

Abstract
The ability of cyanoxime-based synthons to act as versatile synthetic tools for the construction of co-crystals is demonstrated through the preparation and structural characterization of seven co-crystals. Cyanoximes can bind effectively to both five-membered and six-membered N-heterocyclic hydrogen-bond acceptors, and they also display selectivity towards the best acceptor (as determined using semi-empirical AM1 calculations) in ditopic compounds.
Introduction
The hydrogen bond is the synthetic tool of choice in crystal engineering in the organic solid state.1 Although halogen bonds, π···π interactions, and other non-covalent forces undoubtedly influence solid-state assembly, the hydrogen bond remains of prime importance due to its abundance, strength and directionality.2 As crystal engineering moves from fundamental science to the deliberate design and synthesis of functional organic materials with specific pharmaceutical,3 electronic,4 or optical5 properties it is imperative that we continue to identify and map out the reliability and robustness of supramolecular synthons involving known functional groups.6 However, in order to expand the existing synthetic tool box, it is desirable to develop the supramolecular chemistry of new, or less well-known, chemical functionalities.
We previously reported7 that aromatic mono cyanoximes readily form co-crystals with symmetric ditopic imidazole-based hydrogen-bond acceptors, producing discrete, trimeric supermolecules. However, before cyanoximes can be fully embraced as a family of reliable and competitive hydrogen-bond donors, considerably more structural data is required. In this study, we set out to examine (a) the versatility of cyanoximes as hydrogen-bond donors and (b) whether a cyanoxime moiety will preferentially bind to the best hydrogen-bond acceptor when faced with an asymmetric ditopic N-heterocyclic molecule. Four different cyanoximes were employed, Scheme 1, all of which can be prepared in good yields by allowing the relevant phenylacetonitrile to react with methylnitrite.7,8 The ready accessibility of suitable starting materials, and the relative ease of synthesis of cyanoximes, add to the potential usefulness of this functional group as a supramolecular synthetic tool.
Scheme 1.

These four hydrogen-bond donors, A–D, were subsequently allowed to react with a series of N-heterocyclic hydrogen-bond acceptors, Scheme 2.
Scheme 2.

The group of hydrogen-bond accetors, 1–7, contains both five-membered and six-membered, symmetric and asymmetric, monotopic and ditopic compounds in order to determine if there are any particular acceptors that are unsuitable as partners for the cyanoximes.
One asymmetric ditopic acceptor, 7, was employed in order to test the best-donor/best-acceptor guideline for hydrogen-bonding in a competitive situation.9,10 The relative ranking of the two acceptor sites, the pyridine moiety and the benzimidazole moiety, can be accomplished using semi-empirical AM1 calculations. 11,12,13
Seven of the co-crystallization reactions between A–D and 1–7 produced crystals that were suitable for single-crystal X-ray diffraction.
Experimental
5 was purchased from Aldrich and used without further purification. The remaining supramolecular reagents 1,14 2–4,10 6,15 711 were synthesized according to previously-reported methods (or slight modifications thereof).
Synthesis of co-crystals
Synthesis of 4-bromophenylcyanoxime 3-bromo-5-methoxypyridine (1:1) A1
4-Bromophenylcyanoxime (20 mg, 0.089 mmol) was dissolved in 1:1 ethanol/ethyl acetate in a 50 mL beaker. A 1:1 ethyl acetate/ethanol solution of 1 (15 mg, 0.080 mmol) was added. Colourless prisms suitable for X-ray crystallography were obtained after four days; mp: 110–115 °C.
Synthesis of 4-bromophenylcyanoxime 1-[(4-bromophenyl)methyl]-2-methylbenzimidazole (1:1) A2
4-Bromophenylcyanoxime (10 mg, 0.044 mmol) was dissolved in ethyl acetate in a 50 mL beaker. A 1:1 ethyl acetate/ethanol solution of 2 (13 mg, 0.043 mmol) was added. Colourless prisms suitable for X-ray crystallography were obtained after one day; mp: 142–145 °C.
