

Abstract
Understanding the mechanism by which single-stranded oligonucleotides (ODNs) elicit targeted nucleotide exchange (TNE) is imperative to achieving optimal correction efficiencies and medical applicability. It has been previously shown that introduction of an ODN into cells results in the activation of DNA damage response pathways, but there has been no evaluation of the damage created at the level of the DNA. The activation of H2AX, a hallmark protein of DNA breakage, suggests that a double-strand break (DSB) could be occurring during the targeted gene alteration (TGA) reaction. Using the human HCT116 cell line with a single integrated mutant eGFP gene as our model system, we demonstrate that the DNA strand breakage occurs when a specific ODN, designed to direct TGA, is transfected into the cells. Both single and double-stranded DNA cleavage is observed dependent on the level of ODN added to the reaction. Possible mechanisms of ODN-dependent DSB formation, as a function of TGA, are discussed herein.
1. Introduction
Single-stranded DNA oligonucleotides (ODNs) can direct the exchange of a single base in genomic DNA and alter cellular phenotype in a reaction known as targeted gene alteration (TGA) [1, 2 and references therein]. In the proposed mechanism, ODNs align in homologous register with the complementary sequence in the chromosome but create a single mismatched base pair with the nucleotide targeted for change. Once paired at this site, the ODN is hybridized to one strand of the helix, while the other strand is displaced forming a structure known as a D-loop [2-4]. This helical distortion attracts enzymes involved in genome maintenance and activates members of the homologous recombinational repair pathway [5, 6]. Evidence from studies in yeast [7, 8] supports the involvement of proteins such as Nbs1, Rad51, Rad54, and Mre11 in the TGA reaction pathway. Some of these enzymes process and resolve the D-loop restoring the integrity of the helix [9, 10].
The efficiency of nucleotide exchange is elevated dramatically if TGA is directed to active regions of DNA synthesis [11-15]. By utilizing 2′,3′-dideoxycytidine (ddC), an inhibitor of elongation, Engstrom and Kmiec [11] synchronized cells in different cell cycle phases and demonstrated that the concentration correlating to the highest level of cells synchronized in S-phase prior to addition of the ODN correlated with highest level of correction efficiency. These workers were also able to demonstrate the same phenomenon using aphidicolin, a potent inhibitor of polymerase α, δ, and ε. Similar findings using a thymidine block and hydroxyurea, both of which affect the ribonucleotide reductase pathway, corroborated earlier findings in HCT116 cells [5, 16]. It has been observed that blocking the ends of the targeting ODN significantly decreases the level of correction [17]. Finally, Wu et al. [15] demonstrated that inhibition of S-phase progression after addition of ODN completely abolished gene correction. Considering that the presence of the ODN initiates a replication delay [18], evidence strongly supports a model where cells stalled in S-phase exhibit significantly higher levels of correction efficiency than cells stalled in any other phase of the cell cycle.
Since the initial step in TGA is the creation of a D-loop, we wondered if the ODN paired at the target site physically impedes the activity around or within the replication fork. It is possible that this stalling explains why ODN-treated cells experience replication delay and a retardation in cell cycle. Stalled replication forks result in exposed single-stranded regions which are subject to mechanical breakage and nucleolytic attack [19]. A stalled replication fork could be a substrate for single-stranded nicking, vis-à-vis cleavage of DNA secondary structures that eventually appear as strand breaks [20]. In prokaryotes, inhibition or blockage of helicase DnaB, which is necessary for replication, leads to DNA breakage by creation of entry sites for the RecBCD complex. This complex harbors nucleolytic activity and acts in the area of the initial steps of homologous recombination [20, 21]. Seigneur et al. [22] predicted that Holliday junction formation at stalled replication forks are resolved by DNA breakage through the activity of the RuvABC protein complex [22]. Both observations link stalled replication fork resolution by double strand break (DSB) formation with homologous recombination and replication fork re-initiation. In a eukaryotic system, Saintigny et al. observed that when replication inhibitors, such as hydroxyurea, were added to mammalian cell cultures, accumulation of DSBs was observed [23]. Since there seems to be a strong correlation between DNA replication and elevated TGA activity, we asked whether DNA breakage also occurs during the process of gene correction, perhaps as a result of stalled replication activity. Finding such breaks, perhaps only transitory in nature, may provide some insight into the events going on at the site of nucleotide exchange. In this paper, we describe an analysis of DNA cleavage activity as a function of targeted gene alteration.
2. Materials and Methods
2.1 Cell Line and Culture Conditions
HCT116 cells were acquired from ATCC (American Type Cell Culture, Manassas, VA). The integrated HCT116 clone 19 (HCT116-19) was created through the integration of pEGFP-N3 vector (Clontech, Palo Alto, CA) containing a mutated eGFP gene, as described by Hu et al. [24] and clones isolated by selection. The mutated eGFP gene has a nonsense mutation at position +67 resulting in nonfunctional eGFP protein. For these experiments, HCT116-19 cells were cultured in McCoy's 5A Modified medium (Sigma-Aldrich, St. Louis, MO) supplemented with 10% fetal bovine serum, 2 mM L-Glutamine, and 1% Penicillin/Streptomycin. Cells were maintained at 37 °C and 5% CO2.
