm) at room temperature, resuspended in DMEM with 2% fetal calf serum (FCS) and 5 mM HEPES and syringed to break clumps of Mtb.
4.2. Surfactants and subfractions (WLS, CLSE, PPL, and SP-A)
Whole lung surfactant (WLS) was obtained by centrifugation of lavage fluid (0.15M NaCl) from the intact lungs of freshly killed calves (ONY, Inc, Amherst, NY). Centrifugation was done at 12,000 × g at room temperature to pellet the active large aggregate surfactant fraction. WLS prepared by the same methods used here contains approximately 85% phospholipids, 5% cholesterol, and 8% protein plus a small amount of other neutral lipids (e.g., [59,60]). The protein content of WLS has been shown to contain four apoproteins (SP-A, B, C, D) [4,45], with the first three of these having activity important in biophysical function [4]. The specific presence of hydrophilic SP-A and SP-D in WLS was directly verified here (Fig. 1), because these two hydrophilic proteins represent the only significant compositional difference between this preparation and CLSE [59,60]. CLSE was prepared by extraction of WLS into chloroform-methanol by the method of Bligh and Dyer [61], as described in our prior work [59,60]. CLSE is conceptually identical to the substance of the clinical surfactant Infasurf® [4,62–64]. The composition of CLSE contains the same lipids as WLS, but only 1.5% protein [59,60]. N-terminal amino acid analysis and SP-B ELISA have documented that the protein content of CLSE is made up of SP-B and SP-C [59,60], and reflects the hydrophobic apoprotein content of WLS. Mixed lung surfactant phospholipids (PPL) were isolated from CLSE by column chromatography with an elution solvent of 1:1 (v/v) chloroform-methanol plus 5% 0.1 N HCl [59]. Two passes through a Sephadex LH-20 column (Pharmacia-LKB Biotechnology, Piscataway, NJ) separated phospholipids away from hydrophobic surfactant apoproteins and neutral lipids [59,60]. Acid remaining in PPL fractions was removed by a second chloroform-methanol extraction. Final PPL had a protein content below the limits of detection of both the Folin-phenol assay of Lowry et al.[65] and the amido black assay of Kaplin and Pedersen [66], modified by the addition of 15% sodium dodecyl sulfate (SDS) to allow accurate quantitation in the presence of lipid. Human SP-A was a gift from Dr. J. R. Wright at Duke University, and was isolated from bronchoalveolar lavage from patients with alveolar proteinosis as described previously [67]. For exposure studies with Mtb, amounts of WLS, CLSE, and PPL used per experiment were 3.3 mg in a total volume of 4 ml. In preliminary studies, this concentration was shown to induce a transcriptional response in Mtb without affecting viability after a 2 h treatment as determined by serial dilutions compared to untreated Mtb. SP-A was studied at a level of 0.165 mg based on its putative concentration of approximately 5% in endogenous surfactant [4,12].
4.3. Immunoblot methods for identifying SP-A and SP-D in WLS
Aliquots of WLS and CLSE (negative control) were separated by 12% SDS-PAGE and electro-transferred onto a PVDF membrane for initial Western blot analyses for SP-A and SP-D. Equal loading and transfer were confirmed with Ponceau S staining (Sigma, St Louis, MO). The membrane was blocked with 5% casein and 0.1% Tween in tris-buffered saline (blocking solution) for 1 h at room temperature. Western blot analyses in the presence and absence of primary antibodies confirmed specificity, and WLS and CLSE were then directly loaded on a PVDF membrane using a minifold dot blot apparatus (Schleicher and Schuell Inc, Keene, NH) for subsequent immunoblot analyses. For detection of SP-A, the membrane was incubated with goat anti-SP-A antibody N-19 (Santa Cruz Biotechnology, Santa Cruz, CA) at a 1:1000 dilution in blocking solution. The membrane was washed 3 times in 0.1% Tween in TBS (TBS-T), incubated for 1 h in horse radish peroxidase-conjugated donkey anti-goat antibody (Santa Cruz Biotechnology, Santa Cruz CA) at a 1:1000 dilution in blocking solution, and washed 3 times in TBS-T. The membrane was then incubated in Supersignal (Pierce, Rockford, IL) for 5 min, and exposed to Biomax MR X-ray film (Kodak, Rochester, NY) to quantitate SP blot -A. For detection of SP-D, rabbit polyclonal anti-SP-D antibody H-120 (Santa Cruz Biotechnology) was used for the primary antibody at 1:1000 dilution, and goat anti-rabbit antibody (Santa Cruz Biotechnology, Santa Cruz CA) was used as the secondary antibody at 1:1000 dilution.