Synthesis of 4-bromophenylcyanoxime 1-[(4-bromophenyl)methyl]-benzimidazole (1:1) A3
4-Bromophenylcyanoxime (10 mg, 0.044 mmol) was dissolved in ethyl acetate in a 50 mL beaker. A 1:1 ethyl acetate/ethanol solution of 3 (13 mg, 0.045 mmol) was added. Colourless prisms suitable for X-ray crystallography were obtained after five days; mp: 92–96 °C.
Synthesis of 4-bromophenylcyanoxime 1-[(3-cyanophenyl)methyl]-5,6-dimethylbenzimidazole (1:1) A4
4-Bromophenylcyanoxime (10 mg, 0.044 mmol) was dissolved in ethyl acetate in a 50 mL beaker. A 1:1 ethyl acetate/ethanol solution of 4 (12 mg, 0.046 mmol) was added. Colourless prisms suitable for X-ray crystallography were obtained after five days; mp: 136–140 °C.
Synthesis of 2-fluorophenylcyanoxime 1,2-bis(4-pyridyl)ethylene (2:1) B5
2-Fluorophenylcyanoxime (13 mg, 0.082 mmol) and 5 (13 mg, 0.082 mmol) were dissolved separately in acetonitrile. Slow evaporation over 5 days yielded very thin needles not suitable for X-ray diffraction. Consequently the needles were redissolved in methanol, which produced suitable colorless plates; mp: 167–170 °C.
Synthesis of 3-chlorophenylcyanoxime 1,4-bis[(benzimidazol-1-yl)methyl]benzene (2:1) C6
3-Chlorophenylcyanoxime (8.0 mg, 0.045 mmol) was dissolved in 1:1 ethanol/ethyl acetate in a 50 mL beaker. A 1:1 ethyl acetate/ethanol solution of 6 (15 mg, 0.045 mmol) was added. Fibrous colorless crystals were obtained after 2 days. These were subsequently redissolved in methanol and colorless plates were obtained via slow evaporation after ~ 2 weeks; mp: 175–179 °C.
Synthesis of 4-fluorophenylcyanoxime 1-[(4-pyridyl)methyl]-5,6-dimethylbenzimidazole (1:1) D7
7 (20 mg, 0.084 mmol) was dissolved in ethanol with slight heat. A solution of 4-fluorophenylcyanoxime (15 mg, 0.084 mmol) in ethanol was added and the solution was allowed to evaporate at room temperature. Colourless plates suitable for X-ray crystallography were obtained within 2 weeks; mp: 114–116 °C.
Semi-empirical calculations
In order to rank the hydrogen-bond acceptor sites of 7, the molecular structure was constructed using Spartan ’04 (Wavefunction, Inc. Irvine, CA), and the geometry was optimized using AM1. The results show that the benzimidazole moiety is likely to be a better hydrogen-bond acceptor than the pyridyl site. The former gives gives an energy of −299 kJ/mol and the latter −274 kJ/mol when the electrostatic potential surface (0.002 e/au isosurface) around each nitrogen atom is allowed to interact with a positive point charge in vacuum as a probe.
Results
The driving force for the synthesis of the co-crystal A1 is the expected hydrogen bond between the oxime moiety and the pyridyl hydrogen-bond acceptor site, O(17)···N(21): 2.667(6) Å, Figure 1. There is also a relatively short Br(py)···O(oxime) contact of 2.93 Ă. The oxime moiety is almost perpendicular to the pyridyl ring, which rules out the presence of any ancillary C–H···N interactions involing the oxime nitrogen atom as an acceptor. A secondary interaction is frequently observed in the synthon comprising a carboxylic acid and a pyridyl moiety.16
Figure 1.

Dimer connected through an O-H···N hydrogen bond in the crystal structure of A1.
In the crystal structure of A2, the main non-covalent interaction is the hydrogen bond between R-OH (oxime) and the benzimidazole acceptor; O···N 2.627(2) Ă, Figure 2.
Figure 2.