2.2 Oligonucleotide design and eGFP gene targeting
DNA oligonucleotides (ODNs) were synthesized by Integrated DNA Technologies (Coralville, IA). The targeting ODNs were designed as 72-mers that complemented either the non-transcribed (NT) or transcribed (T) strand of the target mutant eGFP gene. Each ODN has three phosphorothioate linkages on either end to help prevent nuclease degradation. The ODNs that were designed to elicit correction of the gene (72NT and 72T) have a central mismatch that would direct conversion of the mutant stop codon to the wild-type eGFP tyrosine, thereby allowing expression of functional eGFP.
Prior to eGFP targeting, cells were treated with 2 μM aphidicolin for 24 hours in complete growth medium. Cells were then trypsinized and harvested by centrifugation. Cells were resuspended to a concentration of 2.5×107 cells/mL in serum-free medium and 100 μL transferred to a 4 mm gap cuvette (Fisher Scientific, Pittsburgh, PA). The respective ODN was added to a final concentration of 8 μM (unless otherwise noted) and the cells were electroporated (250 V, 13 ms, 2 pulses, 1 s interval) using a BTX Electro Square Porator™ ECM 830 (BTX Instrument Division, Holliston, MA). The electroporated cells were then transferred to a 100 mm dish and allowed to recover in complete growth medium for 24 hours (unless otherwise noted) at 37°C.
2.3 Cell-based analyses: flow cytometry and immunofluorescence
eGFP fluorescence was measured by a Becton Dickinson FACScalibur flow cytometer (Becton Dickinson, Franklin Lakes, NJ) 24 hours after electroporation. Cells were harvested by trypsinization and resuspended in FACS buffer (0.5% BSA, 2 mM EDTA, 2 μg/mL propidium iodide in PBS). Correction efficiency was then calculated as the percentage of eGFP positive cells out of the live cell population.
Targeted cells were immediately plated in 8-well chambers (5×104 cells/well) and allowed to recover for 24 hours. Control cells were treated with 300 nM camptothecin (CPT) for 24 hours. H2AX: Cells were then washed for 5 minutes at 37°C in PBS. Cells were fixed in 4% paraformaldehyde for 30 minutes at room temperature. After washing with PBS, cells were permeabilized with 0.5% Triton X-100 in PBS for 10 minutes at room temperature. Cells were blocked with 10% normal goat serum for 10 minutes and then for 30 minutes in a 10% milk solution. pH2AX antibody was added at a dilution of 1:100 in 10% milk and kept at 4°C overnight (Cell Signaling, Boston, MA). The next day, cells were washed 4 times for 10 minutes each in 0.1% Triton X-100. The secondary antibody (α-rabbit, goat-Cy3) was then added for 1 hour at room temperature. Cells were again washed 4 times for 10 minutes each in 0.1% Triton X-100. Anti-fade with DAPI was added and then cells were visualized with a Zeiss LSM 510 NLO attached to an Axiovert 200M.
2.4 DNA damage analyses: COMET assay
Targeted cells were allowed to recover for 24 hours. Cells were harvested by trypsinization and 1×105 cells were isolated and pelleted. The cell pellet was washed with PBS and then suspended in 20 uL of PBS. Cells were combined in a 1:10 ratio with low melt agarose and immediately pipetted onto a CometSlide. Cell mixture was allowed to cool for 10 minutes at 4°C in the dark before immersing in lysis solution at 4°C for 60 minutes. Cells were then immersed in a freshly prepared alkaline solution, pH 13.0, for 60 minutes in the dark at room temperature. Slides were rinsed in TBE buffer and transferred to a horizontal electrophoresis apparatus. TBE was added to cover slides and the unit was run at 1 volt/cm for 15 minutes. Slides were submerged in 70% ethanol for 5 minutes and allowed to air dry in the dark overnight. The next day a few drops of SYBR Green I were added to each sample and the slides were covered with a coverslip and sealed. Slides were visualized on a Zeiss LSM 510. At least 12 images were captured per treatment. Percent of COMETs was determined for each field and averaged for each treatment.