4.4. Surfactant treatment and RNA isolation
To determine the effects of lung surfactant on the transcriptional response of Mtb, preparations of WLS, CLSE, or PPL (in 650 μl PBS) were initially coated onto the bottom surface of a 25 cm2 cell culture flask for 30 min at 37°C to mimic a surfactant layer. A minimum of two independent biological replicates were studied for each surfactant preparation. Purified human SP-A was also examined for its effects on Mtb transcription, and phosphate buffered saline (PBS) was used as a “no treatment” control. In experiments with SP-A (in 650 μl PBS), the pure apoprotein was either pre-coated on the flask as done with the surfactant preparations, or was directly added and incubated with Mtb during the study. Mtb was added to the flask in 3.35 ml of DMEM (supplemented with 2% FCS and 5 mM HEPES) and exposed to coated surfactant/SPA or PBS control for 30 min or 2h at 37°C, followed by RNA isolation as detailed by Rohde et al. [16]. GTC lysis/stabilization buffer was added, and pelleted Mtb was then washed to remove residual GTC and digested with lysozyme. TRIzol (Invitrogen) was added, and organisms were disrupted using glass beads in a Mini-Bead Beater (BioSpec Products, Inc. Bartlesville, OK). After extraction of Mtb lysates with chloroform, ethanol was added to the aqueous phase before loading samples onto RNeasy mini columns (Qiagen, Valencia, CA). Subsequent steps were then performed according to the manufacturer’s instructions. Following elution with RNase-free water, the RNA samples were treated with DNase (Turbo DNAfree, Applied Biosystems, Foster City, CA), and stored at −70°C until analysis.
4.5. Microarray hybridization and data analysis
The MessageAmp™II-Bacteria system (Applied Biosystems) was used to amplify mycobacterial RNA for all samples [16]. Total RNA was reverse transcribed to yield double-stranded amino-allyl modified aRNA, which was labeled with Alexa Fluor 555 and Alexa Fluor 647 (Invitrogen). Excess reactive dyes were removed with a Mega Clear purification kit (Applied Biosystems). Next, Alexa-labeled aRNA from paired samples was dried using a Speedvac, and re-suspended in hybridization buffer (5x SSC, 25% formamide, 0.15% SDS, and 25 μg salmon sperm DNA). The mixed samples were denatured by being heated to 95°C for and then cooled to 50°C prior to loading them onto microarrays. The arrays were hybridized for approximately 15 h at 45°C, washed and then dried by centrifugation [16]. Microarrays were used that consisted of 4295 ORF (open reading frames that encode a protein)-specific 70-mers (Operon) representing 3924 ORFs from strain H37Rv plus 371 ORFs from strain CDC1551 with <97% homology to corresponding H37Rv genes spotted in duplicate on glass slides. Fluorescence intensity data from each array were collected with a GenePix 4000B scanner (Axon Instruments, Union City, CA) using GenePixPro3.0 software (Axon Instruments). Image analysis, i.e. spot identification, background measurements, quality assessment and flagging were conducted with Imagene software (Version 6.0, BioDiscovery, Inc., El Segundo, CA). Poor quality spots were filtered and removed from the analysis. Subsequent normalization, statistical analysis (i.e. t-test), and visualization of array data was conducted using Genespring (Version 7, Agilent Technologies, Palo Alto, CA). Complete microarray data and analyses are included in Supplemental Tables 1–6 on-line, and all transcriptional profile files have also been submitted to the GEO database at NCBI (accession number GSE14005).
