

The crystal structure of human dual-specificity phosphatase 14, DUSP14 (MKP6), in complex with a phosphate ion has been determined and refined to 1.88 Å resolution.
Keywords: dual-specificity phosphatases, MAPK phosphatases, DUSP14, MKP6
Abstract
Dual-specificity phosphatases (DUSPs) are enzymes that participate in the regulation of biological processes such as cell growth, differentiation, transcription and metabolism. A number of DUSPs are able to dephosphorylate phosphorylated serine, threonine and tyrosine residues on mitogen-activated protein kinases (MAPKs) and thus are also classified as MAPK phosphatases (MKPs). As an increasing number of DUSPs are being identified and characterized, there is a growing need to understand their biological activities at the molecular level. There is also significant interest in identifying DUSPs that could be potential targets for drugs that modulate MAPK-dependent signaling and immune responses, which have been implicated in a variety of maladies including cancer, infectious diseases and inflammatory disorders. Here, the overproduction, purification and crystal structure at 1.88 Å resolution of human dual-specificity phosphatase 14, DUSP14 (MKP6), are reported. This structural information should accelerate the study of DUSP14 at the molecular level and may also accelerate the discovery and development of novel therapeutic agents.
1. Introduction
Reversible protein phosphorylation, mediated by a plethora of protein kinases and phosphatases, is an important regulatory mechanism in a wide variety of biological processes such as cell development, differentiation and transformation in higher eukaryotes (Hunter, 1995 ▶; Pawson, 1995 ▶; Pawson & Nash, 2000 ▶; Alonso et al., 2004 ▶). Dual-specificity phosphatases (DUSPs) remove phosphate groups from critical phosphothreonine, phosphoserine and phosphotyrosine residues on kinases, which generally leads to their deactivation (Denu et al., 1996 ▶; Camps et al., 2000 ▶; Patterson et al., 2009 ▶). Because timely coordination of the activities of kinases and phosphatases is of paramount importance in many signal transduction pathways (Owens & Keyse, 2007 ▶), any disruption of or abnormalities associated with these coordinated events often form the molecular basis of serious diseases and biological disorders (Levitzki, 1994 ▶; Cohen, 2002 ▶; Tonks, 2006 ▶; Patterson et al., 2009 ▶).
Some DUSPs preferentially dephosphorylate threonine and tyrosine residues within the signature motif pTXpY in the activation loop of mitogen-activated protein kinases (MAPKs) and thus are also referred to as MAPK phosphatases (MKPs; Theodosiou & Ashworth, 2002 ▶; Farooq & Zhou, 2004 ▶). MAPKs are involved in transducing extracellular signals from hormones, growth factors, cytokines and environmental stresses to elicit effects that regulate cell growth and differentiation, immune responses, oncogenesis or apoptosis (Chang & Karin, 2001 ▶; Pearson et al., 2001 ▶; Kaminska, 2005 ▶; Dickinson & Keyse, 2006 ▶; Boutros et al., 2008 ▶). Consequently, these MKPs are being investigated as potential molecular targets for drugs that would modulate MAPK-dependent signaling pathways (Zhang et al., 2002 ▶) associated with cancer (Wu, 2007 ▶; Keyse, 2008 ▶), immune responses to infectious diseases (Lang et al., 2006 ▶; Salojin & Oravecz, 2007 ▶), inflammatory disorders (Jeffrey et al., 2007 ▶; Ralph & Morand, 2008 ▶) and diabetes (Xu et al., 2003 ▶). However, the precise roles of DUSPs in many of these biological processes remain poorly understood and require further study. Structural information is also needed to assist in the design of selective inhibitors of specific DUSPs (Hoffman et al., 2004 ▶; Almo et al., 2007 ▶; Pulido & Hooft van Huijsduijnen, 2008 ▶).
DUSP14 (MKP6) has been classified as a type 1 dual-specificity mitogen-activated kinase phosphatase (DUSP-MKP), a class of DUSPs that are approximately 200 amino-acid residues in length and are composed of a single catalytic phosphatase domain (Farooq & Zhou, 2004 ▶). Other classes of DUSP-MKPs include additional domains that may contribute to higher order regulatory control (Farooq & Zhou, 2004 ▶; Nordle et al., 2007 ▶). DUSP14 is expressed ubiquitously, although elevated expression has been observed in certain types of cells and tissues (Boutros et al., 2008 ▶; Patterson et al., 2009 ▶). DUSP14 preferentially dephosphorylates c-Jun NH2-terminal kinase (JNK) and extracellular signal-regulated kinase (ERK) (Bai et al., 2004 ▶; Marie-Claire et al., 2008 ▶) and has been proposed to function as a negative regulator of CD28 co-stimulation in intact T cells (Marti et al., 2001 ▶). DUSP14 contributes to the regulation of CD28 signaling by interacting with the cytoplasmic tail of CD28, thereby co-localizing DUSP14 with recently activated MAP kinases at the plasma membrane and facilitating their deactivation prior to nuclear translocation. A report by Nakano also identified DUSP14 as a nonspecific regulatory molecule for delayed-type hypersensitivity, indicating that DUSP14 may itself have some utility as a therapeutic agent (Nakano, 2007 ▶). DUSP14 has also been shown to contribute to the proliferation of pancreatic β-cells by affecting ERK activity, which raises further questions as to the possible roles of DUSP14 in cellular growth, proliferation and differentiation in other cell types such as cancer (Klinger et al., 2008 ▶). As information about the biological roles of DUSP14 continues to emerge (Patterson et al., 2009 ▶), knowledge of its three-dimensional structure should facilitate further studies at the molecular level. To this end, we have crystallized human DUSP14 and determined its structure at a resolution of 1.88 Å.