2.5 Pulse-Field Gel Electrophoresis (PFGE)
Targeted cells were allowed to recover for 24 hours. Cells were harvested by trypsinization and 1×106 cells were isolated and pelleted by centrifuging at 1500 rpm for 5 minutes. Cells were then resuspended in PBS, pelleted again, and finally suspended in 50 mM EDTA. Cells were combined in a 1:1 ratio with 1% low melt agarose (GIBCO, Invitrogen, Carlsbad, CA) in 50 mM EDTA and transferred to plug molds. Plugs were allowed to cool at room temperature for approximately 30 minutes before being transferred to lysis solution (50 mM EDTA, 1% N-laurosylsarcosine, 1 mg/mL proteinase K). Cells were kept in lysis solution at 50°C for 24 hours while shaking. Plugs were then washed four times in 1X TE buffer before being inserted into a 1% pulse field certified agarose gel (Bio-Rad, Hercules, CA). The gel was run for 24 hours using a 120° field angle, 60 to 240 s switch time, 4 V/cm at 14°C. The next day the gel was stained for 1 hour in ethidium bromide prior to imaging on an AlphaImager 2200 (Alpha Innotech Corp., San Leandro, CA).
2.6 BrdU Incorporation
Targeted cells were allowed to recover for specified time in 96-well tissue-culture treated plates at 15,000 cells/well. Cells were incubated in the presence of BrdU for two hours prior to analysis with BrdU ELISA kit (Roche Diagnostics, Mannheim, Germany). Absorbances at 450nm and 690nm were monitored with a Victor X3 2030 Multilabel Reader (Perkin Elmer, Waltham, Massachusetts).
2.7 XL-PCR
Targeted cells were allowed to recover for 24 hours prior to DNA extraction with an Epicentre MasterPure™ Complete DNA and RNA Purification Kit (Epicentre Biotechnologies, Madison, WI). Isolated DNA was quantified with a NanoDrop ND-1000 spectrophotometer (Thermo Scientific, Wilmington, DE) and then 120 ng was used in the XL-PCR reaction. Mitochondrial primers 15149 and 14841 were used to generate a 16.2 kb fragment, while nuclear primers 62007 and 48510 were used to generate a 13.5 kb fragment [25].
3. Results
The correction of a single base mutation in an integrated copy of the eGFP gene results in the emergence of green fluorescence in transgenic HCT116-19 cells [11]. This cell line was created to serve as a model system for the rescue of a point mutation in a single copy gene directed by single-stranded oligonucleotides (ODNs). As such, targeted gene alteration (TGA) can be evaluated in a mechanistic fashion using a validated system wherein both genotypic and phenotypic changes can be confirmed. Fig. 1 displays the sequence surrounding the target base (in bold) in the mutant EGFP gene. Conversion of the G to a C changes a stop codon to one encoding tyrosine, thereby restoring normal protein function. Oligonucleotides, EGFP3S/72NT (72NT) and EGFP3S/72T (72T), are 72 bases in length containing three phosphorothioate linkages between the terminal four bases to protect against nucleolytic attack [see 24]. 72NT hybridizes to the non-transcribed strand while 72T binds to the transcribed strand; both create the appropriate mismatched base pair at the target site (72NT, G•G; 72T, C•C). In contrast, EGFP3S/72NT-PM (72NT-PM) binds with perfect complementarity to the NT strand of the target gene; it should not stimulate TGA. Oligonucleotides designated as NS1 and NS2 have a nonspecific sequence that bears no complementarity to the target sites.
Fig. 1.

The target eGFP sequence has a single base mutation (bold) that codes for a premature stop codon and produces truncated, non-functional eGFP protein. Specific oligonucleotides (ODNs) were designed to bind the non-transcribed (NT) strand of the target eGFP gene (72NT and 72NT-PM) or the transcribed (T) strand (72T), (*) denotes phosphorothioate linkages. 72NT-PM is a perfect match oligonucleotide that complements the NT strand exactly, whereas, 72NT and 72T have one designed central mismatch to induce targeted gene alteration in the eGFP gene (bold). There were two nonspecific (NS) ODNs utilized throughout the experiments, neither bind the target eGFP gene.
The experimental strategy for targeting mutant reporter genes such as eGFP has been widely reported and extensively reviewed [see 6, 26]. Briefly, cells (2.5×106) are electroporated in the presence of the ODN after 24 hours of pretreatment with 2 μM aphidicolin. This drug is used to synchronize cells in early S phase; upon washout and release progression through S phase takes place. This protocol has been shown previously to enhance TGA activity [5, 12, 16, 18, 24]. Twenty-four hours later, the cells are processed for FACS with the emergence of green fluorescence indicating the presence of cells containing a wild-type (corrected) eGFP gene. Corrected cells are quantitated by counting live fluorescent cells and dividing by the total number of live cells [24]. This determination generates a value referred to as the correction efficiency (CE), which is often presented as a percentage (%). This experiment was carried out using three ODNs designed to target mutant eGFP in HCT116-19 cells as illustrated in Fig. 1. The results are depicted in Fig. 2. A dose-response is evident from 2-8 μM when 72NT is used but the two other ODNs promoted either no TGA or at levels not detectable by FACS. The dramatic increase in CE when 2 μM ODN is raised to 4 μM (72NT), represents what is likely to be a mass action effect in which a certain threshold level of ODN must be present in order to initialize TGA [26, 27]. Correction bias favoring the NT strand is also evident, a phenomenon reported previously by many investigators [24, 28-30], some of whom hypothesize that the transcriptional activity on the T strand leads to a dislodging of the 72T ODN [29]. Taken together, these data confirm that TGA activity leads to the correction of a single copy mutant eGFP gene by the 72NT ODN in HCT116-19 cells.