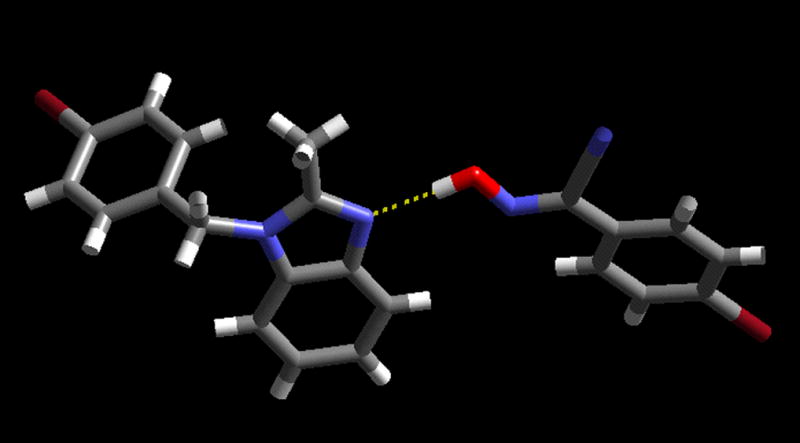
The primary O-H···N hydrogen bond between the oxime and the benzimidazole moieties in the crystal structure of A2.
This time, the oxime nitrogen atom is in the plane of the benzimidazole ring but the distance to the nearest potential C–H donor (H15) is too large to be considered as a structure-directing interaction, C···N; 3.69 Ă. Neither of the two bromine atoms is engaged in a well-defined non-covalent interaction, and the same is true for the nitrile nitrogen atom. Despite very close similarities in chemical composition between A2 and A3, they belong to different crystal systems and space groups, monoclinic P21/c and triclinic P−1, respectively. Nevertheless, the driving force for the co-crystal formation in A3 is still the O-H···N, oxime/benzimidazole, hydrogen bond, Figure 3.
Figure 3.
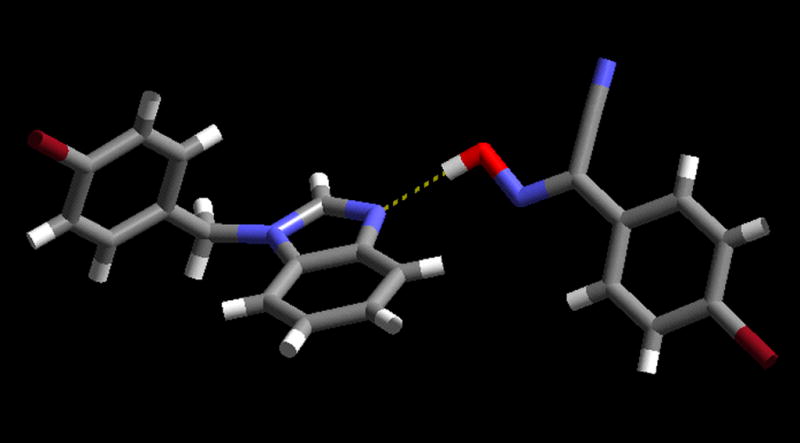
The primary O-H···N hydrogen bond between the oxime and the benzimidazole moiety in the crystal structure of A3.
In both A2 and A3, the primary N···O hydrogen bond is short, 2.63 Ă, and close to linear. Neither bromine atoms, nor the nitrile nitrogen atom, are engaged in any noteworthy intermolecular interactions. The torsion angle between oxime moiety and benzimidazole ring is approximately 25°.
The crystal structure of the 1:1 co-crystal A417 shows that the primary intermolecular interaction is the oxime O-H···N(benzimidazole) hydrogen bond, O(47)···N(13); 2.656(7) Ă, Figure 4.
Figure 4.