2. Materials and methods
2.1. Cloning, expression and purification
Human DUSP14 (Ser2–His191) was amplified from cDNA (IMAGE clone ID 2819474, American Type Culture Collection, Manassas, Virginia, USA) by the polymerase chain reaction (PCR) using the following oligodeoxyribonucleotide primers: 5′-GAG AAC CTG TAC TTC CAG AGC TCC CGT GGT CAC AGC ACG CTA CCA AG-3′ and 5′-GGG GAC CAC TTT GTA CAA GAA AGC TGG GTT ATT AGT GTC GGG ACT CCT TCT CAT AGA C-3′ (primer R). The PCR amplicon was subsequently used as template for a second PCR with the following primers: 5′-GGG GAC AAG TTT GTA CAA AAA AGC AGG CTC GGA GAA CCT GTA CTT CCA G-3′ and primer R. The amplicon from the second PCR was inserted by recombinational cloning into the entry vector pDONR221 (Invitrogen, Carlsbad, California, USA) and the nucleotide sequence was confirmed experimentally. The open reading frame of DUSP14 (Ser2–His191) containing a recognition site (ENLYFQ/S) for tobacco etch virus (TEV) protease at its N-terminus was moved by recombinational cloning into the destination vector pDEST-HisMBP to produce pJT92 (Tropea et al., 2007 ▶). This plasmid directs the expression of human DUSP14 as a fusion to the C-terminus of Escherichia coli maltose-binding protein (MBP) with an intervening TEV protease recognition site. The MBP contains an N-terminal hexahistidine tag for affinity purification by immobilized metal-affinity chromatography. The fusion protein was expressed in E. coli strain Rosetta 2 (DE3) (Novagen, Madison, Wisconsin, USA). Cells containing pJT92 were grown to mid-log phase (OD600 of ∼0.5) at 310 K in Luria broth containing 100 µg ml−1 ampicillin, 30 µg ml−1 chloramphenicol and 0.2% glucose. Overproduction of fusion protein was induced with isopropyl β-d-1-thiogalactopyranoside at a final concentration of 1 mM for 4 h at 303 K. The cells were pelleted by centrifugation and stored at 193 K.
All procedures were performed at 277–281 K. 10 g of E. coli cell paste was suspended in 150 ml ice-cold 50 mM MES pH 6.5, 200 mM NaCl, 25 mM imidazole, 10%(v/v) glycerol buffer (buffer A) containing 1 mM benzamidine–HCl (Sigma Chemical Company, St Louis, Missouri, USA) and Complete EDTA-free protease-inhibitor cocktail tablets (Roche Molecular Biochemicals, Indianapolis, Indiana, USA). The cells were lysed with an APV-1000 homogenizer (Invensys APV Products, Albertslund, Denmark) at 69 MPa and centrifuged at 30 000g for 30 min. The supernatant was filtered through a 0.22 µm polyethersulfone membrane and applied onto a 12 ml Ni–NTA Superflow column (Qiagen, Valencia, California, USA) equilibrated in buffer A. The column was washed to baseline with buffer A and eluted with a linear gradient of imidazole to 250 mM. Fractions containing recombinant His6-MBP-DUSP14 were pooled, concentrated using an Amicon YM10 membrane (Millipore Corporation, Bedford, Massachusetts, USA), diluted with 50 mM MES pH 6.5, 200 mM NaCl, 10%(v/v) glycerol buffer (buffer B) to reduce the imidazole concentration to about 25 mM and digested overnight at 277 K with His6-tagged TEV protease (Kapust et al., 2001 ▶). The digest was applied onto a 12 ml Ni–NTA Superflow column equilibrated in buffer A. The column was washed with 40 column volumes of buffer A and 20 column volumes of buffer B containing 50 mM imidazole. Recombinant DUSP14 emerged in both column effluents. The combined effluents were incubated overnight with 10 mM dithiothreitol, concentrated as above and applied onto a HiPrep 26/60 Sephacryl S-100 HR column (GE Healthcare Biosciences Corporation, Piscataway, New Jersey, USA) equilibrated in 25 mM MES pH 6.5, 150 mM NaCl, 2 mM tris(2-carboxyethyl) phosphine (TCEP), 10%(v/v) glycerol buffer. Fractions containing recombinant protein were pooled, concentrated and re-chromatographed on the HiPrep 26/60 Sephacryl S-100 HR column. The peak fractions containing DUSP14 were pooled and concentrated to 15–25 mg ml−1 (estimated at 280 nm using a molar extinction coefficient of 26 930 M −1 cm−1). Aliquots were flash-frozen in liquid nitrogen and stored at 193 K. The final product was judged to be >95% pure by SDS–PAGE (Fig. 1 ▶ a). The molecular weight was confirmed by electrospray ionization mass spectroscopy.