Fig. 2.

Correction efficiency of integrated eGFP gene in HCT116-19s using FACS. HCT116-19 cells were pre-treated with 2 μM aphidicolin and then electroporated with 2, 4, or 8 μM of respective ODN. Cells were allowed to recover for 24 hours prior to FACS analysis. Correction efficiency was determined as number of GFP positive cells over number of live cells in the population.
High levels of correction are attained when approximately 4.82×1014 molecules of ODN are transfected into 2.5×106 cells. This large excess of ODN appears to be required so that enough targeting molecules can enter the cell, traverse the cytoplasm and pass into the nucleus. Previously, we and others have reported that single-stranded ODNs activate a DNA damage signaling pathway leading to an activation and accumulation of a group of proteins associated with DNA recombinational repair [5, 12, 14]. We confirmed those observations in HCT116-19 cells first by visualizing the penetration of the ODN into the cells using confocal microscopy, then, we looked for the activation of the DNA damage response protein, pH2AX, in cells that had taken up the ODN. Fig. 3A displays typical images of transfected cells bearing fluorescently-tagged ODNs. The majority of ODNs localize in the cytoplasm at the 24 hours post-electroporation time point; DAPI is used as a stain to demarcate the nucleus. Single-stranded ODNs do penetrate the nucleus and apparently lead to the activation pH2AX, as seen in the images presented in Fig. 3B. H2AXγ is a universally accepted protein marker of DNA damage, often in the form of double-strand breaks [31-33]. The anticancer drug, CPT, which introduces irreversible strand breaks, is used as a positive control and, in fact, H2AX activation is readily observed under these reaction conditions. As seen in Fig. 3A and 3B, the number of cells bearing Cy3-ODN roughly corresponds to the level of pH2AX induction within the entire population. Induction of pH2AX is followed by the activation of cell cycle checkpoint proteins, Chk1 and Chk2, which together contribute to the stalling of DNA replication [5, 12, 18], and the elevation of TGA activity [11]. Here, the introduction of single-stranded ODNs activates a signaling pathway that can severely impact cell cycle progression and cell metabolism via the response to free ends.
Fig. 3.
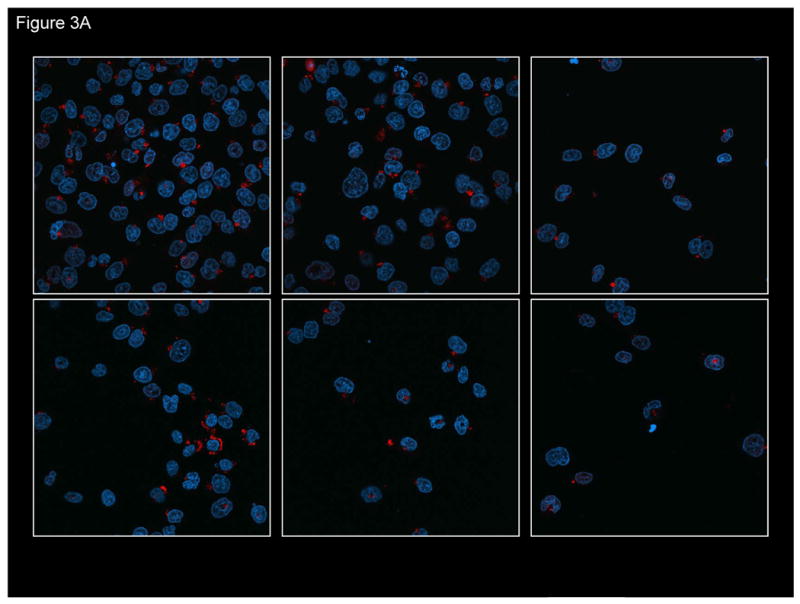
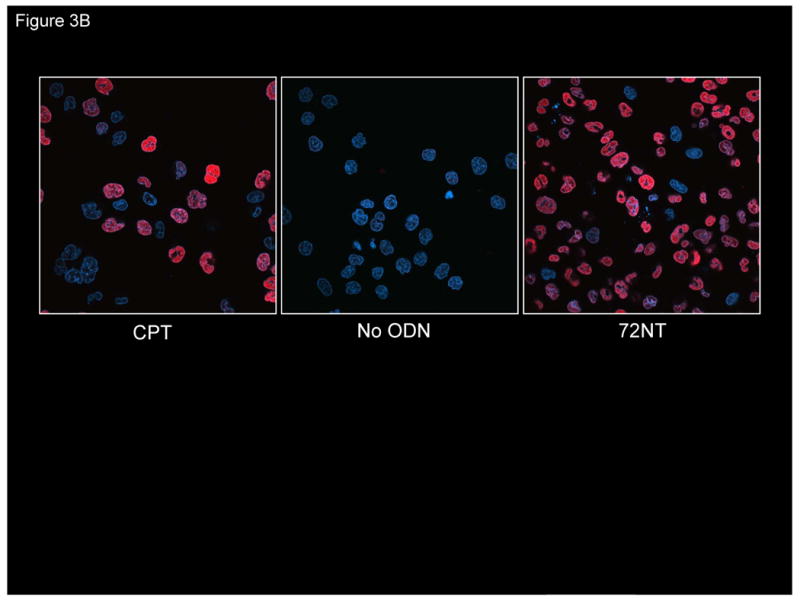
(A) Confocal image of fluorescently-tagged oligonucleotide in and around the cell nucleus. HCT116-19 cells were electroporated with 8 μM of Cy3-labeled ODN (red) and allowed to recover for 24 hours. DAPI was used to label nucleus (blue). (B) Presence of ssODN stimulates H2AX activation. Confocal image of pH2AX in HCT116-19 cells in the presence of 8 μM 72NT. Camptothecin (CPT) (300 nM) treatment serves as positive control.