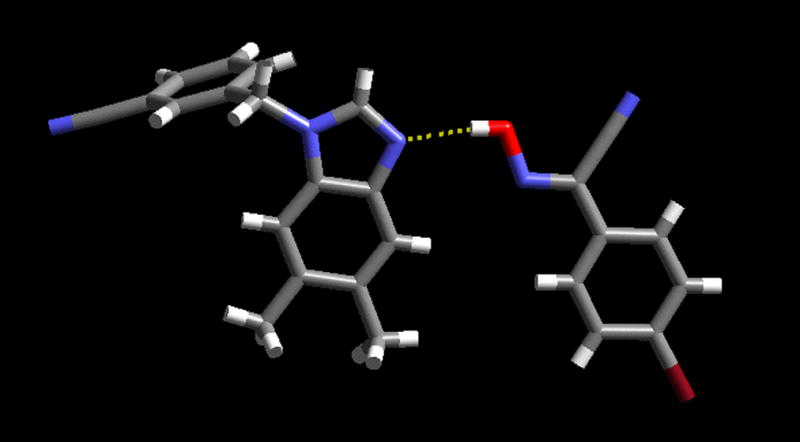
The primary O-H···N hydrogen bond between the oxime and the benzimidazole moiety in the crystal structure of A4.
There are no noteworthy short intermolecular distances to either of the two CN moieties or to the bromide atom. The oxime moiety is co-planar with the adjacent benzimidazole ring.
The crystal structure determination of B5, shows that the outcome is a 2:1 co-crystal with one oxime interacting with each of the two hydrogen-bond sites in the symmetric ditopic acceptor, 1,2-bis(4-pyridyl)ethylene, Figure 5. The heterocycle is located on an inversion centre with the symmetry related oxime moities rotated approx. 30° out of the plane with respect to the adjacent pyridyl ring. Again, the O-H···N hydrogen bond is short, (O(17)···N(21): 2.628(3) Å) and close to linear.
Figure 5.
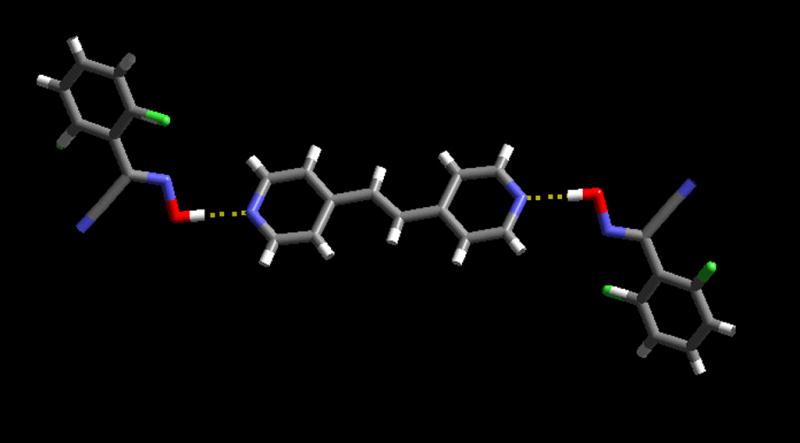
Trimeric supermolecule connected through O-H···N hydrogen bonds in the crystal structure of B5.
Neither the nitrile nitrogen atom, nor the oxime nitrogen atom participate in any significant intermolecular interactions.
The co-crystal C6, also comprises an aromatic cyanoxime and a symmetric ditopic hydrogen-bond acceptor, but this time each pyridyl moiety is replaced with a five-membered analogue, a benzimidazole site. Again, the crystal structure determination shows, Figure 6, that the outcome of the reaction is a co-crystal with a 2:1 stoichiometry, as was the case in B5.
Figure 6.
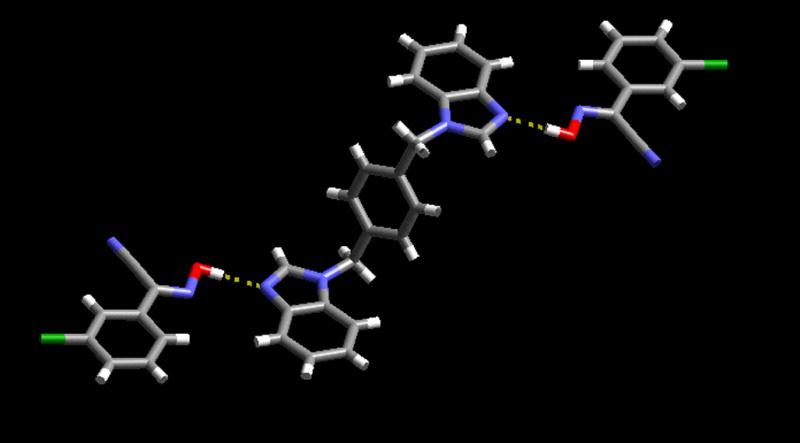
Trimeric supermolecule in the crystal structure of C6.