4.6. Confirmation of microarray data with real-time RT-PCR
To validate the gene expression profiles, we performed real-time RT-PCR of selected genes using the same aRNA samples as were used for the microarray analysis. Total aRNA (250ng) from either control or surfactant treated Mtb was converted to cDNA using the iScript™cDNA Synthesis kit according to the manufacturer’s instructions (BioRad, Hercules, CA). Triplicate reaction mixtures of each primer pair with cDNA and iTaq™ SYBR Green Supermix (with Rox; Bio-Rad) were heated to 95°C for 2 min and then cycled 40 times at 95°C for 15 sec and 60°C for 45 sec in an ABI 7700 real-time PCR instrument (Applied Biosystems). The threshold cycles (CT), i.e. the cycle number at which the reporter dye emission intensities rises above background noise was identified and normalized against the housekeeping gene sigA using the comparative CT method [68]. The levels of sigA were constant by microarray analysis in surfactant-treated and –untreated samples at each time point. Fold regulation of individual genes following WLS exposure was expressed as 2−ΔΔCT, where ΔΔCT= (CT, Target − CT, sigA)WLS-treated − (CT, Target − CT, sigA)Untreated. Real-time RT-PCR experiments were repeated at least twice with similar results. The gene-specific primers used for these experiments are listed in Table 1.
Supplementary Material
Supplemental Tables 1–6 (Gene lists)
Acknowledgments
The authors wish to thank Paul Debbie (Center for Gene Expression Profiling [CGEP], Boyce Thompson Institute, Ithaca, NY) for array printing, and Dr. David Lin (Cornell Big Red Spots Core Facility) for array scanning. This work was supported by grant 5 R01 HL55936 from the National Institutes of Health to D.G.R.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.World Health Organization (WHO) WHO report 2006 (Document WHO/HTM/TB/2006.362) Geneva, Switzerland: 2006. Global tuberculosis: Control, surveillance, planning, financing; pp. 1–242. [Google Scholar]
- 2.Russell DG. Mycobacterium tuberculosis: here today, and here tomorrow. Nat Rev Mol Cell Biol. 2001;2:569–77. doi: 10.1038/35085034. [DOI] [PubMed] [Google Scholar]
- 3.Creuwels LA, van Golde LM, Haagsman HP. The pulmonary surfactant system: biochemical and clinical aspects. Lung. 1997;175:1–39. doi: 10.1007/PL00007554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Notter RH. Lung surfactants: Basic science and clinical applications. New York: Marcel Dekker, Inc; 2000. [Google Scholar]
- 5.Notter RH, Wang Z. Pulmonary surfactant: Physical chemistry, physiology and replacement. Rev Chem Eng. 1997;13:1–118. [Google Scholar]
- 6.Gaynor CD, McCormack FX, Voelker DR, McGowan SE, Schlesinger LS. Pulmonary surfactant protein A mediates enhanced phagocytosis of Mycobacterium tuberculosis by a direct interaction with human macrophages. J Immunol. 1995;155:5343–51. [PubMed] [Google Scholar]
- 7.Ferguson JS, Martin JL, Azad AK, McCarthy TR, Kang PB, Voelker DR, et al. Surfactant protein D increases fusion of Mycobacterium tuberculosis-containing phagosomes with lysosomes in human macrophages. Infect Immun. 2006;74:7005–9. doi: 10.1128/IAI.01402-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ferguson JS, Voelker DR, McCormack FX, Schlesinger LS. Surfactant protein D binds to Mycobacterium tuberculosis bacilli and lipoarabinomannan via carbohydrate-lectin interactions resulting in reduced phagocytosis of the bacteria by macrophages. J Immunol. 1999;163:312–21. [PubMed] [Google Scholar]
- 9.Ferguson JS, Voelker DR, Ufnar JA, Dawson AJ, Schlesinger LS. Surfactant protein D inhibition of human macrophage uptake of Mycobacterium tuberculosis is independent of bacterial agglutination. J Immunol. 2002;168:1309–14. doi: 10.4049/jimmunol.168.3.1309. [DOI] [PubMed] [Google Scholar]