Figure 1.

Expression, purification and crystallization of human DUSP14. (a) Purification of DUSP14 as monitored by SDS–PAGE. Lane M, molecular-weight markers (kDa); lane 1, soluble intracellular protein (crude extract); lane 2, eluate from first Ni–NTA column; lane 3, products of His6-TEV protease digest; lane 4, flowthrough fraction from second Ni–NTA column; lane 5, final product after gel-filtration chromatography. (b) Crystals of DUSP14 viewed under polarized light.
2.2. Crystallization and data collection
DUSP14 [14 mg ml−1 in 25 mM MES pH 6.5, 150 mM NaCl, 2 mM TCEP and 10%(v/v) glycerol] was initially screened for crystals using the Phoenix crystallization robot (Art Robbins Instruments, Sunnyvale, California, USA) and commercially available sparse-matrix screens from Qiagen (Valencia, California, USA), Hampton Research (Aliso Viejo, California, USA) and Axygen Biosciences (Union City, California, USA). The first crystals were obtained at 293 K with condition F12 (0.1 M citrate pH 5.6, 1.0 M ammonium phosphate monobasic) from the Qiagen JCSG Core III suite and were subsequently optimized using the Hampton Research Additive kit. All crystallization reagents used during optimization were purchased from Hampton Research. Crystals suitable for data collection were obtained by mixing 5 µl DUSP14 [10 mg ml−1 in 25 mM MES pH 6.5, 150 mM NaCl, 2 mM TCEP and 10%(v/v) glycerol] with 4 µl well solution (0.1 M sodium citrate pH 5.4, 1.1 M ammonium phosphate monobasic) and 1 µl NDSB-256 (1.0 M). The drops were sealed over 1 ml of crystallization well solution and incubated at room temperature. Crystals appeared within approximately 1–2 weeks (Fig. 1 ▶ b) and were cryoprotected by supplementing the mother liquor with 20%(v/v) glycerol and flash-freezing in liquid nitrogen. Native X-ray diffraction data were collected from a single crystal with a MAR2300 detector mounted on a Rigaku RU-H3R rotating-anode X-ray generator (Rigaku Corporation, The Woodlands, Texas, USA) operated at 50 kV and 100 mA. The Cu Kα radiation was focused by an MSC/Osmic mirror system. 180 frames of data were collected using an oscillation angle of 1°, a crystal-to-detector distance of 120 mm and an exposure time of 5 min. The data were integrated and scaled with HKL-3000 (Minor et al., 2006 ▶). The crystals belonged to space group I4, with unit-cell parameters a = b = 85.0, c = 115.1 Å. The Matthews coefficient of 2.46 Å3 Da−1 and the solvent content of 49.6% suggested that there were two molecules in the asymmetric unit (Matthews, 1968 ▶).
2.3. Structure solution and refinement
The structure of DUSP14 was solved by molecular replacement using the coordinates of DUSP18 (PDB code 2esb; 50% sequence identity; Jeong, Cho et al., 2006 ▶) as a search model after removing all solvent molecules and mutating non-identical residues to alanine. Molecular replacement was performed using MOLREP from the CCP4 suite of programs (Potterton et al., 2002 ▶). Cross-rotation and translational searches for two molecules in the asymmetric unit were performed with data from 15 to 3.0 Å resolution followed by rigid-body refinement to 3.0 Å resolution with REFMAC5 (Murshudov et al., 1997 ▶). The model was rebuilt manually using σA-weighted 2F o − F c and F o − F c electron-density maps (Read, 1997 ▶) and Coot (Emsley & Cowtan, 2004 ▶), and refined with REFMAC5. Water molecules were located using Coot and refined with REFMAC5. The model was refined to a final working R factor of 0.175 and an R free of 0.221 (calculated with 5% of the reflections) at 1.88 Å resolution (Brünger, 1992 ▶). Model validation was performed with MolProbity (Davis et al., 2004 ▶). The all-atom contacts clash score was 5.81 (95th percentile) and the MolProbity score was 1.33 (98th percentile). The Ramachandran plots were prepared with PROCHECK (Laskowski et al., 1993 ▶). X-ray diffraction data-collection and refinement statistics are presented in Table 1 ▶. All structural alignments and figures were prepared with PyMOL (DeLano Scientific LLC, Palo Alto, California, USA). Sequence alignments were performed with ClustalW (Larkin et al., 2007 ▶). The coordinates and structure factors were submitted to the Protein Data Bank under accession code 2wgp.
