

Abstract
Ubiquitin C-terminal hydrolase L1 (UCH-L1) is a deubiquitinating enzyme that is selectively and abundantly expressed in the brain, and its activity is required for normal synaptic function. Here, we show that UCH-L1 functions in maintaining normal synaptic structure in hippocampal neurons. We found that UCH-L1 activity is rapidly upregulated by NMDA receptor activation, which leads to an increase in the levels of free monomeric ubiquitin. Conversely, pharmacological inhibition of UCH-L1 significantly reduces monomeric ubiquitin levels and causes dramatic alterations in synaptic protein distribution and spine morphology. Inhibition of UCH-L1 activity increases spine size while decreasing spine density. Furthermore, there is a concomitant increase in the size of presynaptic and postsynaptic protein clusters. Interestingly, however, ectopic expression of ubiquitin restores normal synaptic structure in UCH-L1-inhibited neurons. These findings point to a significant role of UCH-L1 in synaptic remodeling, most likely by modulating free monomeric ubiquitin levels in an activity-dependent manner.
Introduction
The ubiquitin proteasome system (UPS) is a major cellular pathway for protein degradation in eukaryotic cells. The UPS is involved in the development, maintenance, and remodeling of synaptic connections in the mammalian CNS (Patrick, 2006; Yi and Ehlers, 2007). Ubiquitin C-terminal hydrolase L1 (UCH-L1) belongs to a family of deubiquitinating enzymes (DUBs) comprising UCH-L1–5. It is a highly conserved protein that is selectively and abundantly expressed in neurons, representing 1–2% of total soluble protein in the brain (Wilkinson et al., 1989). UCH-L1 is known to generate free monomeric ubiquitin from ubiquitin precursors (Finley et al., 1989; Larsen et al., 1998). Recent in vitro studies have shown that UCH-L1 possesses ubiquitin ligase activity (Liu et al., 2002). In addition to its enzymatic activities, UCH-L1 associates with ubiquitin to inhibit its degradation and therefore maintain monomeric ubiquitin levels (Osaka et al., 2003).
Numerous lines of evidence have linked UCH-L1 to neurodegenerative disorders. The gracile axonal dystrophy (gad) mice have a naturally occurring, spontaneous mutation in the Uch-l1 gene that causes a loss of detectable UCH-L1 expression (Saigoh et al., 1999). Gad mice exhibit severe sensory ataxia at early stages of pathogenesis caused by axonal degeneration in the gracile tract, followed by motor paresis at later stages (Kikuchi et al., 1990). The I93M mutation in the UCH-L1 gene, which was reported in a German family with autosomal dominant Parkinson's disease (Leroy et al., 1998), leads to a 50% reduction in catalytic of UCH-L1 activity in vitro, implying that loss of UCH-L1 activity may reduce the availability of free ubiquitin and contribute to an impaired clearance of proteins by the UPS. Furthermore, transgenic mice, which express the I93M mutant, exhibit a significant reduction in dopaminergic neurons in the substantia nigra and the dopamine content in the striatum (Setsuie et al., 2007).
UCH-L1 is also commonly found in the neurofibrillary tangles observed in Alzheimer's Disease (AD) brains in which the levels of soluble UCH-L1 are decreased (Choi et al., 2004). A recent study revealed that pharmacological inhibition of UCH-L1 activity leads to an impairment in synaptic transmission and maintenance of long-term potentiation (LTP) (Gong et al., 2006), a form of synaptic plasticity that is involved in learning and memory in the hippocampus. Moreover, transduction of UCH-L1 protein into the hippocampus restored both synaptic and behavioral defects observed in the APP/PS1 mouse model of AD (Gong et al., 2006).
Here, we investigated the role of UCH-L1 at synapses. We find that UCH-L1 activity is regulated by synaptic activity. Synaptic activation of UCH-L1 is correlated with an increase in the levels of free monomeric ubiquitin. Pharmacological suppression of UCH-L1 activity reduces monomeric ubiquitin levels and leads to dramatic alterations to synaptic structure. Strikingly, overexpression of ubiquitin rescues the effects of UCH-L1 inhibition. These data suggest that UCH-L1 is one of the major DUBs in the brain that controls ubiquitin homeostasis. Moreover, our findings indicate altered UCH-L1 activity leads to deleterious effects on synapse structure and function.
Materials and Methods
Reagents.
UCH-L1 [LDN-57444 (LDN)] and UCH-L3 (4,5,6,7-tetrachloroindan-1,3-dione) inhibitors were purchased from Calbiochem. NMDA and d(−)-2-amino-5-phosphonopentanoic acid (APV) (NMDA receptor antagonist) were purchased from Tocris Bioscience. The hemagglutinin (HA)-tagged ubiquitin probe (HAUb-VME; vinyl methyl ester functionalized probe) was synthesized as described previously (Borodovsky et al., 2002) and was provided by Dr. H. Ovaa (The Netherlands Cancer Institute, Amsterdam, The Netherlands).
UCH-L1-deficient mice.
The UCH-L1-deficient and wild-type littermate mouse (Uch-L1nm3419) brains (8 weeks of age) were obtained from Dr. Scott Wilson (University of Alabama, Birmingham, AL). This is a spontaneous mouse mutation that arose at The Jackson Laboratory and subsequently mapped by the Scott Wilson group (Walters et al., 2008).
Antibodies.
The following antibodies were used in this study: mouse anti-Myc, rabbit anti-CDK5, rabbit anti-green fluorescent protein (GFP), and rabbit anti-guanylate kinase-associated protein (GKAP) antibodies (purchased from Santa Cruz Biotechnology); mouse anti-postsynaptic density-95 (PSD-95) and rabbit anti-GluR1 (obtained from Calbiochem); rabbit anti-Shank antibody (a generous gift from Dr. Eunjoon Kim, Korea Advanced Institute of Science and Technology, Daejeon, Korea); rabbit anti-GluR1, mouse anti-NR1, and rabbit anti-NR2A antibodies; rat anti-Homer and rabbit anti-Synapsin I (Millipore Bioscience Research Reagents); mouse anti-Bassoon (Nventa Biopharmaceuticals); chicken anti-Map2 (Abcam); rabbit anti-UCH-L1 (BIOMOL); rabbit anti-ubiquitin (Dako); and rabbit anti-Vamp2 (Synaptic Systems).
Primary neuronal cultures.
Hippocampal neuron cultures were prepared from postnatal day 1 (P1) or P2 rat hippocampi as previously described (Patrick et al., 2003). Briefly, for immunostaining experiments, rat hippocampi were dissected, dissociated by papain treatment and mechanical trituration, and plated at medium density (45,000 cells/cm2) on poly-d-lysine-coated coverslips (12 mm in diameter) or glass-bottom dishes (MatTek; 35 mm). For biochemical experiments, mixed hippocampal and cortical neurons were plated at high density on six-well plates (∼500,000 cells per well) coated with poly-d-lysine. Cultures were maintained in B27 supplemented Neurobasal media (Invitrogen) until 14–21 d in vitro (DIV).
Fractionations and DUB labeling assay.
Fractions from rat brains were prepared as previously described (Carlin et al., 1980; Cho et al., 1992). The DUB activity assay was done by incubating 20 μg of lysates from neuronal cultures or rat brain fractions with the HAUb-VME substrate in labeling buffer (50 mm Tris, pH 7.4, 5 mm MgCl2, 250 mm sucrose, 1 mm DTT, and 1 mm ATP) for 1 h at 37°C. Proteins were then resolved by SDS-PAGE 4–20% gradient gels, and blots were subsequently probed with anti-HA monoclonal antibody. Labeled proteins were identified based on their migration on SDS-PAGE gels, and by comparison with previously published data in which the specific bands were analyzed by mass spectroscopy (Borodovsky et al., 2002).
Recombinant DNA and Sindbis constructs.
The Sindbis enhanced green fluorescent protein (EGFP) viral construct was made by cloning the EGFP (Clontech) open reading frame directly into pSinRep5 (Invitrogen). GFPu (in pEGFP-C1 plasmid backbone; Clontech), a fusion of the CL1 degron (degradation signal) on the C terminus of GFP, was kindly provided by Dr. Ron Kopito (Stanford University, Palo Alto, CA). GFPu is ubiquitinated and specifically degraded by the UPS (Gilon et al., 1998; Bence et al., 2001, 2005). The AgeI–BsrGI fragment from photoactivatable (pa) GFP (a kind gift provided by Jennifer Lipponcott-Schwartz, National Institutes of Health, Bethesda, MD) was subcloned into the GFPu plasmid. paGFPu was then subcloned into pSinRep5 (Invitrogen). Orientation was verified by restriction digest and constructs were confirmed by DNA sequencing. The His6-Myc-ubiquitin was provided by Dr. Ron Kopito and was cloned into pSinRep5. The yellow fluorescent protein (YFP)-actin pSinRep5 plasmids was kindly provided by Dr. E. Schuman (California Institute of Technology, Pasadena, CA). For production of recombinant Sindbis virions, RNA was transcribed using the SP6 mMessage mMachine Kit (Ambion) and electroporated into BHK cells using a BTX ECM 600 at 220 V, 129 Ω, and 1050 μF. Virions were collected after 24–32 h and stored at −80°C until use. For UCH-L1 expression constructs, the UCH-L1 open reading frame was obtained from Incyte full-length human cDNA clone (Open Biosystems) encoding wild-type UCH-L1 and was amplified by PCR with a 5′-oligo containing an XhoI site and a 3′-oligo containing an AgeI site, and subsequently cloned in the pEGFP-N1 vector. The single point mutations in the UCH-L1 DNA were introduced by PCR-based site-directed mutagenesis of template plasmid cDNA using primers designed to introduce specific mutations (C90S, 5′-CCATTGGGAATTCCTCTGGCATCGGAC-3′, and D30A, 5′-TTCGTGGCCCTGGGGCTG-3′). All constructs were verified by sequencing and by expression of proteins of the expected molecular weight in HEK 293T cells.
Drug treatments and virus infections.