- 10.McCormack FX. New concepts in collectin-mediated host defense at the air-liquid interface of the lung. Respirology. 2006;11 (Suppl):S7–10. doi: 10.1111/j.1440-1843.2006.00798.x. [DOI] [PubMed] [Google Scholar]
- 11.McCormack FX, Whitsett JA. The pulmonary collectins, SP-A and SP-D, orchestrate innate immunity in the lung. J Clin Invest. 2002;109:707–12. doi: 10.1172/JCI15293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wright JR. Immunomodulatory functions of surfactant. Physiol Rev. 1997;77:931–62. doi: 10.1152/physrev.1997.77.4.931. [DOI] [PubMed] [Google Scholar]
- 13.Crouch E, Wright JR. Surfactant proteins A and D and pulmonary host defense. Annu Rev Physiol. 2001;63:521–54. doi: 10.1146/annurev.physiol.63.1.521. [DOI] [PubMed] [Google Scholar]
- 14.Chimote G, Banerjee R. Lung surfactant dysfunction in tuberculosis: effect of mycobacterial tubercular lipids on dipalmitoylphosphatidylcholine surface activity. Colloids Surf B Biointerfaces. 2005;45:215–23. doi: 10.1016/j.colsurfb.2005.08.014. [DOI] [PubMed] [Google Scholar]
- 15.Wang Z, Schwab U, Rhoades E, Chess PR, Russell DG, Notter RH. Peripheral cell wall lipids of Mycobacterium tuberculosis are inhibitory to surfactant function. Tuberculosis (Edinb) 2008;88:178–86. doi: 10.1016/j.tube.2007.11.003. [DOI] [PubMed] [Google Scholar]
- 16.Rohde KH, Abramovitch RB, Russell DG. Mycobacterium tuberculosis invasion of macrophages: linking bacterial gene expression to environmental cues. Cell Host Microbe. 2007;2:352–64. doi: 10.1016/j.chom.2007.09.006. [DOI] [PubMed] [Google Scholar]
- 17.Manganelli R, Voskuil MI, Schoolnik GK, Smith I. The Mycobacterium tuberculosis ECF sigma factor sigmaE: role in global gene expression and survival in macrophages. Mol Microbiol. 2001;41:423–37. doi: 10.1046/j.1365-2958.2001.02525.x. [DOI] [PubMed] [Google Scholar]
- 18.Sherman DR, Voskuil M, Schnappinger D, Liao R, Harrell MI, Schoolnik GK. Regulation of the Mycobacterium tuberculosis hypoxic response gene encoding alpha -crystallin. Proc Natl Acad Sci U S A. 2001;98:7534–9. doi: 10.1073/pnas.121172498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Waddell SJ, Butcher PD. Microarray analysis of whole genome expression of intracellular Mycobacterium tuberculosis. Curr Mol Med. 2007;7:287–96. doi: 10.2174/156652407780598548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Butcher PD, Mangan JA, Monahan IM. Intracellular gene expression. Analysis of RNA from mycobacteria in macrophages using RT-PCR. Methods Mol Biol. 1998;101:285–306. doi: 10.1385/0-89603-471-2:285. [DOI] [PubMed] [Google Scholar]
- 21.Fisher MA, Plikaytis BB, Shinnick TM. Microarray analysis of the Mycobacterium tuberculosis transcriptional response to the acidic conditions found in phagosomes. J Bacteriol. 2002;184:4025–32. doi: 10.1128/JB.184.14.4025-4032.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Deb C, Daniel J, Sirakova TD, Abomoelak B, Dubey VS, Kolattukudy PE. A novel lipase belonging to the hormone-sensitive lipase family induced under starvation to utilize stored triacylglycerol in Mycobacterium tuberculosis. J Biol Chem. 2006;281:3866–75. doi: 10.1074/jbc.M505556200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cascioferro A, Delogu G, Colone M, Sali M, Stringaro A, Arancia G, et al. PE is a functional domain responsible for protein translocation and localization on mycobacterial cell wall. Mol Microbiol. 2007;66:1536–47. doi: 10.1111/j.1365-2958.2007.06023.x. [DOI] [PubMed] [Google Scholar]