Table 1. Data-collection and model-refinement statistics.
Values in parentheses are for the highest resolution shell.
| Data collection | |
| X-ray source | Rigaku RU-H3R |
| Space group | I4 |
| Unit-cell parameters | |
| a = b () | 85.0 |
| c () | 115.1 |
| Wavelength () | 1.5418 |
| Resolution () | 501.88 (1.951.88) |
| Total reflections | 230493 |
| Unique reflections | 32950 |
| Completeness (%) | 99.3 (93.4) |
| Redundancy | 7.0 (4.6) |
| R sym † | 0.078 (0.649) |
| I/(I) | 22.8 (1.9) |
| Model-refinement statistics | |
| Resolution () | 501.88 |
| Unique reflections used | 31259 |
| R work/R free ‡ | 0.175/0.221 |
| Molecules per AU§ | |
| Protein | 2 [residues 24191] |
| Water | 347 |
| Phosphate | 2 |
| Mean B factor (2) | |
| Protein | 26.9 [A]/26.7 [B] |
| Water | 40.8 |
| Phosphate | 18.7 [A]/19.5 [B] |
| R.m.s.d. from ideal | |
| Bond length () | 0.014 |
| Bond angle () | 1.4 |
| Ramachandran plot | |
| Most favored (%) | 90.4 |
| Additionally allowed (%) | 9.2 |
| Generously allowed (%) | 0.3 |
| Disallowed (%) | 0 |
| PDB code | 2wgp |
R
sym = 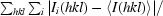
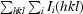 , where I(hkl) is the mean intensity of multiply recorded reflections.
, where I(hkl) is the mean intensity of multiply recorded reflections.
R = 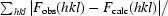
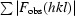 |. R
free is the R value calculated for 5% of the data set not included in the refinement.
|. R
free is the R value calculated for 5% of the data set not included in the refinement.
Asymmetric unit.
3. Results and discussion
3.1. Overall structure
Full-length human DUSP14 (residues 2–198) was poorly soluble when overproduced in E. coli (data not shown). However, a truncated polypeptide consisting of residues 2–191 (DUSP142–191) was generated by limited proteolysis with thermolysin and identified by liquid chromatography–electrospray mass spectrometry. This truncated form of DUSP14 could be produced in a soluble form and was well behaved during purification. DUSP142–191 was crystallized and its structure was solved by molecular replacement at 1.88 Å resolution. Two molecules of DUSP14 were found in the asymmetric unit, but they do not form an extensive dimer interface. Rather, the principal interactions between them consist of two salt bridges [Arg163(A)–Asp160(B) and Asp160(A)–Arg163(B)]. The buried surface area at the interface of the two molecules is only 209 Å2, which suggests that the monomer is likely to be the biologically relevant unit. A previous report by Nakano indicated that DUSP14 exists as a mixture of monomers and dimers in solution under nonreducing conditions, but is driven toward the monomeric state under reducing conditions (Nakano, 2007 ▶). All but one of the Cys residues in DUSP14 are buried from the solvent and Cys133, the only exposed Cys residue, is not located near any other symmetry-related Cys residue. The core catalytic domain of DUSP14 is formed by a central five-stranded β-sheet that is surrounded by helices α1 and α2 on one side and four α-helices (α3–α6) on the other side (Fig. 2 ▶). The C-terminal end of DUSP14 (residues 166–191) folds into a two-stranded antiparallel β-sheet (β6 and β7) with a single α-helix (α7) extending from it. No electron density was observed for residues 2–23 of DUSP14, suggesting that this region of the protein is disordered. However, the electron-density maps did reveal well resolved density in the active site near the catalytic Cys111, which was modeled as a phosphate ion. The Ramachandran plot obtained from analysis of the coordinates indicates that 90% of the residues fall within the most favored region and no residues are found in the disallowed region.
Figure 2.

Ribbon representation of the three-dimensional structure of monomeric DUSP14.
3.2. The active site
Fortuitously, the active site contained a bound phosphate ion from the crystallization solution that enabled us to examine its interactions with the catalytic residues of DUSP14 (Fig. 3 ▶). The removal of phosphate groups by protein tyrosine phosphatases proceeds by a two-step mechanism (Fauman & Saper, 1996 ▶; Denu & Dixon, 1998 ▶; Camps et al., 2000 ▶). The phosphate-binding pocket is formed by a loop containing the active-site motif HCXXGXXRS(T), where X is a variable residue. Catalysis is initiated by a conserved cysteine thiolate anion that attacks the tyrosine phosphate to form a cysteinyl-phosphate intermediate. This is followed by the release of the tyrosine, which occurs by the donation of a proton from an invariant aspartic acid that serves as a general acid. This same aspartic acid then serves as a general base by removing a proton from a water molecule that attacks the phospho-enzyme intermediate to eliminate phosphate and regenerate the active enzyme. The dephosphorylation of serine and threonine residues by DUSPs has also been proposed to occur through a similar mechanism (Denu & Dixon, 1995 ▶).