We wondered if the activation of pH2AX and its downstream partners arise not only from the presence of single-stranded (ODN) free ends but also from nascent DNA-strand breaks caused by the TGA reaction itself. Hence, an analysis of genomic DNA integrity in the target cells was carried out under the same reaction conditions that support the highest level of TGA, i.e. 8 μM 72NT for 24 hours. Single-stranded breaks were monitored by the COMET and XL-PCR assays. The COMET assay is based on the appearance of a tailing halo in cells reflecting single-strand interruptions in genomic DNA (Trevigen, Gaithersburg, MD). In this assay, cells and agarose are combined and then exposed to alkaline which causes membrane degeneration and DNA denaturation. After the extract is electrophoresed on a slide, SYBR green is used to stain for DNA; the appearance of a cone-shaped tail indicates breakage. The results of the COMET assay, shown in Fig. 4, demonstrate that the targeting ODNs induce single-stranded DNA breaks. Fig. 4A shows a representative time course experiment where HCT116-19 cells, treated with 8 μM of a particular ODN, were allowed to recover for either 8, 12, 16, or 24 hours post-electroporation (after introduction of the ODN). HCT116-19 cells all show a similar pattern of COMET appearance 12 hours post-electroporation and with an apparent recovery after 24 hours as judged by a severe reduction in halos. Percent COMETs per treatment are quantified in Fig. 4B and suggest an ODN-independent single-strand break activity since the control (No ODN) exhibits the same pattern described above. However, it is pertinent to consider that cells treated with ODN experience a significant delay in replication; a process that can generate transient single-stranded breaks [13, 16, 18, 34, 35]. Therefore, the demonstrated lag in replication in ODN-treated cells, as seen with both the BrdU incorporation assay (Figure 4C) and cell counts after recovery (data not shown), implicate that ODN treatment is responsible for some degree of single-stranded break occurrence independent of normal cellular replication. For example, by 16 hours of recovery the amount of replication is significantly lessened in ODN-treated cells, but the amount of single-strand breaks is similar between 72NT, 72T, and control (No ODN) treated cells. This is indicative of the targeting ODNs having a positive effect on single-strand break occurrence since the level of single-strand breaks in ODN-treated cells cannot be explained by replication alone. This is more clearly demonstrated when the percent of COMETs is compared to the number of cells present at time of collection, a number representative of the level of cellular replication (Figure 4D). Here, the amount of single-stranded DNA breaks in cells treated with targeting ODN appears significantly higher than in samples where cells were treated with nonspecific ODN or without ODN. And since the single-stranded breaks seen at 16 hours are resolved by 24 hours without the ODN-treated cells replicating (Fig. 4C) it follows that the level of single-stranded DNA breaks induced by the targeting ODNs is independent of single-stranded breaks contributed by DNA replication.
Fig. 4.