- 24.Perez E, Constant P, Laval F, Lemassu A, Laneelle MA, Daffe M, et al. Molecular dissection of the role of two methyltransferases in the biosynthesis of phenolglycolipids and phthiocerol dimycoserosate in the Mycobacterium tuberculosis complex. J Biol Chem. 2004;279:42584–92. doi: 10.1074/jbc.M406134200. [DOI] [PubMed] [Google Scholar]
- 25.Rao A, Ranganathan A. Interaction studies on proteins encoded by the phthiocerol dimycocerosate locus of Mycobacterium tuberculosis. Mol Genet Genomics. 2004;272:571–9. doi: 10.1007/s00438-004-1088-3. [DOI] [PubMed] [Google Scholar]
- 26.Kana BD, Gordhan BG, Downing KJ, Sung N, Vostroktunova G, Machowski EE, et al. The resuscitation-promoting factors of Mycobacterium tuberculosis are required for virulence and resuscitation from dormancy but are collectively dispensable for growth in vitro. Mol Microbiol. 2008;67:672–84. doi: 10.1111/j.1365-2958.2007.06078.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.MacGurn JA, Raghavan S, Stanley SA, Cox JS. A non-RD1 gene cluster is required for Snm secretion in Mycobacterium tuberculosis. Mol Microbiol. 2005;57:1653–63. doi: 10.1111/j.1365-2958.2005.04800.x. [DOI] [PubMed] [Google Scholar]
- 28.Notter RH, Finkelstein JN. Pulmonary surfactant: An interdisciplinary approach. J Appl Physiol. 1984;57:1613–24. doi: 10.1152/jappl.1984.57.6.1613. [DOI] [PubMed] [Google Scholar]
- 29.Onwueme KC, Vos CJ, Zurita J, Ferreras JA, Quadri LE. The dimycocerosate ester polyketide virulence factors of mycobacteria. Prog Lipid Res. 2005;44:259–302. doi: 10.1016/j.plipres.2005.07.001. [DOI] [PubMed] [Google Scholar]
- 30.Cole ST, Brosch R, Parkhill J, Garnier T, Churcher C, Harris D, et al. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature. 1998;393:537–44. doi: 10.1038/31159. [DOI] [PubMed] [Google Scholar]
- 31.Russell DG. Phagosomes, fatty acids and tuberculosis. Nat Cell Biol. 2003;5:776–8. doi: 10.1038/ncb0903-776. [DOI] [PubMed] [Google Scholar]
- 32.Grabner R, Meerbach W. Phagocytosis of surfactant by alveolar macrophages in vitro. Am J Physiol. 1991;261:L472–7. doi: 10.1152/ajplung.1991.261.6.L472. [DOI] [PubMed] [Google Scholar]
- 33.Wright JR, Youmans DC. Degradation of surfactant lipids and surfactant protein A by alveolar macrophages in vitro. Am J Physiol. 1995;268:L772–80. doi: 10.1152/ajplung.1995.268.5.L772. [DOI] [PubMed] [Google Scholar]
- 34.Poelma DL, Zimmermann LJ, Scholten HH, Lachmann B, van Iwaarden JF. In vivo and in vitro uptake of surfactant lipids by alveolar type II cells and macrophages. Am J Physiol Lung Cell Mol Physiol. 2002;283:L648–54. doi: 10.1152/ajplung.00478.2001. [DOI] [PubMed] [Google Scholar]
- 35.Bouhafs RK, Jarstrand C. Lipid peroxidation of lung surfactant by bacteria. Lung. 1999;177:101–10. doi: 10.1007/pl00007629. [DOI] [PubMed] [Google Scholar]
- 36.Beatty AL, Malloy JL, Wright JR. Pseudomonas aeruginosa degrades pulmonary surfactant and increases conversion in vitro. Am J Respir Cell Mol Biol. 2005;32:128–34. doi: 10.1165/rcmb.2004-0276OC. [DOI] [PubMed] [Google Scholar]
- 37.Crystal RJ. Alveolar macrophages. New York, NY: Raven Press; 1991. [Google Scholar]
- 38.Cox JS, Chen B, McNeil M, Jacobs WR., Jr Complex lipid determines tissue-specific replication of Mycobacterium tuberculosis in mice. Nature. 1999;402:79–83. doi: 10.1038/47042. [DOI] [PubMed] [Google Scholar]
- 39.Camacho LR, Constant P, Raynaud C, Laneelle MA, Triccas JA, Gicquel B, et al. Analysis of the phthiocerol dimycocerosate locus of Mycobacterium tuberculosis. Evidence that this lipid is involved in the cell wall permeability barrier. J Biol Chem. 2001;276:19845–54. doi: 10.1074/jbc.M100662200. [DOI] [PubMed] [Google Scholar]