For protein expression analysis by Western blotting or immunofluorescence staining experiments, cultured neurons were treated with 10 μm UCH-L1 (LDN) or UCH-L3 inhibitor for 24 h. In experiments in which neurons were subjected to LDN treatment and infections, neurons were first treated with LDN and then infected by adding virions directly to the culture medium and allowing protein expression for 12–14 h. The total time of exposure to LDN was kept constant (24 h). Activity stimulation experiments were performed by treating cultures with NMDA and glycine at 50 and 10 μm, respectively, for 10 min at 37°C. Where indicated, neurons were pretreated with UCH-L1 inhibitor (10 μm) for 24 h or APV (50 μm) for 10 min before addition of NMDA/glycine to the culture media.
Immunostaining.
At the end of each experiment, hippocampal neurons plated on coverslips or 35 mm glass-bottom dishes were rinsed briefly in PBS and fixed with 4% paraformaldehyde (PFA) and 4% sucrose in PBS-MC (PBS with 1 mm MgCl2 and 0.1 CaCl2) for 10 min at room temperature. Neurons were then rinsed three times with PBS-MC and subsequently blocked and permeabilized with blocking buffer containing 2% BSA and 0.2% Triton X-100 in PBS-MC for 20 min. After rinsing neurons three times with PBS-MC, primary antibodies were added in blocking buffer and cultures were incubated overnight at 4°C. The following antibodies and dilutions were used for immunofluorescence stainings: mouse anti-PSD-95 (1:500), rabbit anti-Synapsin I (1:2000), mouse anti-Bassoon (1:2000), rabbit anti-Shank (1:2000), rabbit anti-GluR1 (1:20), chicken anti-Map2 (1:5000), and mouse anti-Myc (1:1000). After three washes with PBS-MC, neurons were incubated in goat anti-rabbit, -mouse, or -chicken secondary antibodies conjugated to Alexa 488, Alexa 568, or Alexa 678 (1:500 each; Invitrogen) at room temperature for 1 h. Neurons were washed three times with PBS-MC and mounted on slides with Aqua Poly/Mount (Polysciences). For live labeling of surface GluR1, the anti-GluR1 antibody against the N terminus extracellular epitope of the receptor was added to neurons in culture medium at 1:20 dilution for 10 min before washing out excess antibody and fixing with 4% PFA/4% sucrose.
Electron microscopy.
Mature hippocampal neurons (>21 DIV) were plated in 35 mm glass-bottom dishes and treated with DMSO (control) or LDN (10 μm). After 24 h, cells were fixed in 2% paraformaldehyde and 1% glutaraldehyde, and then fixed in osmium tetraoxide and embedded in Epon Araldite. Once the resin hardened, blocks with the cells were detached from each dish and mounted for sectioning with an ultramicrotome (Leica). Grids were stained with 1% uranyl acetate and analyzed with a Zeiss OM 10 electron microscope as previously described (Rockenstein et al., 2001). Manual analysis of presynaptic terminal diameter, vesicle number, and synaptic contact zone was performed. A total of 10 micrographs was obtained and from each grid (nine grids per condition) for a total of 90 electron micrographs analyzed per condition. Analysis was performed using ImageQuant. Magnification was 30,000×. Statistical significance was determined by unpaired two-tailed Student's t test.
Live imaging of paGFPu degradation.
Cultured hippocampal neurons (>21 DIV) on 35 mm glass-bottom dishes were incubated for 24 h before imaging in media containing either DMSO (control) or LDN (10 μm). paGFPu virions were added directly to culture media after 12 h of LDN treatment and protein expression was allowed to continue for 12–14 h. Culture media was then replaced with warm HBS [HEPES-buffered saline solution containing the following (in mm): 119 NaCl, 5 KCl, 2 CaCl2, 2 MgCl2, 30 glucose, 10 HEPES]. Cells were maintained at ∼35°C using a ceramic heat lamp (ZooMed), and bath temperature was continually monitored by a digital probe thermometer. Infected neurons (identified by mCherry expression) were then photoactivated for 10–15 s with 100 W Hg2+ lamp and a D405/40× with 440 DCLP dichroic filter set (Chroma). For live imaging, pyramidal-like neurons were selected in a random manner. Confocal z-stack images (with 0.5 μm sections) were acquired with a 63× objective every 2 min.
In vitro proteasome activity assay.
Proteasome activity was measured as previously described with slight modifications (Kisselev and Goldberg, 2005). Briefly, cultured neurons were incubated for 24 h in media containing either DMSO (control) or LDN (10 μm). Neurons were then lysed in proteasome assay buffer (50 mm Tris-HCl, pH 7.5, 250 mm sucrose, 5 mm MgCl2, 0.5 mm EDTA, 2 mm ATP, 1 mm DTT, and 0.025% digitonin) for 15 min on ice. Lysates were cleared by centrifugation at 14,000 rpm for 15 min. One hundred micromolar fluorogenic proteasome peptide substrate N-succinyl-Leu-Leu-Val-Tyr-7-amido-4-methylcoumarin (Suc-LLVY-AMC) (BIOMOL International) was then added to equal amounts of cleared lysates in a 96-well microtiter plate. Fluorescence (380 nm excitation; 460 nm emission) was monitored on a microplate fluorometer (HTS 7000 Plus; PerkinElmer) every 5 min for 2 h at room temperature.
Western blot analysis.
Cultured neurons were lysed in radioimmunoprecipitation assay (RIPA) lysis buffer (50 mm Tris, pH 7.4, 150 mm NaCl, 1% Triton X-100, 0.1% SDS, 1 mm EDTA) containing protease inhibitors (Roche). Rat or mouse brains were homogenized in RIPA buffer at 900 rpm in Teflon-glass homogenizers. Neuronal cell lysates or brain homogenates were centrifuged at 14,000 rpm, and supernatants were removed and protein concentration was determined by BSA protein assay kit (Pierce) using bovine serum albumin as a standard. Protein samples were resolved by SDS-PAGE and electrophoretically transferred to nitrocellulose membranes. Membranes were then blocked for 1 h in TBST blocking buffer (TBS, 0.1% Tween 20, and 5% milk) at room temperature and then incubated with primary antibodies in blocking buffer overnight at 4°C. The antibodies used were at the following dilutions: mouse anti-PSD-95 (1:5000), rabbit anti-Synapsin I (1:10,000), rabbit anti-GluR1 (1:5000), rabbit anti-Shank (1:10,000), rabbit anti-GFP (1:10,000), rabbit anti-UCH-L1 (1:5000), rabbit anti-GKAP (1:2000), rat anti-Homer (1:2000), mouse anti-NR1 (1:2000), rabbit anti-NR2A (1:2000), rabbit anti-ubiquitin (1:2000), rabbit anti-Vamp2 (1:5000), and rabbit anti-CDK5 (1:10,000). Blots were then washed three times in TBST washing buffer (TBS, 0.1% Tween 20) and incubated with goat anti-rabbit, -mouse, or -rat IgG conjugated to horseradish peroxidase (1:5000). Protein bands were visualized by chemiluminescence plus reagent (PerkinElmer) and were digitized and quantified using NIH ImageJ software. For statistical analysis, unpaired Student's t test was performed between any two conditions.
Image acquisition and quantification.
Confocal images were acquired using a Leica DMI6000 inverted microscope outfitted with a Yokogawa spinning disk confocal head, an Orca ER high-resolution black-and-white cooled CCD camera (6.45 μm/pixel at 1×) (Hamamatsu), Plan Apochromat 40×/1.25 numerical aperture (NA) and 63×/1.4 NA objective, and a Melles Griot argon/krypton 100 mW air-cooled laser for 488/568/647 nm excitations. Exposure times were held constant during acquisition of all images for each experiment. Pyramidal-like cells were chosen in a random manner. Confocal z-stacks were taken at 0.4–0.5 μm depth intervals in all experiments. For image analysis, maximum z-projections were used. Images were thresholded equally 1.5–2 times above background. Dendrites from individual neurons were then straightened and used for analysis. Fluorescence intensity associated with presynaptic and postsynaptic protein puncta was measured to determine the size and number of puncta (normalized to dendritic length) in control and LDN-treated neurons. GFP-expressing hippocampal neurons were used for spine morphology analysis using the Edgefitter NIH ImageJ plug-in (Ghosh Laboratory, University of California, San Diego, La Jolla, CA). To determine the length of a spine, the distance from the tip of the protrusion to the dendritic shaft was measured manually. To measure the width of a spine, the maximal length of the spine head perpendicular to the long axis of the spine neck was measured. The number of spines visible along the dendrite was counted manually per 1 μm dendritic length. Measurements were then automatically logged from NIH ImageJ into Microsoft Excel. Statistical significance was determined by unpaired two-tailed Student's t test. All imaging and analysis of ubiquitin rescue experiments (see Fig. 8) were performed in a blinded manner. For quantification of the proteasome reporter degradation, images were thresholded above background equally between conditions. Total integrated fluorescence intensity was measured from dendrites at each time interval and expressed as the percentage change from time 0. Grouped analysis of dendritic fluorescence decay over time from each treatment group was plotted as line graphs (mean ± SEM). The degradation rate of reporter fluorescence decay was obtained by taking the difference of total fluorescence loss [arbitrary units (AU)] over time (Fi − Fn/timen) from individual experiments. The mean rate ± SEM per treated group was then normalized to the control rates. For statistical analysis, grouped degradation rates were analyzed by unpaired two-tailed t tests. One-half of the paGFPu reporter degradation experiments were performed in a blinded manner. There was no significant difference between experiments performed blind and unblind, and therefore the data were combined.
Figure 8.