The heterocycle is located on an inversion centre, and the trimeric supermolecule is constructed via two symmetry related O-H···N hydrogen bonds (O(37)···N(13): 2.615(2)) The oxime moiety is co-planar with the benzimidazole ring, and no additional short intermolecular contacts are observed. Finally, when one equivalent of a D is allowed to react with one equivalent of an asymmetric ditopic acceptor, 7, the result is a 1:1 co-crystal, D7. The crystal structure determination allows us to establish binding preference for the single donor moiety; the oxime hydrogen-bond donor interacts with the benzimidazole nitrogen atom, (O(47)-H(47)···N(13) 2.6047(10) Å), Figure 7.
Figure 7.
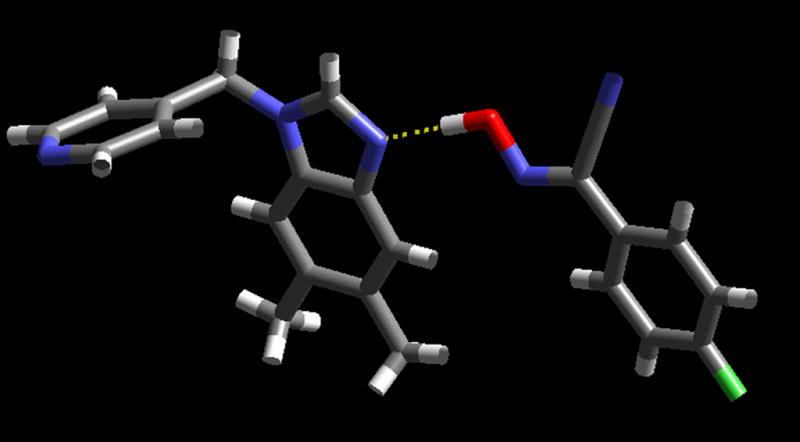
Dimer in the crystal structure of D7.
The cyanoxime moiety, the only strong hydrogen-bond donor in this system, selects the better of the two hydrogen-bond acceptors (as determined by a calculation of the electrostatic potential around each atom), whereas the remaining pyridyl moiety, a perfectly good hydrogen-bond acceptor, is left without a partner.
Discussion
This structural study of co-crystals of N-heterocycles and cyanoximes has furnished information that allows us to begin to draw some conclusions about the structural behaviour of the latter functional group. There are four phenylcyanoximes in the CSD,18 and all of them contain O-H···N (nitrile) hydrogen-bonded catemers. This is in contrast to the structural preference of aromatic aldoxime and acetyloximes which show a preference for dimers that utilize the O-H moiety as a donor, and the oxime nitrogen atom as the hydrogen-bond acceptor. 25 of 32 structures show this arrangement and seven of 32 structures contain OH···N(oxime) catemers.19 This change in pattern preference may come about because the nitrile moiety is a better hydrogen-bond acceptor, −224 kJ/mol (N-nitrile) vs. −125 kJ/mol (N-oxime) as indcated by AM1 calculated electrostatic potential, Scheme 3). This makes the cyanoxime hydrogen-bond donor more inclined to interact with the nitrile moiety, than with the oxime nitrogen atom. This behaviour is in accordance with the best-donor/best-acceptor guideline.
Scheme 3.

Molecular electrostatic potential data (maxima and minima) for three generic oximes (a) acetyloximes, (b) aldoximes, and (c) cyanoximes.
There is only one example of a non-solvated pyridylcyanoxime,20 and in this case, the O-H···N hydrogen bond involves the pyridyl nitrogen atom as the acceptor site. In addition to the seven structures presented herein, there are a further eight published examples of co-crystals that combine a phenylcyanoxime hydrogen-bond donor and a N-heterocyclic compound as the hydrogen-bond acceptor (ten benzimidazoles, three imidazoles, and two pyridines). The cyanoxime hydrogen-bond donor seems equally adept at interacting with a N-heterocyclic compound regardless of the geometry (five-membered vs. six-membered) of the acceptor site. Again, this is not necessarily surprising since the oxime moiety is a single-point donor (it does not rely on an ancillary acceptor atom), which makes it less sensitive to the precise geometry of the acceptor site, as long as the lone-pair on the nitrogen atom is accessible.