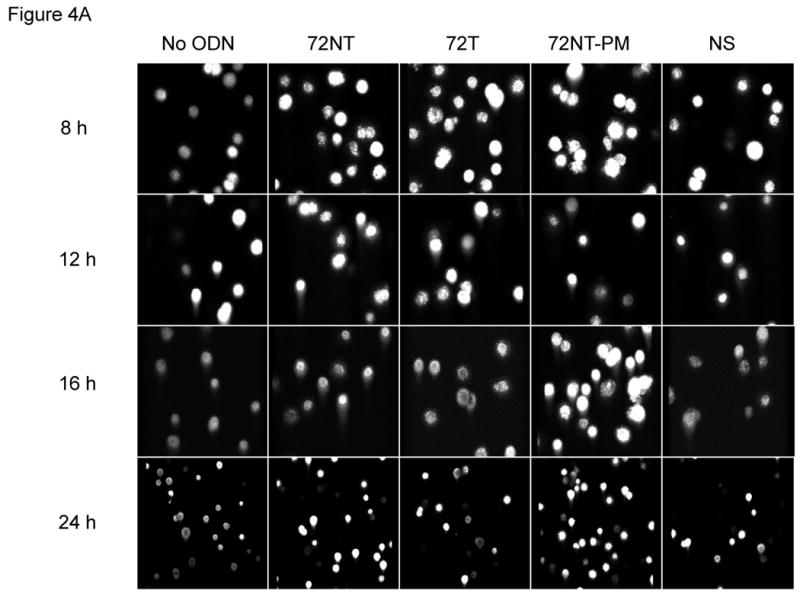




(A) COMET assay shows single-stranded DNA breaks with and without the presence of 8 μM ODN. CPT (300 nM for 24 h) is used as a positive control. (B) Quantification of COMET time course. Average percent COMETs per field was used to compare between treatments over time. Error bars are standard error. (C) BrdU incorporation in ODN-treated and untreated cells over time demonstrates that ODN inhibits the level of cellular replication. Cells without ODN present are able to replicate at a higher level. Error bars are standard error. (D) Normalizing the average percent COMET data (B) to the cell counts for each treatment over time highlights the significant level of single-strand breaks present in cells treated with targeting ODN at 16 hours post-electroporation that are independent of replication. (E) Mixing experiment shows that addition of NS ODN significantly reduces the amount of single-strand breaks associated with targeting ODN presence in cells. 72NT and NS ODNs were mixed in molar ratios totaling 4 μM ODN. As in (D), average percent COMETs are normalized to cell count.
To our surprise, the NS ODN elicits less single-stranded breaks than the targeting ODNs and so the specificity of the targeting ODN reaction was investigated by mixing the targeting ODN 72NT with NS ODN in different molar ratios with a final concentration of 4 μM respectively. As seen in Fig. 4E, diluting the 72NT ODN with NS ODN drastically decreases the amount of single-strand DNA breaks induced. Interestingly, in the HCT116-19 cell model system, dilution of targeting ODN with NS ODN concurrently decreases correction efficiency (data not shown), in contrast to some evidence suggesting that addition of NS ODN raises correction efficiency either by sequestering factors inhibitory to the TGA reaction or mass action [36, 37].
Next, we employed a gene specific Q-PCR assay, XL-PCR, to look for breaks in genomic (nuclear and mitochondrial) DNA under conditions that enable TGA. This assay is based on the concept that genomic DNA containing lesions will amplify less effectively than genomic DNA with fewer lesions [25]. As such, DNA containing strand breaks will produce a lower number of amplicons than intact DNA samples. Using this rationale, it is possible to discriminate between host nuclear and mitochondrial DNA by using a set of primers unique to each genome. Importantly, this assay can be used to compare nonspecific effects of the ODN on nuclear DNA (nDNA) and mitochondrial DNA (mtDNA). For our analysis of mtDNA, we chose primers that amplify a mitochondrial specific fragment of 16.2 kb; for nuclear DNA we used primers that amplify a 13.5 kb fragment of the β-globin gene [25]. Here, we ask whether random DNA breaks are elevated in the presence of ODN. Since the targeting ODNs show an increased level of single-stranded breaks, the 72NT, 72T and the 72NT-PM ODNs were electroporated into HCT116-19 cells and after 24 hours, mitochondrial and genomic DNA were amplified by the specific primers described above to determine if the single-stranded breaks are being induced randomly throughout the genome. As shown in Table I, the mtDNA sample does not exhibit a statistically significant difference in DNA breaks as compared to the mock-transfected cells. The same is true for the nDNA samples except for one point; 72NT-PM does produce a statistically significant difference, albeit at a low confidence level. But, the 72NT ODN, which produces the highest level of TGA when introduced into HCT116-19 cells, does not induce random single-stranded breaks in mtDNA or nDNA. Taking the results of the COMET and XL-PCR assays together, we believe that the reaction conditions at which the ODN that promotes TGA at discernable levels does increase the number of single-stranded breaks in HCT116-19 cells, most likely at or near the target gene site.
Table 1.
Results of extra-long nuclear and mitochondrial PCR amplification using 8 μM of designated oligonucleotide presented with the corresponding correction efficiencies.
| Oligonucleotide | Relative Amplification of nDNA | Relative Amplification of mtDNA | Correction Efficiency (%)a |
|---|---|---|---|
| 72NT | 0.75 ± 0.25 | 0.99 ± 0.29 | 2.01 ± 0.10 |
| 72T | 0.74 ± 0.24 | 1.18 ± 0.27 | 0.01 ± 0.00 |
| 72NT-PM | 0.38 ± 0.20 * | 1.61 ± 0.59 | 0.00 ± 0.00 |
Errors are standard error.
p = 0.036.