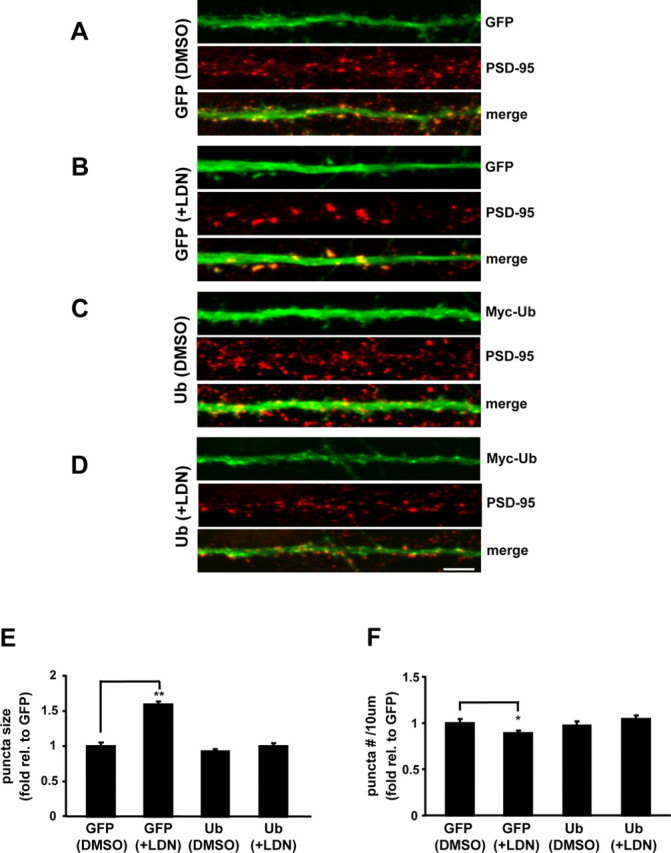
Overexpression of ubiquitin restores synaptic structure in LDN-treated neurons. Cultured neurons were treated with vehicle (DMSO) or LDN for 24 h. EGFP or Myc-ubiquitin Sindbis virions were added directly to culture media after 12 h of treatment, and protein expression was allowed to continue for 12 h. At the end of treatments, neurons were fixed, permeabilized, and immunolabeled with anti-PSD-95 or colabeled with anti-PSD-95 and -Myc antibodies. A–D, Representative maximum z-projected confocal images of straightened dendrites from GFP (DMSO), GFP (+LDN), myc-ubiquitin (DMSO), and myc-ubiquitin (+LDN), respectively. E, F, PSD-95 protein puncta size (E) and number (F) were analyzed in neurons that expressed EGFP or myc-ubiquitin. The mean puncta size and number from three independent experiments were normalized to the GFP (DMSO) control. The number of puncta was calculated per 10 μm dendrite length. Measurements for PSD-95 stainings were made on >60 dendrites per condition. Scale bar, 5 μm. Mean values ± SEM are shown. *p < 0.05, **p < 0.001, unpaired Student's t test.
Results
Characterization of UCH-L1 distribution and activity in neurons
To determine the subcellular localization of UCH-L1, cultured hippocampal neurons were immunostained for endogenous UCH-L1 and the postsynaptic marker PSD-95 (Fig. 1A,B). In some experiments, GFP was used as a cell-filling marker to visualize spines (Fig. 1B). UCH-L1 expression was detected in both soma and dendrites of hippocampal neurons. UCH-L1 is distributed in a micropunctate manner and partially colocalized with PSD-95 (Fig. 1A,B). Moreover, UCH-L1 was found to be localized to dendritic spines of neurons (Fig. 1B). To further characterize the subcellular distribution of UCH-L1, we used differential and density gradient centrifugation to enrich for various synaptic compartments from rat brain. In addition, we used a novel hemagglutinin-tagged ubiquitin-vinyl methyl ester-derived active site-directed probe (HAUb-VME) that covalently modifies DUBs including UCHs (Borodovsky et al., 2002) to profile active DUBs present in the brain. This assay provides a highly sensitive approach for detecting UCH-L1 activity as our probe is specifically targeted to UCH-L1 when it is in an active form. Using this probe, we monitored the activity of UCH-L1 in lysates generated from rat brain fractions. The profile of active DUBs present in these various rat brain fractions is shown in Figure 1C. A lower exposure of the blot presented in Figure 1C is given in supplemental Figure 1A (available at www.jneurosci.org as supplemental material). As observed, UCH-L1 is highly expressed in all fractions compared with the other DUBs detected and is present in an active form. A comparison between active UCH-L1 modified by the substrate (upper band) and the unmodified, inactive UCH-L1 (lower band) is demonstrated in Figure 1D and indicates that ∼50% of UCH-L1 is present in active form in the total homogenate (Fig. 1D, lane 1). Interestingly, we found that UCH-L1 associated with the PSD is primarily in an active form (Fig. 1D, lanes 6, 7). To verify that the HAUb-VME labeling of the DUB at ∼35 kDa is primarily modified UCH-L1 and not UCH-L3, we performed the DUB labeling assay on lysates from UCH-L1-deficient mice (nm3419) (Walters et al., 2008). Here, we found virtually no labeling of any other DUB that runs at or near the same molecular weight of UCH-L1 (supplemental Fig. S1B, top panel, available at www.jneurosci.org as supplemental material). Together, our data demonstrate that UCH-L1 is expressed ubiquitously in neurons with a subpopulation distributed to spines and postsynaptic densities.
Figure 1.
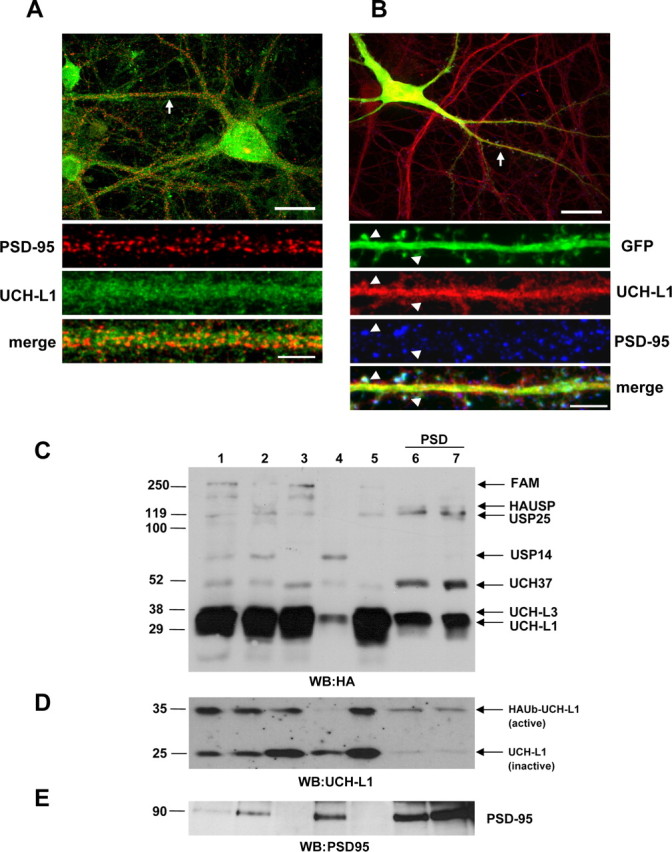
UCH-L1 expression and activity levels in rat brain and mature cultured hippocampal neurons. A, Representative image of a hippocampal neuron immunolabeled with anti-PSD-95 and anti-UCH-L1 antibodies. B, GFP-expressing hippocampal neurons immunostained with anti-PSD-95 and anti-UCH-L1 antibodies. The straightened dendrites in A and B correspond to the regions indicated by arrows in the whole-cell images. The arrowheads indicate selected regions in which UCH-L1 colocalizes with PSD-95 and GFP-filled spines. Representative maximum z-projected confocal images (cell and straightened dendrites) are depicted. Whole-cell scale bar, 20 μm; dendrite scale bar, 5 μm. C, Active DUB profiling with HAUb-VME in subcellular fractions of rat brain. The immunoblot was probed with anti-HA antibody to detect active DUBs. D, Comparison of labeled (active) and unlabeled (inactive) UCH-L1 (HAUb-UCH-L1 and UCH-L1, respectively) in rat brain fractions in an immunoblot that was probed with anti-UCH-L1 antibody. E, The postsynaptic marker, PSD-95, was used as control for fractionation. Numbers correspond to the following fractions: (1) whole-brain homogenate (H); (2) crude synaptosomal fraction (P2); (3) cytosol plus light membranes (S2); (4) light membranes (Golgi, ER) (P3); (5) cytosolic fraction (S3); (6) PSD-1T (one Triton X-100 extraction); (7) PSD-2T (two Triton X-100 extraction).
NMDA receptor activation upregulates UCH-L1 activity
A recent study has demonstrated that pharmacological inhibition of UCH-L1 activity by 40% is sufficient to significantly attenuate LTP in rat hippocampal slices (Gong et al., 2006). This suggests that UCH-L1 function may be required for synaptic plasticity and may itself be regulated by synaptic activity. Indeed, depolarization-dependent Ca2+ influx into synaptosomes has been shown to produce a rapid decrease in ubiquitin conjugates (Chen et al., 2003). To test the possibility that UCH-L1 might be regulated by synaptic activity, we stimulated neuronal cultures (DIV 21) with NMDA receptor agonist. We found that stimulation of synaptic activity by NMDA/glycine significantly upregulated UCH-L1 activity in cultured neurons (Fig. 2A, top panel). A comparison of labeled (active) and unlabeled (inactive) UCH-L1 in response to NMDA receptor stimulation is shown in Figure 2A, bottom panel. On average, the activity of UCH-L1 increased by ∼1.5-fold in response to NMDA receptor stimulation (Fig. 2B,C) (control, 1.0 ± 0.07; NMDA/Gly, 1.63 ± 0.16; p = 0.003, one-way ANOVA). Moreover, the NMDA-induced upregulation of UCH-L1 activity was efficiently blocked by pretreatment of neurons with the NMDA receptor antagonist APV demonstrating the specificity of our treatment (Fig. 2B,C). This novel and potentially important finding demonstrates that synaptic activity can modulate the activity of UCH-L1.
Figure 2.

Activity-dependent upregulation of free monomeric ubiquitin levels in neurons is dependent on UCH-L1 activity. A–C, Cultured neurons were treated with 50 μm NMDA/10 μm glycine or 50 μm NMDA/10 μm glycine plus 50 μm APV for 10 min. Lysates from control and treated neurons were labeled with HAUb-VME substrate and subjected to Western blot analysis. A, A representative immunoblot probed with anti-HA antibody demonstrates the levels of labeled (active) UCH-L1 in control and treated neurons in the presence or absence of the DUB labeling reagent (top panel). The blot was subsequently stripped and reprobed with anti-UCH-L1 antibody to demonstrate the levels of labeled and unlabeled UCH-L1 (HAUb-UCH-L1 and UCH-L1, respectively) (bottom panel). B, A representative immunoblot probed with anti-HA antibody demonstrates activity levels of UCH-L1 in response to drug treatments (top panel). The bottom panel of B shows equal amounts of UCH-L1 between treatments. C, Densitometry analysis of six independent experiments from Dub labeling assays is shown. D–F, Cultured neurons were treated with 50 μm NMDA/10 μm glycine for 10 min (D) or 10 μm UCH-L1 inhibitor (LDN) for 24 h with or without an additional 10 min treatment with 50 μm NMDA/10 μm glycine (E, F), respectively. Lysates were resolved on 15% SDS-PAGE and immunoblots were probed with anti-ubiquitin antibody. Representative blots from each experiment are shown. Relative band intensities of corresponding monomeric ubiquitin were quantified and are depicted in the bottom panel of figure F. Mean values ± SEM of four to five independent experiments are shown. For statistical analysis, one-way ANOVA with post hoc Bonferroni's multiple-comparison test was used. *p < 0.05; **p < 0.01; ***p < 0.001.