Based on existing structural information from the CSD,18 the carboxylic acid moiety is more likely than an oxime site to be co-planar with an aromatic N-heterocycle (when they form an O-H···N hydrogen bond). This indicates that the C=O moiety of the carboxylic acid is a better hydrogen-bond acceptor (for a C–H donor) than is the nitrogen atom of the oxime moiety. An alternative explanation could be that oximes are more likely to be sterically hindered from approaching an aromatic heterocycle in a co-planar manner. However, a space-filling representation of the co-planar supermolecule in C6 suggests that there is no obvious steric factor that would make it less likely for an oxime to be co-planar with a heterocycle, than it would be for an aromatic carboxylic acid, Fig. 8
Figure 8.
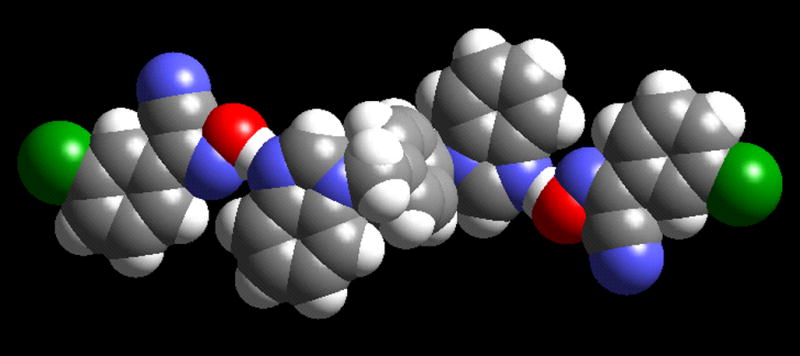
The oximes on either side of the bis-benzimidazole molecule in the crystal structure of C6 are able to approach in a co-planar manner.
Although there are very few structures available that probe the potential hydrogen-bond selectivity of cyanoximes, both D7 and the previously reported co-crystal of D and the 3-pyridyl isomer of 7, form 1:1 co-crystals with an asymmetic ditopic ligand where the oxime binds to the better of the two hydrogen-bond acceptors, the benzimidazole moiety. More information is obviously required before we can confidently discuss the possible limitations of the best-donor/best-acceptor for predicting synthon-preferences of cyanoximes but, at this, stage, the simple charge-based approach seems to offer a useful guideline.
In a comparative structural and spectroscopic study of co-crystals between phenyloximes R-C=N-OH (where R = H, Me, or CN) and N-heterocyclic hydrogen-bond acceptors, cyanoximes were shown to be more effective co-crystallizing agents,7 and this could be directly related to the superior hydrogen-bond donor ability of the oxime –OH moiety in the latter family.
In summary, cyanoximes have considerable potential as effective co-crystallizing agents. As single-point hydrogen-bond donors they should be, based on the electrostatic potential surfaces around their acidic protons, comparable to, (if not better than) carboxylic acids, they are synthetically readily available, and they also tend to be soluble in a wide range of solvents, which facilitates solution-based co-crystal synthesis.
Supplementary Material
Acknowledgments
We gratefully acknowledge financial support from the Terry C. Johnson Center for Basic Cancer Research, ACS-PRF (46011-AC1) and a Grant (P20 RR015563) from the National Center for Research Resources, a component of the National Institutes of Health, and the State of Kansas. This manuscript is solely the responsibility of the authors and does not necessarily represent the official view of the NCRR or NIH.
Footnotes
Electronic Supplementary Information (ESI) available: CIF files and experimental crystallographic data.