Figure 3.

The active site of DUSP14. The secondary-structure elements are illustrated in cyan ribbon format and the residues in the phosphate-binding loop are illustrated in stick format with carbon in gray, nitrogen in blue, oxygen in red, sulfur in yellow and phosphorus in orange. Hydrogen bonds are highlighted as dashed red lines. The OMIT F o − F c difference electron-density map (green) contoured at the 3.5σ level (1.88 Å resolution) is shown superimposed on residues Asp80, Cys111, Arg117 and phosphate, which were removed from the model before map calculation.
The phosphate-binding pocket of DUSP14 is formed by residues 111–117, which are located on a loop (the phosphate-binding loop) between helix α4 and β-strand β5. The α4 helix is capped by the positively charged Arg117 residue, which forms a stabilizing interaction with the negatively charged phosphate ion and also makes two hydrogen bonds between its side-chain guanidinium and two of the phosphate O atoms. The dipole moment of this helix also contributes to the binding of phosphate (Zhang, 2002 ▶). The phosphate is also bound in the active site by hydrogen bonds between the phosphate O atoms and the backbone amides of Ala113, Val115, Ser116 and Arg117. The catalytic Cys111 is located 3.8 Å away from the phosphate. The side chain of Asp80, which is located 2.8 Å from one of the phosphate O atoms and is found on the loop between helix α3 and strand β4, is positioned appropriately to act as a general acid/base. Sequence and structural alignments with DUSP18 and the vaccinia H1-related phosphatase (VHR) show that both Cys111 and Asp80 are conserved (Fig. 4 ▶; Yuvaniyama et al., 1996 ▶; Jeong, Cho et al., 2006 ▶). The conserved aspartic acid, in particular, has been observed to undergo a large conformational shift from the apo to the substrate-bound enzyme in many protein tyrosine phosphatases, underscoring the importance of the proper structural context of this residue during catalysis (Denu et al., 1995 ▶, 1996 ▶; Zhang, 2002 ▶).
Figure 4.

Sequence alignment of DUSP14, DUSP18 and VHR performed with ClustalW. The critical residues involved in catalysis are highlighted in yellow.
Inspection of the surface of the DUSP14 active site reveals that the binding pocket for the phospho-amino acid is shallow (approximately 7.0 Å deep as measured from Asp80 to Cys111), which would allow the enzyme to accommodate either phosphotyrosine or phosphothreonine/serine (Fig. 5 ▶ a). Indeed, it has previously been demonstrated that the depth of this pocket is a probable structural determinant of phosphotyrosine or dual-specificity substrate-specificity (Denu et al., 1996 ▶). Protein tyrosine phosphatases that have deeper pockets are typically unable to accommodate the shorter phosphothreonine/phosphoserine residue and position it properly for efficient catalysis and thus exhibit greater selectivity for phosphotryosine substrates (Zhang, 2002 ▶). The electrostatic surface potential for the region around the binding pocket is predominantly hydrophobic on one side with small negatively charged patches and has a distinctly positively charged region on the other (Figs. 5 ▶ a and 5 ▶ b).
Figure 5.

The electrostatic potential mapped onto the surface area of DUSP14. Positively charged regions are highlighted in blue and negatively charged regions in red. White areas represent neutral and hydrophobic residues. (a) shows a view of the phosphate-binding pocket with phosphate illustrated in stick format (P atom in orange and O atoms in red) and (b) shows a view of the molecule after a 90° rotation with respect to the vertical axis. (c) A view of the crystal-packing lattice, illustrating the interactions of the C-terminal motif of DUSP14 with neighboring symmetry mates.
3.3. Comparison with structural homologs
To identify similar structures, we entered the coordinates of DUSP14 into the Secondary Structure Matching (SSM) server (http://www.ebi.ac.uk/msd-srv/ssm; Krissinel & Henrick, 2004 ▶). As expected, the closest structural homolog was DUSP18 (Z score = 12.0; r.m.s.d. of 0.65 Å over 160 common Cα atoms; PDB code 2esb). The core catalytic phosphatase domain is also very similar to a number of other reported dual-specificity phosphatases, as indicated by additional high Z scores (Table 2 ▶). A structural alignment of DUSP14 and DUSP18 confirms that the overall folds of the proteins are highly similar, with only a few regions exhibiting small deviations (Fig. 6 ▶; Jeong, Cho et al., 2006 ▶). For example, the loops between strands β3 and β4 and strands β6 and β7 exhibit slight structural deviations when the two structures are compared. Furthermore, in the N-terminal region of the proteins there is a minor structural deviation in the helix that follows strand β2. While this is a long contiguous helix in DUSP18, the analogous segment of DUSP14 forms a single-turn helix (α1, residues 39–42) followed by a short loop and then helix α2. A distinguishing feature of DUSP14 and DUSP18 is that both proteins possess a small extended C-terminal motif that is absent from other DUSPs. In both proteins this C-terminal motif folds into a two-stranded antiparallel β-sheet. However, the C-terminal motif of DUSP14 differs from DUSP18 in that it is followed by an α-helix (α7; Figs. 2 ▶ and 5 ▶ c). Curiously, the amino acids that form the α7 helix in DUSP14 are highly conserved in DUSP18 but are unstructured in the latter protein. A report by Jeong and coworkers suggested that this region of DUSP18 may contribute to its observed thermostability (Jeong, Cho et al., 2006 ▶).