UCH-L1 activity is required for NMDA-induced upregulation of free monomeric ubiquitin
Multiple functions have been ascribed to UCH-L1 in vitro and in vivo. It is known that UCH-L1 can act as a ubiquitin hydrolase and generate free ubiquitin species from precursor ubiquitin polypeptides (Wilkinson et al., 1989). UCH-L1 can also bind to ubiquitin and act as a ubiquitin stabilizer to prevent its degradation by lysosomes (Osaka et al., 2003). Therefore, we were interested to determine whether NMDA receptor stimulation had any effect on the levels of free monomeric ubiquitin (also referred to as monoubiquitin) and whether UCH-L1 activity played a role in modulating ubiquitin levels in response to NDMA receptor activation. We found that stimulation with NMDA/glycine (10 min) increased free monomeric ubiquitin levels approximately twofold in cultured neurons (Fig. 2D) (control, 1.0 ± 0.11; NMDA, 1.8 ± 0.14). We next asked whether inhibiting UCH-L1 activity blocks the upregulation of monoubiquitin in NMDA receptor-stimulated neurons. To assess this, we used a previously described UCH-L1 inhibitor, LDN, which specifically inhibits UCH-L1 while having no effect on other UCH family members (Liu et al., 2003; Gong et al., 2006). To demonstrate the efficacy of UCH-L1 inhibition by LDN, neuronal lysates were preincubated with increasing amounts of LDN before DUB labeling assays. We found that 10 μm LDN significantly inhibited UCH-L1 activity in vitro (supplemental Fig. S2A, top panel, available at www.jneurosci.org as supplemental material). Neurons treated with LDN at 10 μm had significantly decreased monoubiquitin levels (Fig. 2E). On average, we observed a 40% reduction in monoubiquitin levels in LDN-treated neurons (Fig. 2E) (control, 1.0 ± 0.11; LDN-treated, 0.59 ± 0.01). Moreover, the levels of free monomeric ubiquitin in neuronal cultures pretreated with LDN before NMDA receptor stimulation were reduced to the levels observed in control untreated neurons (Fig. 2F) (control, 1.0 ± 0.02; LDN-treated, 0.64 ± 0.05; NMDA/Gly-treated, 2.0 ± 0.13; NMDA/Gly plus LDN-treated, 1.04 ± 0.08; p < 0.001, one-way ANOVA). This suggests that NMDA receptor activation in neurons increases free monomeric ubiquitin in an UCH-L1-dependent manner. In addition, we tested whether LDN affected the ability of UCH-L1 to bind ubiquitin. We performed in vitro ubiquitin binding assays with either bacterially expressed glutathione S-transferase (GST) or GST-UCH-L1 and ubiquitin. We found that pretreatment of GST-UCH-L1 with LDN greatly diminished its ability to bind ubiquitin (supplemental Fig. S2B, top panel, available at www.jneurosci.org as supplemental material). Together, this indicates LDN affects both the catalytic and ubiquitin binding activities of UCH-L1.
Inhibition of UCH-L1 activity affects synaptic protein clusters
Recent electrophysiological studies on gad mice and hippocampal slices treated with LDN have demonstrated that UCH-L1 is required for LTP and maintenance of memory (Gong et al., 2006; Sakurai et al., 2008). The brains of gad mice show no gross structural abnormalities; however, it is possible that discrete alterations in neuronal morphology or synaptic structure occur because of the lack of UCH-L1 activity. To examine this possibility, we compared the immunocytochemical distribution of synaptic proteins in control and LDN-treated neurons, several of which are targets for ubiquitination and degradation by the UPS (Colledge et al., 2003; Ehlers, 2003; Guo and Wang, 2007; Lee et al., 2008). Moreover, the postsynaptic proteins we examined are major components of the PSD, which is a highly dynamic structure and its molecular composition and biochemical stability is very responsive to changes in synaptic activity. Furthermore, these activity-dependent molecular changes are regulated in part by the UPS (Ehlers, 2003). We found that exposure of mature hippocampal neurons to LDN leads to dramatic alterations in synaptic structure (Fig. 3). We observed a significant increase in the size of several synaptic protein puncta in LDN-treated neurons compared with those in control neurons. On average, the size of postsynaptic proteins PSD-95, Shank, and surface GluR1 puncta increased by 77, 70, and 39%, respectively (Fig. 3A–D) (PSD-95 puncta, control, 1.0 ± 0.03; LDN-treated, 1.77 ± 0.04; Shank puncta, control, 1.0 ± 0.04; LDN-treated, 1.69 ± 0.04; surface GluR1 puncta, control, 1.0 ± 0.07; LDN-treated, 1.39 ± 0.08). We also detected an increase in the size of presynaptic protein puncta as measured by immunolabeling for presynaptic nerve terminals with Synapsin I and Bassoon. We found that, on average, there was a 34 and 25% increase in the size of Synapsin I and Bassoon puncta, respectively (Fig. 3A,B,D) (Synapsin I puncta, control, 1.0 ± 0.05; LDN-treated, 1.34 ± 0.07; Bassoon puncta, control, 1.0 ± 0.03; LDN-treated, 1.25 ± 0.02). Interestingly, we found no observable difference in the dendritic protein marker MAP2 staining between control and LDN-treated neurons (Fig. 3C). This indicates that UCH-L1 inhibition preferentially affects synapses while having no effect on the overall integrity of dendrites. We also examined whether there was a concomitant alteration in the density of synaptic protein puncta. We found that the density of PSD-95 puncta was decreased by 30% (Fig. 3E) (control, 1.0 ± 0.04; LDN-treated, 0.8 ± 0.3). However, we did not observe any changes in the number of Shank, surface GluR1, Synapsin I, and Bassoon puncta (Fig. 3E). To demonstrate the specificity of our UCH-L1 inhibitor, neurons were treated with a UCH-L3 inhibitor (LDN-L3). UCH-L3 is a closely related DUB and has also been shown to be involved in generating free monomeric ubiquitin. However, we did not find any alterations in synaptic structures in LDN-L3-treated neurons (supplemental Fig. S3A–C, available at www.jneurosci.org as supplemental material). Together, these data show that UCH-L1 activity is specifically involved in maintenance of synapse structure.
Figure 3.
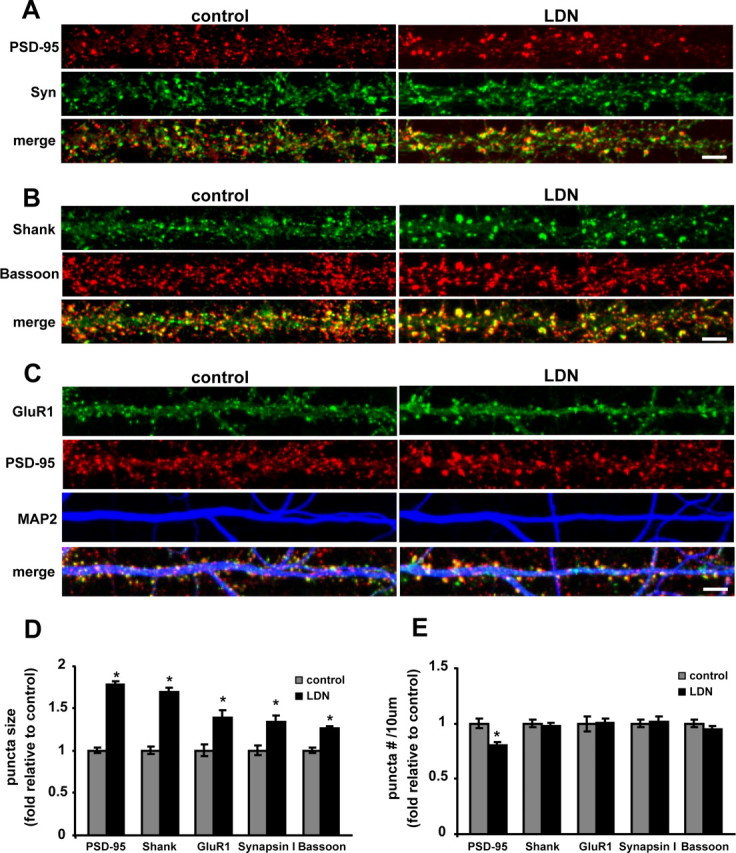
Inhibition of UCH-L1 activity alters synaptic structure. A–C, Cultured neurons were treated with LDN for 24 h. At the end of LDN treatments, neurons were fixed, permeabilized, and immunolabeled with anti-PSD-95 and anti-Synapsin I (A), anti-Shank and anti-Bassoon (B), or live-labeled with anti-GluR1 for surface GluR1 staining, and then fixed and stained with anti-PSD-95 and anti-MAP2 antibodies (C). Representative maximum z-projected confocal images and straightened dendrites from control and LDN-treated neurons are depicted. Scale bar, 5 μm. D, E, Presynaptic and postsynaptic protein puncta size and number were analyzed in control and LDN-treated neurons. The mean puncta size and number in LDN-treated neurons were normalized to those of control neurons from three to four independent experiments. The number of puncta was calculated per 10 μm dendrite length. Measurements for individual stainings were made on the following number of dendrites corresponding to control and LDN-treated neurons, respectively: PSD-95, 84 and 99; Shank, 70 and 102; Synapsin I, 46 and 55; Bassoon, 66 and 93; and GluR1, 51 and 56. Mean values ± SEM are shown. For statistical analysis, unpaired Student's t test was performed between any two conditions. *p < 0.001.