Notes and references
- 1.(a) Bhogala BR, Basavoju S, Nangia A. CrystEngComm. 2005;90:551. [Google Scholar]; (b) Aakeröy CB, Hussain I, Forbes S, Desper J. CrystEngComm. 2007;9:46. [Google Scholar]; (c) Aakeröy CB, Desper J, Urbina JF. Cryst Growth Des. 2005;5:1283. [Google Scholar]; (d) Aakeröy CB, Salmon DJ. CrystEngComm. 2005;7:439. doi: 10.1039/b811322j. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Aakeröy CB, Desper J, Scott BMT. Chem Commun. 2006:1445. doi: 10.1039/b517118k. [DOI] [PubMed] [Google Scholar]; (f) MacGillivray LR. CrystEngComm. 2004;16:77. [Google Scholar]; (g) Desiraju GR. Angew Chem, Int Ed Engl. 1995;34:2311. [Google Scholar]; (h) Hosseini MW. CrystEngComm. 2004;6:318. [Google Scholar]; (i) Brammer L. Chem Soc Rev. 2004;33:476. doi: 10.1039/b313412c. [DOI] [PubMed] [Google Scholar]; (j) Caulder DL, Raymond KN. Acc Chem Res. 1999;32:975. [Google Scholar]; (k) Reinhoudt DN, Crego-Calama M. Science. 2002;295:2403. doi: 10.1126/science.1069197. [DOI] [PubMed] [Google Scholar]; (l) Lehn JM. Science. 2002;295:2400. doi: 10.1126/science.1071063. [DOI] [PubMed] [Google Scholar]; (m) Steiner T, Desiraju GR. The Weak Hydrogen Bond in Structural Chemistry and Biology. Oxford University Press; New York: 1999. An extensive discussion ‘weaker’ or unconventional hydrogen bonds is provided. [Google Scholar]; (n) Braga Dario, Grepioni Fabrizia., editors. Making Crystals by Design; Methods, Techniques and Applications. Wiley-VCH; Weinheim: 2007. [Google Scholar]
- 2.(a) Zheng SL, Pham O, Vande Velde CML, Gembicky M, Coppens P. Chem Commun. 2008;22:2538. doi: 10.1039/b802103a. [DOI] [PubMed] [Google Scholar]; (b) Berry DJ, Seaton CC, Clegg W, Harrington RW, Coles SJ, Horton PN, Hursthouse MB, Storey R, Jones W, Friscic T, Blagden N. Cryst Growth Des. 2008;8:1697. [Google Scholar]; (c) Nugrahani I, Asyarie S, Soewandhi SN, Ibrahim S. Int J Pharmacology. 2007;3:475. [Google Scholar]; (d) Cooke MW, Stanton M, Shimanovich R, Bak A. Am Pharm Rev. 2007;10:54. [Google Scholar]; (e) Morimoto M, Kobatake S, Irie M. Chem Commun. 2008;3:335. doi: 10.1039/b713694c. [DOI] [PubMed] [Google Scholar]; (f) Burchell TJ, Soldatov DV, Enright GD, Ripmeester JA. CrystEngComm. 2007;9:922. [Google Scholar]; (g) Tomasic V, Stefanic Z. CrystEngComm. 2007;9:1124. [Google Scholar]; (h) Broker GA, Tiekink ERT. CrystEngComm. 2007;9:1096. [Google Scholar]; (i) Siegler MA, Fu Y, Simpson GH, King DP, Parkin S, Brock CP. Acta Crystallogr Sect B: Struct Sci. 2007;B63:912. doi: 10.1107/S0108768107054699. [DOI] [PubMed] [Google Scholar]
- 3.(a) Vishweshwar P, McMahon JA, Bis JA, Zaworotko MJ. J Pharm Sci. 2006;95:499. doi: 10.1002/jps.20578. [DOI] [PubMed] [Google Scholar]; (b) Peterson ML, Hickey MB, Zaworotko MJ, Almarsson Ö. J Pharmacy & Pharmaceutical Sciences. 2006;9:317. [PubMed] [Google Scholar]; (c) Shan N, Zaworotko MJ. Drug Disc Today. 2008;13:440. doi: 10.1016/j.drudis.2008.03.004. [DOI] [PubMed] [Google Scholar]; (d) Porter WW, Elie SC, Matzger AJ. Cryst Growth Des. 2008;8:14. doi: 10.1021/cg701022e. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Nair R-H. Mol Pharmaceutics. 2007;4:299. [Google Scholar]