Table 2. Closest structural homologs of DUSP14.
| Protein | PDB code | Z score† | R.m.s.d.‡ () | N align § | Sequence identity (%) |
|---|---|---|---|---|---|
| DUSP18¶ | 2esb | 12.0 | 0.65 | 160 | 50 |
| Slingshot phosphatase 2†† | 2nt2 | 10.9 | 0.94 | 141 | 39 |
| Human tyrosine phosphatase-like serine/threonine/tyrosine-interacting protein‡‡ | 2r0b | 9.8 | 1.16 | 141 | 35 |
| MKP-5† | 1zzw | 10.5 | 0.98 | 143 | 34 |
| DUSP5‡ | 2g6z | 10.9 | 1.03 | 141 | 36 |
| DUSP9§ | 2hxp | 10.0 | 1.25 | 137 | 36 |
The Z score measures the statistical significance of a match in terms of Gaussian statistics. The higher the Z score, the higher the statistical significance of the match.
Root-mean-square deviation.
Number of matched residues.
Jeong, Cho et al. (2006 ▶).
Jung et al. (2007 ▶).
Almo et al. (2007 ▶).
Jeong, Yoon et al. (2006 ▶).
Jeong et al. (2007 ▶).
Almo et al. (2007 ▶).
Figure 6.

Stereoview of the superimposed coordinates of DUSP14 (PDB code 2wgp; cyan) and DUSP18 (PDB code 2esb; red).
A previous study reported that DUSP14 interacts with the cytoplasmic tail of CD28 in intact T cells, which is proposed to control the activation of certain MAP kinases (Marti et al., 2001 ▶). The extended C-terminal motif in DUSP14 may be involved in protein–protein interactions, the regulation of phosphatase activity, or both. The C-terminal motif contains positively and negatively charged patches and is solvent-accessible as a binding site (Figs. 5 ▶ a and 5 ▶ b). Indeed, the crystal packing of the DUSP14 molecules reveals that this C-terminal motif interacts with other symmetry-related molecules in which strand β6 forms hydrogen bonds to β6 of a symmetry-related molecule to form a four-stranded antiparallel β-sheet (Fig. 5 ▶ c). Interestingly, the C-terminal α7 helix of DUSP14 and the N-terminal α1 helix of VHR occupy similar positions when the two structures are superimposed. However, in contrast to the α1 helix in VHR, which forms part of the substrate-recognition motif, the α7 helix in DUSP14 is tilted away from the core catalytic domain and the two helices lack sequence similarity (Yuvaniyama et al., 1996 ▶; Schumacher et al., 2002 ▶). Hence, further studies will be required in order to establish whether or not the α7 helix in DUSP14 plays any role in protein–protein interactions (Nordle et al., 2007 ▶).
The active sites of DUSP14 and DUSP18 are also very highly conserved. In particular, the residues that form the phosphate-binding loop are strictly conserved and the regions near the phosphate-binding loop are nearly invariant. However, DUSP14 does exhibit sequence divergence in the identity of the residues in the phosphate-binding loop and regions near the catalytic site when compared with other dual-specificity phosphatases. For example, while the phosphate-binding loop in DUSP14 is comprised of the sequence H110CAAGVSR117, the same loop in VHR is formed by the sequence H122CREGYSR129 (Fig. 4 ▶; Yuvaniyama et al., 1996 ▶). Thus, this structural information should open opportunities for the rational design of inhibitors exhibiting selectivity for DUSP14 over other dual-specificity phosphatases (Hoffman et al., 2004 ▶). However, as DUSP14 and DUSP18 share highly conserved active-site pockets, it may be more difficult to achieve selectivity for DUSP14 over DUSP18.
4. Conclusions
We have successfully purified recombinant human DUSP14 to homogeneity and solved its crystal structure at a resolution of 1.88 Å. As expected, the overall fold of the core catalytic phosphatase domain is highly similar to many other DUSPs. Additionally, like DUSP18, DUSP14 possesses an extended C-terminal motif that may play a role in protein–protein interactions. The bound phosphate in the active site also enables examination of the interaction of the catalytic residues with phosphate, which should facilitate the rational design of inhibitors targeting DUSP14.