UCH-L1 regulates spine morphology and density
Abundant evidence demonstrates that spines undergo activity-dependent changes in shape and number, and therefore, spines could serve as a cellular substrate for chemical and structural synaptic plasticity (Segal, 2002, 2005). Therefore, we asked whether the observed alterations in the synaptic protein clusters could possibly be accompanied by any changes in spine size and density. To detect alterations in spine morphology, we analyzed spines from GFP-expressing neurons treated with vehicle or LDN. We found striking alterations in the size of spines in LDN-treated neurons. Spines exhibited an enlargement of ∼80% in spine head width and 37% increase in spine length (Fig. 4C,F) (spine head width, control, 1.0 ± 0.09; LDN-treated, 1.8 ± 0.22) (Fig. 4D,G) (spine length, control, 1.0 ± 0.03; LDN-treated, 1.37 ± 0.08). We also observed that blocking UCH-L1 activity dramatically reduced the number of spines (Fig. 4E). LDN-treated neurons had a ∼50% reduction in the number of spines compared with the control untreated neurons (spines/micrometer: control, 0.72 ± 0.05; LDN-treated, 0.35 ± 0.05). These data demonstrate that alterations in synaptic structure induced by inhibition of UCH-L1 activity are also accompanied by changes in spine morphology. Indeed, we observed a 30% decrease in the density of PSD-95 puncta in LDN-treated neurons. This potentially indicates that spine loss precedes the disassembly of the postsynaptic density in UCH-L1-inhibited neurons.
Figure 4.
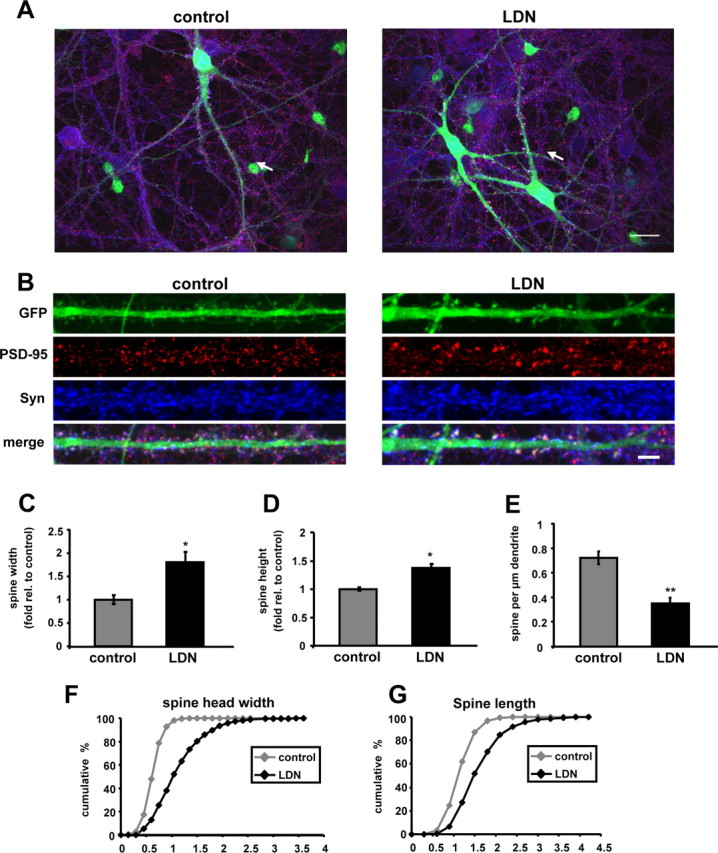
Inhibition of UCH-L1 activity alters spine size and density. A, B, Cultured neurons were treated with LDN for 24 h. EGFP Sindbis virions were added directly to culture media after 10–12 h of LDN treatment, and protein expression was allowed to continue for 12–14 h. At the end of LDN treatments, neurons were fixed, permeabilized, and immunolabeled with anti-PSD-95 and anti-Synapsin I antibodies. The straightened dendrites in B correspond to the regions indicated by arrows in the whole-cell images in A. C–G, Quantification of the number, widths, and lengths of GFP-filled spines in control and LDN-treated neurons is shown. C, D, Measurements for spine head widths and spine lengths were normalized to those of control values. F, G, Cumulative frequency plots showing distribution of spine head width (in micrometers) and spine length (in micrometers) in control and LDN-treated neurons are shown. E, Quantification of spine density is represented as the number of spines per 1 μm dendrite length. The quantified data were obtained from three independent experiments, in which >20 dendrites and >900 spines were analyzed per condition. Mean values ± SEM are shown. *p <0.05, **p < 0.01, unpaired Student's t test.
Filamentous actin (F-actin) accumulates at high concentration in dendritic spines, and actin filaments provide the structural basis for cytoskeletal organization in spines. Actin-based changes in the morphology of spines are regulated by synaptic transmission and are known to contribute to synaptic plasticity (Fischer et al., 1998, 2000; Fukazawa et al., 2003; Okamoto et al., 2004). We examined whether structural changes observed in spines were accompanied by alterations in the actin cytoskeleton. To visualize actin-filled spines, neurons were infected with YFP-actin Sindbis virus (supplemental Fig. S4, available at www.jneurosci.org as supplemental material). In line with our previous observation, we found that YFP-actin-filled spines were enlarged in neurons that were treated with LDN. In addition, neurons were stained with phalloidin to label F-actin in dendritic spines (supplemental Fig. S4B,C, available at www.jneurosci.org as supplemental material). We found that F-actin puncta to also be enlarged in LDN-treated neurons and that they colocalized with synaptic markers PSD-95 (supplemental Fig. S4B, available at www.jneurosci.org as supplemental material) or Shank (supplemental Fig. S4C, available at www.jneurosci.org as supplemental material). Together, our data indicate that alterations to the actin cytoskeleton occur concomitantly with altered presynaptic and postsynaptic protein puncta and spine size in UCH-L1-inhibited neurons.
To further analyze the effects of UCH-L1 inhibition on synaptic structure, we performed electron microscopy on LDN-treated neurons (Fig. 5). In untreated hippocampal cultures, as expected, abundant and well organized neuritic processes were identified (Fig. 5A–D). Most synaptic contacts were synapto-dendritric rather than axo-somatic. On average, presynaptic terminals contained 81.4 ± 6.2 vesicles per synapse and measured 0.89 ± 0.07 μm in diameter, displaying symmetrical postsynaptic densities of ∼0.59 ± 0.06 μm in length (Fig. 5I–K). In contrast, hippocampal cultures treated with LDN displayed abnormal presynaptic and postsynaptic terminals (Fig. 5E–H). The presynaptic terminals contained on average 107.2 ± 6.2 vesicles per synapse and were enlarged, averaging in diameter from 1.64 ± 0.14 μm in diameter, and displayed thick enlarged postsynaptic densities with an average length of 0.64 ± 0.04 μm (Fig. 5I–K). The synaptic terminals were irregular and contained abundant clear synaptic vesicles and coated pits (Fig. 5F). The dendrites were also irregular and displayed enlarged mitochondria, and focal vacuolization (Fig. 5G,H). Together, our EM studies corroborate our immunofluorescence studies and further substantiate a role for UCH-L1 function in maintaining normal synaptic structure.
Figure 5.
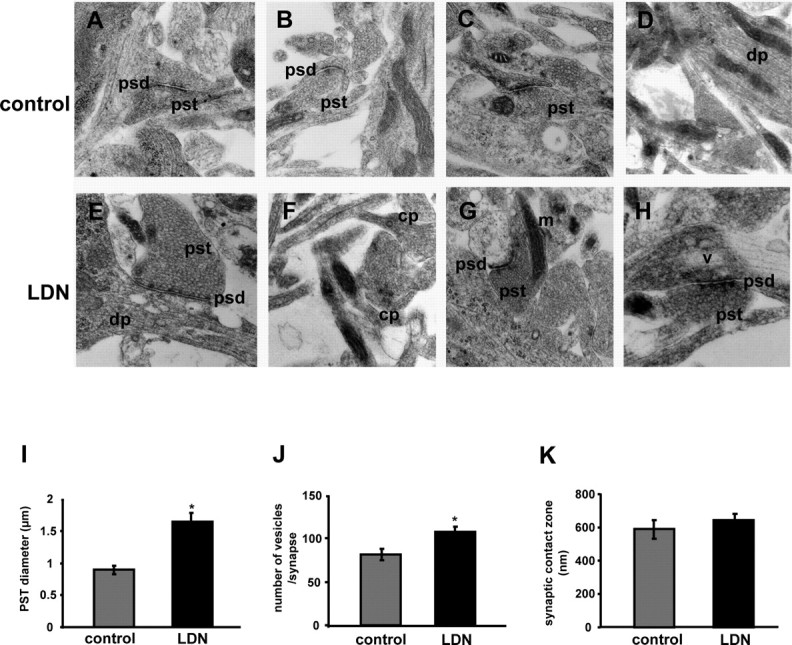
Ultrastructural synaptic alterations in primary neurons treated with LDN. A–H, Representative electron micrographs of cultured neurons treated with vehicle or 10 μm LDN for 24 h. A–D, Control, untreated neurons exhibit normal characteristics for presynaptic terminals (pst), postsynaptic densities (psd), mitochondria (m), and dendritic processes (dp). E–H, LDN-treated neurons display abnormal synaptic structures demonstrated by enlarged presynaptic terminals, irregular postsynaptic densities, abundant coated pits (cp) and clear synaptic vesicles (sv), and vacuolization of dendrites. Magnification, 30,000×. Quantifications of PSD diameter (I), number of vesicles (J), and synaptic zone contact (K) in control and LDN-treated neurons are shown. Averaged results from a total of 10 micrographs for each grid (9 grids per condition) were used for data analysis. Mean values ± SEM are shown. *p < 0.01, unpaired Student's t test.
Altered UCH-L1 activity affects synaptic protein composition
Since inhibition of UCH-L1 activity resulted in a reduction in the levels of free monomeric ubiquitin and structural alterations at synapses, we asked whether these changes were accompanied by alterations in the stability of synaptic proteins (Fig. 6). Of the proteins examined, we only found altered PSD-95 expression levels in LDN-treated neurons. We observed a 60% increase in the levels of PSD-95 in neurons treated with LDN (Fig. 6A,C) (control, 1.0 ± 0.12; LDN-treated, 1.6 ± 0.16). We also examined protein expression levels in brain homogenates obtained from wild-type and UCH-L1-deficient mice (Fig. 6B). Interestingly, we found that protein expression levels of PSD-95 and Shank were increased by 70 and 47% in mouse brain homogenates deficient in UCH-L1 activity (Fig. 6D) (PSD-95, wild type, 1.0 ± 0.15; UCH-L1 null, 1.7 ± 0.19; Shank, wild type, 1.0 ± 0.09; UCH-L1 null, 1.47 ± 0.10). These data show that loss of UCH-L1 activity affects the stability of certain scaffold proteins but not all synaptic proteins.