- 4.(a) Landauro CV, Janssen T. J Non-Cryst Solids. 2007;353:3192. [Google Scholar]; (b) Reshak AH, Auluck S. Solid State Commun. 2008;145:571. [Google Scholar]; (c) Erslev PT, Chiang HQ, Hong D, Wager JF, Cohen JD. Journal of Non-Cryst Solids. 2008;354:2801. [Google Scholar]; (d) Falkowski M, Kowalczyk A, Tolinski T, Chelkowska G. Mat Sci. 2007;25:321. [Google Scholar]; (e) Kawahara T, Kuroda T, Matsui H, Mishima M, Karuppuchamy S, Seguchi Y, Yoshihara M. J Mat Sci. 2007;42:3708. [Google Scholar]
- 5.(a) Vaishnavi TS, Haridoss P, Vijayan C. Mat Lett. 2008;62:1649. [Google Scholar]; (b) Salem AM, El-Gendy YA, Sakr GB, Soliman WZ. J Phys D: App Phys. 2008;41:025311. [Google Scholar]; (c) Meena M, Mahadevan CK. Mat Lett. 2008;62:3742. [Google Scholar]
- 6.Aakeröy CB, Fasulo M, Schultheiss N, Desper J, Moore C. J Am Chem Soc. 2007;129:13772. doi: 10.1021/ja073201c. [DOI] [PubMed] [Google Scholar]
- 7.Aakeröy CB, Salmon DJ, Smith MM, Desper J. Cryst Growth Des. 2006;6:1033. [Google Scholar]
- 8.(a) Robertson D, Barnes C, Gerasimchuk N. J Coord Chem. 2004;57:1205. [Google Scholar]; (b) Robertson D, Barnes C, Gerasimchuk N. Inorg Chem. 2005;44:832. doi: 10.1021/ic050465w. [DOI] [PubMed] [Google Scholar]
- 9.Etter MC. J Phys Chem. 1991;95:4601. [Google Scholar]
- 10.Aakeröy CB, Desper J, Urbina JF. Chem Commun. 2005;22:2820. doi: 10.1039/b503718b. [DOI] [PubMed] [Google Scholar]
- 11.Aakeröy CB, Desper J, Smith MM. Chem Commun. 2007;38:3936. doi: 10.1039/b707518a. [DOI] [PubMed] [Google Scholar]
- 12.Hunter CA. Angew Chem Int Ed. 2000;43:5310. doi: 10.1002/anie.200301739. [DOI] [PubMed] [Google Scholar]
- 13.Aakeröy CB, Desper J, Fasulo ME. CrystEngComm. 2006;8:586. [Google Scholar]
- 14.Aakeröy CB, Schultheiss N, Desper J. Inorg Chem. 2005;44:4983. doi: 10.1021/ic048405y. [DOI] [PubMed] [Google Scholar]
- 15.(a) Abrahams BF, Hoskins BF, Robson R, Slizys DA. Acta Crystallogr, C. 1998;54:1666. [Google Scholar]; (b) Aakeröy CB, Desper J, Leonard B, Urbina JF. Cryst Growth Des. 2004;5:865. [Google Scholar]
- 16.Desiraju GR. Angew Chem Int Ed. 1995;34:2311. [Google Scholar]
- 17.Co-crystal A4 provided only small needle-like crystals. Despite our best efforts, we could not obtain larger crystals, and our intensity data were therefore weak. Fit statistics and atomic coordinate uncertainties (and derived data such as bond lengths and angles) reflect the quality of the dataset (see ESI).
- 18.Allen FH. Acta Crystallogr B. 2002;58:380. doi: 10.1107/s0108768102003890. [DOI] [PubMed] [Google Scholar]
- 19.The search only includes aldoximes and acetyloximes in the absence of solvent molecules, metal ions, and potentially disruptive substituents such as –OH and –NH2.
- 20.Mokhir AA, Domasevich KV, Dalley NK, Kou Xiaolan, Gerasimchuk N, Gerasimchuk OA. Inorg Chim Acta. 1999;284:85. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.