3.1 Induction of double-stranded DNA breakage by TGA
We next examined whether double-stranded DNA breaks (DSBs) are formed during the TGA reaction by carrying out pulse-field gel electrophoresis (PFGE) on treated samples. We have used this technique previously to analyze how anti-cancer agents influence the frequency of TGA [38]. In PFGE, DSBs in DNA are detected by the capacity of shortened chromosomal fragments to migrate through an agarose gel within a constantly changing electric field. We monitored double-strand breakage under conditions that promote efficient TGA activity (4 μM). The reaction was carried out in HCT116-19 cells for 24 hours and the samples processed for PFGE. The results are presented in Fig. 5A and in reactions containing ODNs specific for the target site (72NT, 72T, 72NT-PM), DNA breakage is seen; in contrast, samples containing mock electroporated cells or cells treated with the nonspecific ODN exhibited a very low level of double-stranded breakage. While there is some difference in the degree of breakage among various cells treated with site-specific ODNs, the variance is likely not statistically significant. CPT is used as a positive control and generates DNA breakage vis-à-vis the appearance of a faster migrating band and a smear throughout the lane. Fig. 5B shows that the DSBs induced by the ODNs exhibit a dose-response, similar to the dose response in the correction efficiency of the ODNs; increasing concentration correlating to both increasing DSBs and correction efficiency. However, the NS ODN maintains a lower level of DSBs than the targeting ODN regardless of concentration.
Fig. 5.
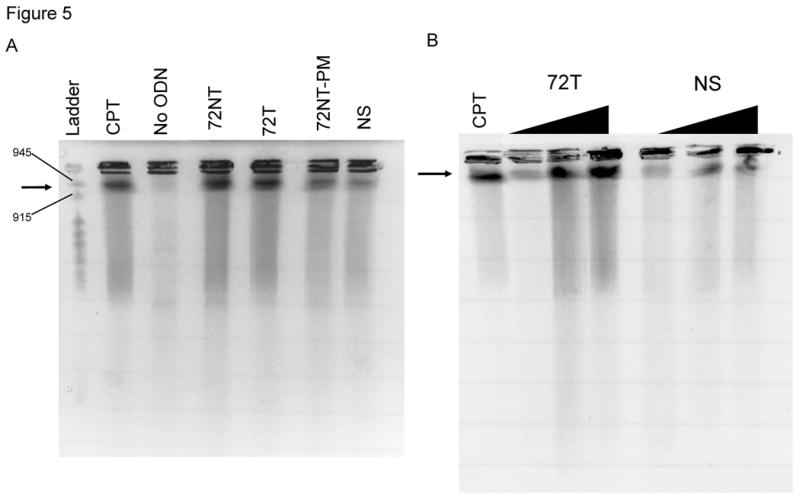
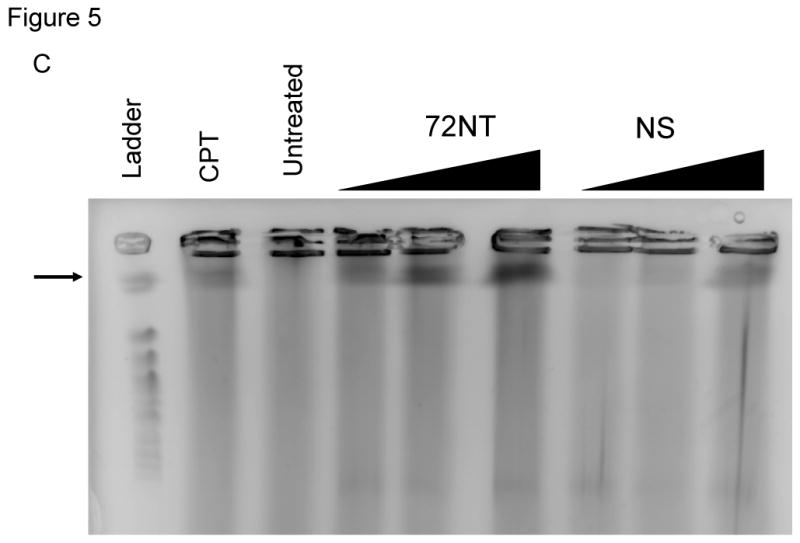
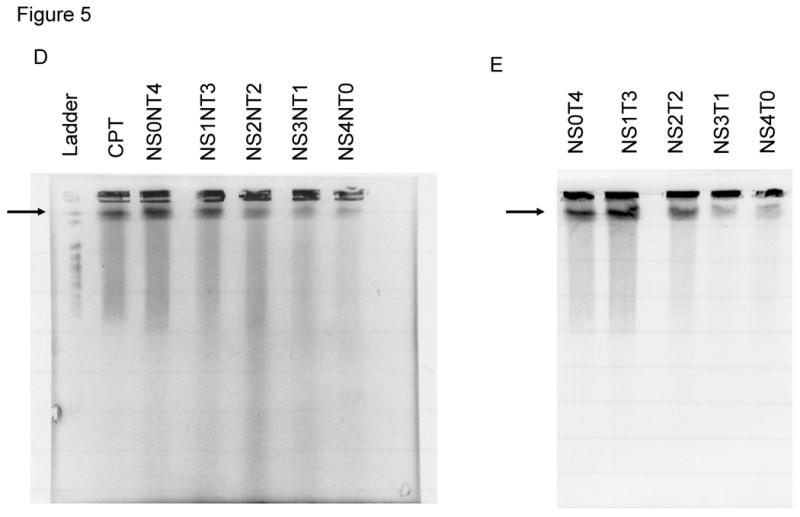
(A) Pulse-field gel electrophoresis (PFGE) of HCT116-19 cells 24 hours after being electroporated with 4 μM of respective ODN after 24 hour pre-treatment with 2 μM aphidicolin. A non-specific (NS) ODN does not induce double-strand breaks, whereas ODNs specific to either the NT or T strand do. (B) PGFE of HCT116-19 cells, 24 hours after electroporation, shows DSB formation is dose-dependent. 72NT and NS ODNs were present at 2, 4, and 8 μM. (C) PFGE of HCT116-19 cells, 24 hours after electroporation, with 4 μM total ODN (72NT and non-specific) in specified molar ratios. (D) Pulse-field gel of HCT116-19 cells, 24 hours after electroporation, with 4 μM total ODN (72T and non-specific) in specified molar ratios. Increasing the molar ratio of NS ODN relative to either the 72NT or 72T ODN decreases the amount of DSBs.
Since the HCT116 cell line is mismatch repair deficient, we wanted to employ the use of another human cell line with no known mismatch repair deficiencies to confirm that the ODN induced DSB occurrence is a more universal mechanism. For this purpose, a DLD1 cell line that has the mutant eGFP gene stably integrated into it (DLD1-1) was treated with increasing amounts of either the 72NT or NS ODN and allowed to recover for 24 hours prior to harvesting for PFGE. As can be seen in Figure 5C, a dose-dependent response is seen when the cells were electroporated with increasing concentrations of the targeting ODN, whereas minimal DSB occurrence is seen in cells treated with an NS ODN. Therefore the mechanism of ODN induced DSB occurrence is not a factor of a lacking mismatch repair response.