Supplementary Material
PDB reference: human dual-specificity phosphatase 14, 2wgp, r2wgpsf
Acknowledgments
This research was supported by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research. Electrospray mass-spectrometry experiments were conducted on the LC/ESMS instrument maintained by the Biophysics Resource in the Structural Biophysics Laboratory, Center for Cancer Research, National Cancer Institute at Frederick.
References
- Almo, S. C. et al. (2007). J. Struct. Funct. Genomics, 8, 121–140. [DOI] [PMC free article] [PubMed]
- Alonso, A., Sasin, J., Bottini, N., Friedberg, I., Friedberg, I., Osterman, A., Godzik, A., Hunter, T., Dixon, J. & Mustelin, T. (2004). Cell, 117, 699–711. [DOI] [PubMed]
- Bai, L., Yoon, S. O., King, P. D. & Merchant, J. L. (2004). Cell Death Differ. 11, 663–673. [DOI] [PubMed]
- Boutros, T., Chevet, E. & Metrakos, P. (2008). Pharmacol. Rev. 60, 261–310. [DOI] [PubMed]
- Brünger, A. T. (1992). Nature (London), 355, 472–475. [DOI] [PubMed]
- Camps, M., Nichols, A. & Arkinstall, S. (2000). FASEB J. 14, 6–16. [PubMed]
- Chang, L. & Karin, M. (2001). Nature (London), 410, 37–40. [DOI] [PubMed]
- Cohen, P. (2002). Nature Cell Biol. 4, E127–E130. [DOI] [PubMed]
- Davis, I. W., Murray, L. W., Richardson, J. S. & Richardson, D. C. (2004). Nucleic Acids Res. 32, W615–W619. [DOI] [PMC free article] [PubMed]
- Denu, J. M. & Dixon, J. E. (1995). Proc. Natl Acad. Sci. USA, 92, 5910–5914. [DOI] [PMC free article] [PubMed]
- Denu, J. M. & Dixon, J. E. (1998). Curr. Opin. Chem. Biol. 2, 633–641. [DOI] [PubMed]
- Denu, J. M., Stuckey, J. A., Saper, M. A. & Dixon, J. E. (1996). Cell, 87, 361–364. [DOI] [PubMed]
- Denu, J. M., Zhou, G., Guo, Y. & Dixon, J. E. (1995). Biochemistry, 34, 3396–3403. [DOI] [PubMed]
- Dickinson, R. J. & Keyse, S. M. (2006). J. Cell Sci. 119, 4607–4615. [DOI] [PubMed]
- Emsley, P. & Cowtan, K. (2004). Acta Cryst. D60, 2126–2132. [DOI] [PubMed]
- Farooq, A. & Zhou, M. M. (2004). Cell. Signal. 16, 769–779. [DOI] [PubMed]
- Fauman, E. B. & Saper, M. A. (1996). Trends Biochem. Sci. 21, 413–417. [DOI] [PubMed]
- Hoffman, B. T., Nelson, M. R., Burdick, K. & Baxter, S. M. (2004). Curr. Pharm. Des. 10, 1161–1181. [DOI] [PubMed]
- Hunter, T. (1995). Cell, 80, 225–236. [DOI] [PubMed]
- Jeffrey, K. L., Camps, M., Rommel, C. & Mackay, C. R. (2007). Nature Rev. Drug Discov. 6, 391–403. [DOI] [PubMed]
- Jeong, D. G., Cho, Y. H., Yoon, T. S., Kim, J. H., Ryu, S. E. & Kim, S. J. (2007). Proteins, 66, 253–258. [DOI] [PubMed]
- Jeong, D. G., Cho, Y. H., Yoon, T.-S., Kim, J. H., Son, J. H., Ryu, S. E. & Kim, S. J. (2006). Acta Cryst. D62, 582–588. [DOI] [PubMed]
- Jeong, D. G., Yoon, T. S., Kim, J. H., Shim, M. Y., Jung, S. K., Son, J. H., Ryu, S. E. & Kim, S. J. (2006). J. Mol. Biol. 360, 946–955. [DOI] [PubMed]
- Jung, S. K., Jeong, D. G., Yoon, T. S., Kim, J. H., Ryu, S. E. & Kim, S. J. (2007). Proteins, 68, 408–412. [DOI] [PubMed]
- Kaminska, B. (2005). Biochim. Biophys. Acta, 1754, 253–262. [DOI] [PubMed]
- Kapust, R. B., Tozser, J., Fox, J. D., Anderson, D. E., Cherry, S., Copeland, T. D. & Waugh, D. S. (2001). Protein Eng. 14, 993–1000. [DOI] [PubMed]
- Keyse, S. M. (2008). Cancer Metastasis Rev. 27, 253–261. [DOI] [PubMed]
- Klinger, S., Poussin, C., Debril, M. B., Dolci, W., Halban, P. A. & Thorens, B. (2008). Diabetes, 57, 584–593. [DOI] [PubMed]
- Krissinel, E. & Henrick, K. (2004). Acta Cryst. D60, 2256–2268. [DOI] [PubMed]
- Lang, R., Hammer, M. & Mages, J. (2006). J. Immunol. 177, 7497–7504. [DOI] [PubMed]
- Larkin, M. A., Blackshields, G., Brown, N. P., Chenna, R., McGettigan, P. A., McWilliam, H., Valentin, F., Wallace, I. M., Wilm, A., Lopez, R., Thompson, J. D., Gibson, T. J. & Higgins, D. G. (2007). Bioinformatics, 23, 2947–2948. [DOI] [PubMed]
- Laskowski, R. A., MacArthur, M. W., Moss, D. S. & Thornton, J. M. (1993). J. Appl. Cryst. 26, 283–291.