- 40.Rousseau C, Winter N, Pivert E, Bordat Y, Neyrolles O, Ave P, et al. Production of phthiocerol dimycocerosates protects Mycobacterium tuberculosis from the cidal activity of reactive nitrogen intermediates produced by macrophages and modulates the early immune response to infection. Cell Microbiol. 2004;6:277–87. doi: 10.1046/j.1462-5822.2004.00368.x. [DOI] [PubMed] [Google Scholar]
- 41.Soliveri JA, Gomez J, Bishai WR, Chater KF. Multiple paralogous genes related to the Streptomyces coelicolor developmental regulatory gene whiB are present in Streptomyces and other actinomycetes. Microbiology. 2000;146:333–43. doi: 10.1099/00221287-146-2-333. [DOI] [PubMed] [Google Scholar]
- 42.Geiman DE, Raghunand TR, Agarwal N, Bishai WR. Differential gene expression in response to exposure to antimycobacterial agents and other stress conditions among seven Mycobacterium tuberculosis whiB-like genes. Antimicrob Agents Chemother. 2006;50:2836–41. doi: 10.1128/AAC.00295-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Morris RP, Nguyen L, Gatfield J, Visconti K, Nguyen K, Schnappinger D, et al. Ancestral antibiotic resistance in Mycobacterium tuberculosis. Proc Natl Acad Sci U S A. 2005;102:12200–5. doi: 10.1073/pnas.0505446102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mason RJ, Greene K, Voelker DR. Surfactant protein A and surfactant protein D in health and disease. Am J Physiol. 1998;275:L1–L13. doi: 10.1152/ajplung.1998.275.1.L1. [DOI] [PubMed] [Google Scholar]
- 45.Kuroki Y, Voelker DR. Pulmonary surfactant proteins. J Biol Chem. 1994;269:25943–6. [PubMed] [Google Scholar]
- 46.Williams MC, Hawgood S, Hamilton RL. Changes in lipid structure produced by surfactant proteins SP-A, SP-B, and SP-C. Am J Respir Cell Mol Biol. 1991;5:41–50. doi: 10.1165/ajrcmb/5.1.41. [DOI] [PubMed] [Google Scholar]
- 47.Persson A, Chang D, Rust K, Moxley M, Longmore W, Crouch E. Purification and biochemical characterization of CP4 (SP-D), a collagenous surfactant-associated protein. Biochemistry. 1989;28:6361–7. doi: 10.1021/bi00441a031. [DOI] [PubMed] [Google Scholar]
- 48.Sidobre S, Nigou J, Puzo G, Riviere M. Lipoglycans are putative ligands for the human pulmonary surfactant protein A attachment to mycobacteria. Critical role of the lipids for lectin-carbohydrate recognition. J Biol Chem. 2000;275:2415–22. doi: 10.1074/jbc.275.4.2415. [DOI] [PubMed] [Google Scholar]
- 49.Beharka AA, Gaynor CD, Kang BK, Voelker DR, McCormack FX, Schlesinger LS. Pulmonary surfactant protein A up-regulates activity of the mannose receptor, a pattern recognition receptor expressed on human macrophages. J Immunol. 2002;169:3565–73. doi: 10.4049/jimmunol.169.7.3565. [DOI] [PubMed] [Google Scholar]
- 50.Ragas A, Roussel L, Puzo G, Riviere M. The Mycobacterium tuberculosis cell-surface glycoprotein apa as a potential adhesin to colonize target cells via the innate immune system pulmonary C-type lectin surfactant protein A. J Biol Chem. 2007;282:5133–42. doi: 10.1074/jbc.M610183200. [DOI] [PubMed] [Google Scholar]
- 51.Henning LN, Azad AK, Parsa KV, Crowther JE, Tridandapani S, Schlesinger LS. Pulmonary surfactant protein A regulates TLR expression and activity in human aacrophages. J Immunol. 2008;180:7847–58. doi: 10.4049/jimmunol.180.12.7847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pasula R, Wright JR, Kachel DL, Martin WJ., 2nd Surfactant protein A suppresses reactive nitrogen intermediates by alveolar macrophages in response to Mycobacterium tuberculosis. J Clin Invest. 1999;103:483–90. doi: 10.1172/JCI2991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wu H, Kuzmenko A, Wan S, Schaffer L, Weiss A, Fisher JH, et al. Surfactant proteins A and D inhibit the growth of Gram-negative bacteria by increasing membrane permeability. J Clin Invest. 2003;111:1589–602. doi: 10.1172/JCI16889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Childs RA, Wright JR, Ross GF, Yuen CT, Lawson AM, Chai W, et al. Specificity of lung surfactant protein SP-A for both the carbohydrate and the lipid moieties of certain neutral glycolipids. J Biol Chem. 1992;267:9972–9. [PubMed] [Google Scholar]