Figure 6.
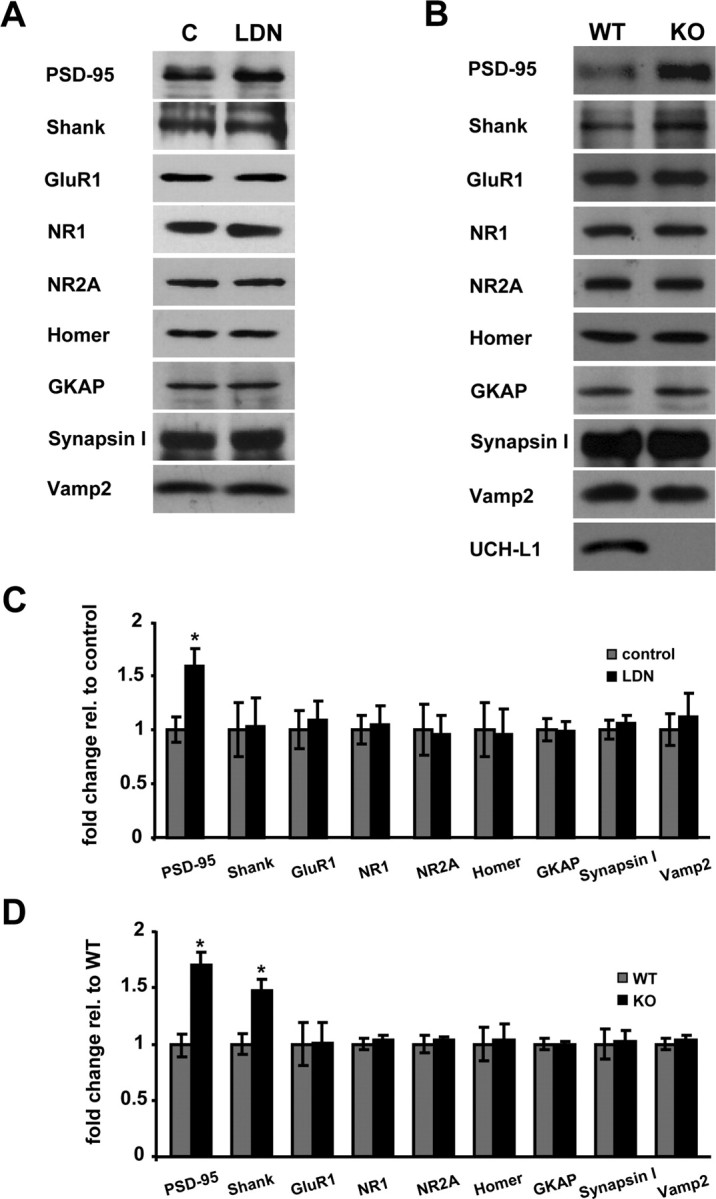
Comparison of synaptic protein expression levels in control and LDN-treated neurons and in wild-type and UCH-L1-deficient mouse brains. A, B, Lysates from control and LDN-treated cultured neurons (A) or homogenates from wild-type and UCH-L1-deficient mouse brains (B) were subjected to Western blot analysis. C, D, Relative band intensities were quantified and normalized to those of control neurons or wild-type mouse brains. The quantified data were obtained from two and four independent experiments for brain homogenates and LDN-treated neurons, respectively. Mean values ± SEM are shown. *p < 0.01, unpaired Student's t test.
Effects of UCH-L1 inhibition on global UPS activity
Since our data clearly demonstrate that blocking UCH-L1 activity reduces the levels of free monomeric ubiquitin, we set out to determine whether inhibiting UCH-L1 activity plays a role in modulating global UPS function. We assessed proteasome activity in vitro by monitoring the cleavage of a synthetic fluorogenic substrate (Suc-LLVY-AMC) in total lysates from control and LDN-treated neurons and detected no differences between conditions (Fig. 7A). Since one of the major functions of UCH-L1 is generating free ubiquitin, we assessed whether altering ubiquitin levels by overexpressing UCH-L1 has any effect on UPS activity. We generated wild-type and mutant GFP-tagged UCH-L1 mammalian expression constructs. All of our constructs were equally expressed in HEK 293T cells (Fig. 7B, top panel). Furthermore, HAUb-VME labeling assay on these lysates showed wild-type UCH-L1-GFP to be active, whereas no activity was detected for either the catalytically inactive C90S or ubiquitin binding-deficient D30A mutants (Fig. 7B, bottom panel). Our data are consistent with previous reports describing these UCH-L1 mutants (Sakurai et al., 2006). We next examined the levels of polyubiquitin conjugates by Western blot analysis as it is known that inhibition of the proteasome activity can directly affect the levels of ubiquitin-conjugated proteins. Interestingly, although we found that overexpression of wild-type or mutant UCH-L1-GFP had no effect on the levels of polyubiquitinated proteins (Fig. 7C, top panel), there was an approximately fourfold to fivefold increase in the levels of free monomeric ubiquitin in cells overexpressing wild-type and the C90S mutant of UCH-L1 (Fig. 7C, middle panel; D) (GFP, 1.0 ± 0.16; wild-type UCH-L1, 4.3 ± 0.23; C90S-UCH-L1, 4.7 ± 0.67; D30A-UCH-L1, 0.97 ± 0.07). These data are quite interesting because although both the C90S and D30A mutants lack hydrolytic activity, the C90S mutant still maintains its ubiquitin binding ability, which is thought to be important for stabilizing ubiquitin levels (Osaka et al., 2003; Sakurai et al., 2006). We also measured proteasome activity in vitro in lysates from wild-type and mutant UCH-L1 overexpressing HEK 293T cells. We did not detect any changes in proteasome activity levels between wild-type and C90S or D30A mutants of UCH-L1 (supplemental Fig. S5A,B, available at www.jneurosci.org as supplemental material). Together, these experiments suggest that altered UCH-L1 activity does not directly affect proteasome function. Furthermore, an increased monomeric ubiquitin levels generated by overexpression of UCH-L1 does not have an effect on the level of polyubiquitin conjugates and proteasome function.
Figure 7.

Effects of altered UCH-L1 activity on UPS function. A, Cultured neurons were treated with vehicle or LDN for 24 h. Proteasome activity was measured in total cell lysates from control and LDN-treated neurons in vitro by fluorimetric assay using a Suc-LLVY-AMC fluorogenic substrate. Relative fluorescence units over time in control and LDN-treated neurons are shown in a line graph. Mean values ± SEM of five independent experiments are shown. B–D, HEK 293T cells were transfected with wild-type, C90S and D30A UCH-L1-GFP constructs for 48 h. B, Expression levels of GFP, wild-type, and mutant UCH-L1-GFP were assessed by Western blot analysis. Immunoblots were probed with anti-GFP antibody (top panel). Activity levels of wild-type and mutant UCH-L1-GFP were assessed by DUB labeling assay (HAUb-VME) of lysates from HEK 293T-transfected cells. A representative Western blot of labeled lysates is demonstrated (bottom panel). C, Expression levels of free monomeric ubiquitin and polyubiquitin conjugates in transfected HEK 293T cells are shown. Western blot probed with anti-UCH-L1 and anti-CDK5 (loading control) antibodies demonstrating equal expression levels of wild-type and mutant UCH-L1-GFP and equal protein loading, respectively. D, Relative intensities of free monomeric ubiquitin levels in GFP, wild-type, and mutant UCH-L1-GFP transfected cells obtained by densitometry analysis of ubiquitin blots from three independent experiments. Mean values ± SEM are shown. *p < 0.01, unpaired Student's t test between control (GFP) and any test group. E–G, Cultured neurons were treated with LDN for 24 h. paGFPu virions were added directly to culture media after 10–12 h of LDN treatment, and protein expression was allowed to continue for 12–14 h. E, Representative straightened dendrites of time-lapse experiments from control or LDN-treated cultured neurons expressing paGFPu are shown. Scale bar, 10 μm. F, Grouped analysis (plotted as line graphs; mean ± SEM) of dendritic paGFPu fluorescence intensity normalized to time 0 for control (paGFPu alone) or LDN-treated neurons. Live imaging of paGFPu degradation was made on 6 and 11 cells, two dendrites per cell, from control and LDN-treated neurons, respectively. RFU, Relative fluorescence unit. The color look-up scale for paGFPu degradation is black (lowest) to white (highest). *p < 0.01, unpaired Student's t test.
However, to better determine whether global UPS function is affected, we monitored the degradation of GFPu in hippocampal neurons using time-lapse confocal microscopy. We used a photoactivatable variant of GFPu (paGFPu), which is a UPS-specific reporter protein substrate and requires polyubiquitination for degradation by the proteasome (Bence et al., 2005). We observed a 50% decrease in the degradation rate of paGFPu in LDN-treated neurons (Fig. 7E,F). Together, this suggests that the catalytic activity of the proteasome is not affected by decreased UCH-L1 activity. However, UCH-L1 inhibition can affect global ubiquitin-dependent UPS function, most likely because of decreased monomeric ubiquitin levels.