- Levitzki, A. (1994). Eur. J. Biochem. 226, 1–13. [DOI] [PubMed]
- Marie-Claire, C., Benturquia, N., Lundqvist, A., Courtin, C. & Noble, F. (2008). Brain Res. 1239, 42–48. [DOI] [PubMed]
- Marti, F., Krause, A., Post, N. H., Lyddane, C., Dupont, B., Sadelain, M. & King, P. D. (2001). J. Immunol. 166, 197–206. [DOI] [PubMed]
- Matthews, B. W. (1968). J. Mol. Biol. 33, 491–497. [DOI] [PubMed]
- Minor, W., Cymborowski, M., Otwinowski, Z. & Chruszcz, M. (2006). Acta Cryst. D62, 859–866. [DOI] [PubMed]
- Murshudov, G. N., Vagin, A. A. & Dodson, E. J. (1997). Acta Cryst. D53, 240–255. [DOI] [PubMed]
- Nakano, Y. (2007). Br. J. Dermatol. 156, 848–860. [DOI] [PubMed]
- Nordle, A. K., Rios, P., Gaulton, A., Pulido, R., Attwood, T. K. & Tabernero, L. (2007). Proteins, 69, 19–31. [DOI] [PubMed]
- Owens, D. M. & Keyse, S. M. (2007). Oncogene, 26, 3203–3213. [DOI] [PubMed]
- Patterson, K. I., Brummer, T., O’Brien, P. M. & Daly, R. J. (2009). Biochem. J. 418, 475–489. [DOI] [PubMed]
- Pawson, T. (1995). Nature (London), 373, 573–580. [DOI] [PubMed]
- Pawson, T. & Nash, P. (2000). Genes Dev. 14, 1027–1047. [PubMed]
- Pearson, G., Robinson, F., Beers Gibson, T., Xu, B. E., Karandikar, M., Berman, K. & Cobb, M. H. (2001). Endocr. Rev. 22, 153–183. [DOI] [PubMed]
- Potterton, E., McNicholas, S., Krissinel, E., Cowtan, K. & Noble, M. (2002). Acta Cryst. D58, 1955–1957. [DOI] [PubMed]
- Pulido, R. & Hooft van Huijsduijnen, R. (2008). FEBS J. 275, 848–866. [DOI] [PubMed]
- Ralph, J. A. & Morand, E. F. (2008). Expert Opin. Ther. Targets, 12, 795–808. [DOI] [PubMed]
- Read, R. J. (1997). Methods Enzymol. 277, 110–128. [DOI] [PubMed]
- Salojin, K. & Oravecz, T. (2007). J. Leukoc. Biol. 81, 860–869. [DOI] [PubMed]
- Schumacher, M. A., Todd, J. L., Rice, A. E., Tanner, K. G. & Denu, J. M. (2002). Biochemistry, 41, 3009–3017. [DOI] [PubMed]
- Theodosiou, A. & Ashworth, A. (2002). Genome Biol. 3, REVIEWS3009. [DOI] [PMC free article] [PubMed]
- Tonks, N. K. (2006). Nature Rev. Mol. Cell Biol. 7, 833–846. [DOI] [PubMed]
- Tropea, J. E., Cherry, S., Nallamsetty, S., Bignon, C. & Waugh, D. S. (2007). Methods Mol. Biol. 363, 1–19. [DOI] [PubMed]
- Wu, G. S. (2007). Cancer Metastasis Rev. 26, 579–585. [DOI] [PubMed]
- Xu, H., Dembski, M., Yang, Q., Yang, D., Moriarty, A., Tayber, O., Chen, H., Kapeller, R. & Tartaglia, L. A. (2003). J. Biol. Chem. 278, 30187–30192. [DOI] [PubMed]
- Yuvaniyama, J., Denu, J. M., Dixon, J. E. & Saper, M. A. (1996). Science, 272, 1328–1331. [DOI] [PubMed]
- Zhang, Z. Y. (2002). Annu. Rev. Pharmacol. Toxicol. 42, 209–234. [DOI] [PubMed]
- Zhang, Z. Y., Zhou, B. & Xie, L. (2002). Pharmacol. Ther. 93, 307–317. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: human dual-specificity phosphatase 14, 2wgp, r2wgpsf