- 55.Gao E, Wang Y, McCormick SM, Li J, Seidner SR, Mendelson CR. Characterization of two baboon surfactant protein A genes. Am J Physiol. 1996;271:L617–30. doi: 10.1152/ajplung.1996.271.4.L617. [DOI] [PubMed] [Google Scholar]
- 56.Johansson J, Curstedt T. Molecular structures and interactions of pulmonary surfactant components. Eur J Biochem. 1997;244:675–93. doi: 10.1111/j.1432-1033.1997.00675.x. [DOI] [PubMed] [Google Scholar]
- 57.McCormack FX. The structure and function of surfactant protein-A. Chest. 1997;111:114S–19S. doi: 10.1378/chest.111.6_supplement.114s. [DOI] [PubMed] [Google Scholar]
- 58.Voss T, Melchers K, Scheirle G, Schafer KP. Structural comparison of recombinant pulmonary surfactant protein SP-A derived from two human coding sequences: implications for the chain composition of natural human SP-A. Am J Respir Cell Mol Biol. 1991;4:88–94. doi: 10.1165/ajrcmb/4.1.88. [DOI] [PubMed] [Google Scholar]
- 59.Hall SB, Wang Z, Notter RH. Separation of subfractions of the hydrophobic components of calf lung surfactant. J Lipid Res. 1994;35:1386–94. [PubMed] [Google Scholar]
- 60.Notter RH, Wang Z, Egan EA, Holm BA. Component-specific surface and physiological activity in bovine-derived lung surfactants. Chem Phys Lipids. 2002;114:21–34. doi: 10.1016/s0009-3084(01)00197-9. [DOI] [PubMed] [Google Scholar]
- 61.Bligh EG, Dyer WJ. A rapid method of total lipid extraction and purification. Can J Biochem Physiol. 1959;37:911–7. doi: 10.1139/o59-099. [DOI] [PubMed] [Google Scholar]
- 62.Willson DF, Thomas NJ, Markovitz BP, DiCarlo JV, Pon S, Jacobs BR, et al. Effect of exogenous surfactant (calfactant) in pediatric acute lung injury: a randomized controlled trial. JAMA. 2005;293:470–76. doi: 10.1001/jama.293.4.470. [DOI] [PubMed] [Google Scholar]
- 63.Hudak ML, Farrell EE, Rosenberg AA, Jung AL, Auten RL, Durand DJ, et al. A multicenter randomized masked comparison of natural vs synthetic surfactant for the treatment of respiratory distress syndrome. J Pediatr. 1996;128:396–406. doi: 10.1016/s0022-3476(96)70291-3. [DOI] [PubMed] [Google Scholar]
- 64.Hudak ML, Martin DJ, Egan EA, Matteson EJ, Cummings J, Jung AL, et al. A multicenter randomized masked comparison trial of synthetic surfactant versus calf lung surfactant extract in the prevention of neonatal respiratory distress syndrome. Pediatrics. 1997;100:39–50. doi: 10.1542/peds.100.1.39. [DOI] [PubMed] [Google Scholar]
- 65.Lowry OH, Rosebrough NJ, Farr AL, Randall RJ. Protein measurement with the Folin phenol reagent. J Biol Chem. 1951;193:265–75. [PubMed] [Google Scholar]
- 66.Kaplin RS, Pedersen PL. Sensitive protein assay in the presence of high levels of lipid. Anal Biochem. 1989;150:97–104. doi: 10.1016/s0076-6879(89)72025-5. [DOI] [PubMed] [Google Scholar]
- 67.Wright JR, Wager RE, Hawgood S, Dobbs L, Clements JA. Surfactant apoprotein Mr = 26,000–36,000 enhances uptake of liposomes by type II cells. J Biol Chem. 1987;262:2888–94. [PubMed] [Google Scholar]
- 68.Schmittgen TD. Real-time quantitative PCR. Methods. 2001;25:383–5. doi: 10.1006/meth.2001.1260. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Tables 1–6 (Gene lists)