Ectopic expression of ubiquitin restores synaptic structure in LDN-treated neurons
The major function of UCH-L1 is thought to maintain the levels of free monomeric ubiquitin used for various cellular processes. To determine whether alteration in synaptic structure induced by inhibition of UCH-L1 were mainly attributable to a reduction in the levels of free monomeric ubiquitin, we performed ubiquitin rescue experiments in LDN-treated neurons (Fig. 8). We found that the expression of ubiquitin for 12 h completely rescued the effects of UCH-L1 inhibition on PSD-95 size and distribution. Although PSD-95 puncta was increased in LDN-treated neurons, the expression of myc-ubiquitin completely blocked the effect of LDN (Fig. 8A–E) (GFP-DMSO, 1.0 ± 0.05; ubiquitin-DMSO, 0.93 ± 0.04; GFP plus LDN, 1.6 ± 0.04; ubiquitin plus LDN, 1.0 ± 0.03). As previously observed, there was a slight decrease in the density of PSD-95 puncta in LDN-treated neurons; however, there was no significant difference in the density of PSD-95 puncta between control (GFP and DMSO-treated neurons) and myc-ubiquitin-expressing neurons (Fig. 8A–D,F) (GFP-DMSO, 1.0 ± 0.04; ubiquitin-DMSO, 0.97 ± 0.05; GFP plus LDN, 0.89 ± 0.03; ubiquitin plus LDN, 1.1 ± 0.03). Moreover, expression of ubiquitin itself had no effect on synaptic structure. Together, our data demonstrate that reductions in the levels of free monomeric ubiquitin because of the lack of UCH-L1 activity lead to major structural synaptic alterations in hippocampal neurons.
Discussion
Activity-dependent remodeling of synaptic connections in the brain is thought to be crucial for modulations in synaptic strength. Recently, the UPS has been shown to be an important factor for this remodeling and for synaptic plasticity. Interestingly, very little is understood about the UPS components involved and their regulation at synapses. In the present study, we set out to understand the function of UCH-L1, the highest expressed DUB in the brain, at synapses to uncover regulatory mechanisms which exist in neurons to control its function. Our data demonstrate that UCH-L1 is distributed throughout somatic and dendritic compartments of hippocampal neurons. Using a specific substrate that can monitor UCH-L1 activity, we show that UCH-L1 is partially active in total lysates from cultured neurons and in the postsynaptic density fractions prepared from rat brain. Strikingly, however, we found that NMDA receptor activation rapidly increases UCH-L1 activity and concomitantly increases free monomeric ubiquitin levels. Furthermore, blocking UCH-L1 activity by LDN significantly reduced the levels of monomeric ubiquitin. Together, these findings suggest that UCH-L1 may be responsible for modulating the levels of free monomeric ubiquitin pools available for various cellular processes. Furthermore, NMDA receptor-dependent activation of UCH-L1 may potentially have a significant effect on synaptic transmission by controlling ubiquitin levels in neurons. This novel finding is the first report demonstrating that the function of UCH-L1 is modulated by neuronal activity.
Many targets of the UPS exist at synapses including scaffold and structural proteins whose function is critical for several forms of plasticity (Patrick, 2006; Yi and Ehlers, 2007). Since decreased UCH-L1 activity is associated with several neurodegenerative disorders, and pharmacological inhibition of UCH-L1 activity substantially reduces LTP, we assessed any structural alterations to synapses in UCH-L1-inhibited neurons. We found that spines became greatly enlarged in LDN-treated neurons while their density significantly decreased. These enlarged spines were associated with presynaptic and postsynaptic proteins, in particular, important scaffold proteins such as PSD-95 and Shank. We found a positive correlation between the size of spines and the size of synaptic protein clusters associated with those spines. Furthermore, we observed alterations to the actin cytoskeleton as there was a significant accumulation of F-actin in spines. The increased spine size together with the striking decrease in spine number potentially reveals heterogeneity in the sensitivity to altered monomeric ubiquitin levels between spines. These alterations in synaptic structure may contribute to the LTP defects observed in UCH-L1-inhibited neurons (Gong et al., 2006). We also analyzed the density of synaptic protein puncta and found a decrease only in PSD-95. This could reflect changes in the stability or trafficking of PSD-95. Indeed, the retention of PSD-95 in individual spines has been shown to be highly dynamic (Gray et al., 2006). We also analyzed protein expression levels for several synaptic proteins to determine whether the observed alterations in the size or density of synaptic puncta correlated with their stability. We only detected changes in the protein expression levels of PSD-95 and Shank. Although the levels of PSD-95 increased in both LDN-treated neurons and brain lysates from UCH-L1-deficient mice, the levels of Shank protein expression were only elevated in the brains of UCH-L1-deficient mice. However, it is quite plausible that the stability of synaptic proteins is not equally sensitive to altered UCH-L1 activity and/or levels of monomeric ubiquitin. Alternatively, UCH-L1 may have specific targets in neurons as other interacting partners for UCH-L1 have been reported (Caballero et al., 2002; Liu et al., 2002; Kabuta et al., 2008). However, it is also feasible to hypothesize that decreased UCH-L1 activity results in decreased monomeric ubiquitin levels, which elicits a pathogenic program that initiates with the loss of unstable spines with a concomitant redistribution and accumulation of synaptic proteins such as PSD-95 and Shank into more stable spines. Strikingly, we found that overexpression of ubiquitin restored normal synaptic structure in LDN-treated neurons. This indicates monomeric ubiquitin levels to be regulatory for normal synaptic structure. Together, these findings point to a significant role of UCH-L1 in synaptic remodeling by modulating free ubiquitin levels most likely in an activity-dependent manner.
Does altered UCH-L1 activity affect UPS function? To address this question, we examined proteasome activity levels in vitro in control and LDN-treated neurons. We did not detect any changes in proteasome activity levels measured in total cell lysates from control and UCH-L1-inhibited neurons. Kyratzi et al. (2008) reported similar findings. They found that the loss of UCH-L1 in neurons derived from gad mice did not affect proteasome activity when assayed in vitro. Furthermore, they also found that overexpression of wild-type UCH-L1, which significantly upregulated free monomeric ubiquitin levels, did not alter proteasome activity in vitro (Kyratzi et al., 2008). This is consistent with our findings (supplemental Fig. 5A,B, available at www.jneurosci.org as supplemental material). This would suggest that the excess availability of monomeric ubiquitin does not increase the proteolytic activity of the proteasome. Interestingly, however, a significant decrease in the degradation rate of the paGFPu reporter was found in neurons that were treated with LDN. This could potentially reflect the sensitivity of our live-imaging reporter assays to altered ubiquitin levels in UCH-L1-inhibited neurons. Although our in vitro measurement of proteasome activity is independent of substrate ubiquitination, paGFPu degradation by the proteasome is dependent on ubiquitination (Bence et al., 2005). Indeed, the stability of PSD-95 and Shank, two PSD scaffolds shown to be regulated by ubiquitin-mediated proteasomal degradation, is significantly increased in UCH-L1-deficient mice.
Together, our findings have uncovered a novel link between neuronal activity, UCH-L1 function, the maintenance of free monomeric ubiquitin levels, and the regulation of synaptic structure. Importantly, this study suggests that altered synapse structure caused by the misregulation of UCH-L1 activity and monomeric ubiquitin homeostasis may serve as an underlying mechanism for defects in synaptic plasticity seen in neurodegenerative disease.
Footnotes
This work was supported by a National Institutes of Health (NIH) postdoctoral training grant (A.E.C.); NIH Grants AG18440, AG10435, and AG22074 (E.M.); The Ray Thomas Edwards Foundation (G.N.P.); and University of California, San Diego startup funds (G.N.P.). We thank Maria Moribito, Darwin Berg, Anirvan Ghosh, and the Patrick Laboratory for advice and critical review of this manuscript. We also thank Alan Okada for technical assistance.
References
- Bence NF, Sampat RM, Kopito RR. Impairment of the ubiquitin-proteasome system by protein aggregation. Science. 2001;292:1552–1555. doi: 10.1126/science.292.5521.1552. [DOI] [PubMed] [Google Scholar]
- Bence NF, Bennett EJ, Kopito RR. Application and analysis of the GFPu family of ubiquitin-proteasome system reporters. Methods Enzymol. 2005;399:481–490. doi: 10.1016/S0076-6879(05)99033-2. [DOI] [PubMed] [Google Scholar]
- Borodovsky A, Ovaa H, Kolli N, Gan-Erdene T, Wilkinson KD, Ploegh HL, Kessler BM. Chemistry-based functional proteomics reveals novel members of the deubiquitinating enzyme family. Chem Biol. 2002;9:1149–1159. doi: 10.1016/s1074-5521(02)00248-x. [DOI] [PubMed] [Google Scholar]
- Caballero OL, Resto V, Patturajan M, Meerzaman D, Guo MZ, Engles J, Yochem R, Ratovitski E, Sidransky D, Jen J. Interaction and colocalization of PGP9.5 with JAB1 and p27(Kip1) Oncogene. 2002;21:3003–3010. doi: 10.1038/sj.onc.1205390. [DOI] [PubMed] [Google Scholar]
- Carlin RK, Grab DJ, Cohen RS, Siekevitz P. Isolation and characterization of postsynaptic densities from various brain regions: enrichment of different types of postsynaptic densities. J Cell Biol. 1980;86:831–845. doi: 10.1083/jcb.86.3.831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Polo S, Di Fiore PP, De Camilli PV. Rapid Ca2+-dependent decrease of protein ubiquitination at synapses. Proc Natl Acad Sci U S A. 2003;100:14908–14913. doi: 10.1073/pnas.2136625100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho KO, Hunt CA, Kennedy MB. The rat brain postsynaptic density fraction contains a homolog of the Drosophila discs-large tumor suppressor protein. Neuron. 1992;9:929–942. doi: 10.1016/0896-6273(92)90245-9. [DOI] [PubMed] [Google Scholar]
- Choi J, Levey AI, Weintraub ST, Rees HD, Gearing M, Chin LS, Li L. Oxidative modifications and down-regulation of ubiquitin carboxyl-terminal hydrolase L1 associated with idiopathic Parkinson's and Alzheimer's diseases. J Biol Chem. 2004;279:13256–13264. doi: 10.1074/jbc.M314124200. [DOI] [PubMed] [Google Scholar]
- Colledge M, Snyder EM, Crozier RA, Soderling JA, Jin Y, Langeberg LK, Lu H, Bear MF, Scott JD. Ubiquitination regulates PSD-95 degradation and AMPA receptor surface expression. Neuron. 2003;40:595–607. doi: 10.1016/s0896-6273(03)00687-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehlers MD. Activity level controls postsynaptic composition and signaling via the ubiquitin-proteasome system. Nat Neurosci. 2003;6:231–242. doi: 10.1038/nn1013. [DOI] [PubMed] [Google Scholar]
- Finley D, Bartel B, Varshavsky A. The tails of ubiquitin precursors are ribosomal proteins whose fusion to ubiquitin facilitates ribosome biogenesis. Nature. 1989;338:394–401. doi: 10.1038/338394a0. [DOI] [PubMed] [Google Scholar]
- Fischer M, Kaech S, Knutti D, Matus A. Rapid actin-based plasticity in dendritic spines. Neuron. 1998;20:847–854. doi: 10.1016/s0896-6273(00)80467-5. [DOI] [PubMed] [Google Scholar]
- Fischer M, Kaech S, Wagner U, Brinkhaus H, Matus A. Glutamate receptors regulate actin-based plasticity in dendritic spines. Nat Neurosci. 2000;3:887–894. doi: 10.1038/78791. [DOI] [PubMed] [Google Scholar]
- Fukazawa Y, Saitoh Y, Ozawa F, Ohta Y, Mizuno K, Inokuchi K. Hippocampal LTP is accompanied by enhanced F-actin content within the dendritic spine that is essential for late LTP maintenance in vivo. Neuron. 2003;38:447–460. doi: 10.1016/s0896-6273(03)00206-x. [DOI] [PubMed] [Google Scholar]
- Gilon T, Chomsky O, Kulka RG. Degradation signals for ubiquitin system proteolysis in Saccharomyces cerevisiae. EMBO J. 1998;17:2759–2766. doi: 10.1093/emboj/17.10.2759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong B, Cao Z, Zheng P, Vitolo OV, Liu S, Staniszewski A, Moolman D, Zhang H, Shelanski M, Arancio O. Ubiquitin hydrolase Uch-L1 rescues beta-amyloid-induced decreases in synaptic function and contextual memory. Cell. 2006;126:775–788. doi: 10.1016/j.cell.2006.06.046. [DOI] [PubMed] [Google Scholar]
- Gray NW, Weimer RM, Bureau I, Svoboda K. Rapid redistribution of synaptic PSD-95 in the neocortex in vivo. PLoS Biol. 2006;4:e370. doi: 10.1371/journal.pbio.0040370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo L, Wang Y. Glutamate stimulates glutamate receptor interacting protein 1 degradation by ubiquitin-proteasome system to regulate surface expression of GluR2. Neuroscience. 2007;145:100–109. doi: 10.1016/j.neuroscience.2006.11.042. [DOI] [PubMed] [Google Scholar]
- Kabuta T, Furuta A, Aoki S, Furuta K, Wada K. Aberrant interaction between Parkinson's disease-associated mutant UCH-L1 and the lysosomal receptor for chaperone-mediated autophagy. J Biol Chem. 2008;283:23731–23738. doi: 10.1074/jbc.M801918200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kikuchi T, Mukoyama M, Yamazaki K, Moriya H. Axonal degeneration of ascending sensory neurons in gracile axonal dystrophy mutant mouse. Acta Neuropathol. 1990;80:145–151. doi: 10.1007/BF00308917. [DOI] [PubMed] [Google Scholar]
- Kisselev AF, Goldberg AL. Monitoring activity and inhibition of 26S proteasomes with fluorogenic peptide substrates. Methods Enzymol. 2005;398:364–378. doi: 10.1016/S0076-6879(05)98030-0. [DOI] [PubMed] [Google Scholar]
- Kyratzi E, Pavlaki M, Stefanis L. The S18Y polymorphic variant of UCH-L1 confers an antioxidant function to neuronal cells. Hum Mol Genet. 2008;17:2160–2171. doi: 10.1093/hmg/ddn115. [DOI] [PubMed] [Google Scholar]
- Larsen CN, Krantz BA, Wilkinson KD. Substrate specificity of deubiquitinating enzymes: ubiquitin C-terminal hydrolases. Biochemistry. 1998;37:3358–3368. doi: 10.1021/bi972274d. [DOI] [PubMed] [Google Scholar]
- Lee SH, Choi JH, Lee N, Lee HR, Kim JI, Yu NK, Choi SL, Lee SH, Kim H, Kaang BK. Synaptic protein degradation underlies destabilization of retrieved fear memory. Science. 2008;319:1253–1256. doi: 10.1126/science.1150541. [DOI] [PubMed] [Google Scholar]
- Leroy E, Boyer R, Auburger G, Leube B, Ulm G, Mezey E, Harta G, Brownstein MJ, Jonnalagada S, Chernova T, Dehejia A, Lavedan C, Gasser T, Steinbach PJ, Wilkinson KD, Polymeropoulos MH. The ubiquitin pathway in Parkinson's disease. Nature. 1998;395:451–452. doi: 10.1038/26652. [DOI] [PubMed] [Google Scholar]
- Liu Y, Fallon L, Lashuel HA, Liu Z, Lansbury PT., Jr The UCH-L1 gene encodes two opposing enzymatic activities that affect alpha-synuclein degradation and Parkinson's disease susceptibility. Cell. 2002;111:209–218. doi: 10.1016/s0092-8674(02)01012-7. [DOI] [PubMed] [Google Scholar]
- Liu Y, Lashuel HA, Choi S, Xing X, Case A, Ni J, Yeh LA, Cuny GD, Stein RL, Lansbury PT., Jr Discovery of inhibitors that elucidate the role of UCH-L1 activity in the H1299 lung cancer cell line. Chem Biol. 2003;10:837–846. doi: 10.1016/j.chembiol.2003.08.010. [DOI] [PubMed] [Google Scholar]
- Okamoto K, Nagai T, Miyawaki A, Hayashi Y. Rapid and persistent modulation of actin dynamics regulates postsynaptic reorganization underlying bidirectional plasticity. Nat Neurosci. 2004;7:1104–1112. doi: 10.1038/nn1311. [DOI] [PubMed] [Google Scholar]
- Osaka H, Wang YL, Takada K, Takizawa S, Setsuie R, Li H, Sato Y, Nishikawa K, Sun YJ, Sakurai M, Harada T, Hara Y, Kimura I, Chiba S, Namikawa K, Kiyama H, Noda M, Aoki S, Wada K. Ubiquitin carboxy-terminal hydrolase L1 binds to and stabilizes monoubiquitin in neuron. Hum Mol Genet. 2003;12:1945–1958. doi: 10.1093/hmg/ddg211. [DOI] [PubMed] [Google Scholar]
- Patrick GN. Synapse formation and plasticity: recent insights from the perspective of the ubiquitin proteasome system. Curr Opin Neurobiol. 2006;16:90–94. doi: 10.1016/j.conb.2006.01.007. [DOI] [PubMed] [Google Scholar]
- Patrick GN, Bingol B, Weld HA, Schuman EM. Ubiquitin-mediated proteasome activity is required for agonist-induced endocytosis of GluRs. Curr Biol. 2003;13:2073–2081. doi: 10.1016/j.cub.2003.10.028. [DOI] [PubMed] [Google Scholar]
- Rockenstein E, Mallory M, Mante M, Sisk A, Masliaha E. Early formation of mature amyloid-beta protein deposits in a mutant APP transgenic model depends on levels of Abeta(1–42) J Neurosci Res. 2001;66:573–582. doi: 10.1002/jnr.1247. [DOI] [PubMed] [Google Scholar]
- Saigoh K, Wang YL, Suh JG, Yamanishi T, Sakai Y, Kiyosawa H, Harada T, Ichihara N, Wakana S, Kikuchi T, Wada K. Intragenic deletion in the gene encoding ubiquitin carboxy-terminal hydrolase in gad mice. Nat Genet. 1999;23:47–51. doi: 10.1038/12647. [DOI] [PubMed] [Google Scholar]
- Sakurai M, Ayukawa K, Setsuie R, Nishikawa K, Hara Y, Ohashi H, Nishimoto M, Abe T, Kudo Y, Sekiguchi M, Sato Y, Aoki S, Noda M, Wada K. Ubiquitin C-terminal hydrolase L1 regulates the morphology of neural progenitor cells and modulates their differentiation. J Cell Sci. 2006;119:162–171. doi: 10.1242/jcs.02716. [DOI] [PubMed] [Google Scholar]
- Sakurai M, Sekiguchi M, Zushida K, Yamada K, Nagamine S, Kabuta T, Wada K. Reduction in memory in passive avoidance learning, exploratory behaviour and synaptic plasticity in mice with a spontaneous deletion in the ubiquitin C-terminal hydrolase L1 gene. Eur J Neurosci. 2008;27:691–701. doi: 10.1111/j.1460-9568.2008.06047.x. [DOI] [PubMed] [Google Scholar]
- Segal M. Dendritic spines: elementary structural units of neuronal plasticity. Prog Brain Res. 2002;138:53–59. doi: 10.1016/S0079-6123(02)38070-1. [DOI] [PubMed] [Google Scholar]
- Segal M. Dendritic spines and long-term plasticity. Nat Rev Neurosci. 2005;6:277–284. doi: 10.1038/nrn1649. [DOI] [PubMed] [Google Scholar]
- Setsuie R, Wang YL, Mochizuki H, Osaka H, Hayakawa H, Ichihara N, Li H, Furuta A, Sano Y, Sun YJ, Kwon J, Kabuta T, Yoshimi K, Aoki S, Mizuno Y, Noda M, Wada K. Dopaminergic neuronal loss in transgenic mice expressing the Parkinson's disease-associated UCH-L1 I93M mutant. Neurochem Int. 2007;50:119–129. doi: 10.1016/j.neuint.2006.07.015. [DOI] [PubMed] [Google Scholar]
- Walters BJ, Campbell SL, Chen PC, Taylor AP, Schroeder DG, Dobrunz LE, Artavanis-Tsakonas K, Ploegh HL, Wilson JA, Cox GA, Wilson SM. Differential effects of Usp14 and Uch-L1 on the ubiquitin proteasome system and synaptic activity. Mol Cell Neurosci. 2008;39:539–548. doi: 10.1016/j.mcn.2008.07.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkinson KD, Lee KM, Deshpande S, Duerksen-Hughes P, Boss JM, Pohl J. The neuron-specific protein PGP 9.5 is a ubiquitin carboxyl-terminal hydrolase. Science. 1989;246:670–673. doi: 10.1126/science.2530630. [DOI] [PubMed] [Google Scholar]
- Yi JJ, Ehlers MD. Emerging roles for ubiquitin and protein degradation in neuronal function. Pharmacol Rev. 2007;59:14–39. doi: 10.1124/pr.59.1.4. [DOI] [PubMed] [Google Scholar]