To investigate the specificity of ODN induced DSB occurrence, a mixing experiment similar to the COMET mixing experiment (Fig. 4E) was performed. Here, 4 μM of ODN is maintained but the composition of the reaction is changed. To confirm specific ODN activity, we mixed specific and nonspecific ODNs in the ratios indicated (Fig. 5D and 5E). For example, NS1NT3 indicates that 3 times as much ODN targeting the non-transcribed strand is present in the reaction; NS0NT4 means only NT ODN is present. These combinations of ODNs were electroporated into the cells and 24 hours later, the amount of double-strand breakage was evaluated using PFGE. Fig. 5D reveals that the extent of double-strand breakage reflects the level of the specific NT ODN present in the reaction in a dose-dependent fashion. We see the same response from a mixing experiment in which 72T and NS were used (4 μM total, once again) where breakage depends on the level of specific ODN in the reaction (Fig. 5E). In both cases, the capacity to support double-stranded DNA breakage can be diluted by increasing the level of NS ODN transfected into the cells. We have replicated these experiments with ODNs bearing different nonspecific sequences (all of which are noncomplementary to the target gene and human genome) and none of them induced double-stranded DNA breakage (data not shown). Hence, our data suggest that the process of TGA directed by ODNs that target the specific site, results in single- and double-stranded DNA breakage within the cell and the amount of this breakage correlates with correction efficiency.
Another factor known to influence correction efficiency of the TGA reaction is the cell cycle. Previously, we demonstrated that slowing progression through S-phase increases the time spent in S-phase significantly which results in increasing correction efficiency [11, 17, 18]. Since the DSBs induced in the TGA reaction correlate positively to correction efficiency, we wanted to investigate if the DSBs were dependent upon S-phase progression. To accomplish this, HCT116-19 cells were pre-treated with 2 μM aphidicolin for 24 hours prior to electroporation. Cells were electroporated with 8 μM of different ODNs and allowed to recover for 24 hours in full growth media supplemented with 2 μM aphidicolin. Cells were then harvested and part of the reaction processed for PFGE. The remaining part of the sample was resuspended in cell cycle FACS buffer [17] and analyzed using a Becton Dickinson FACScalibur flow cytometer (Becton Dickinson, Franklin Lakes, NJ). Fig. 6A shows that DSB formation is not affected by reduced cell cycle progression of the entire population into and through S-phase. Fig. 6B confirms that targeted cells have been substantially blocked in early S or at the G1/S border by treatment with aphidicolin as predicted. Hence, DSB formation does not appear to be affected by cell cycle progression, but seems to correlate with correction efficiency of the TGA reaction.
Fig. 6.
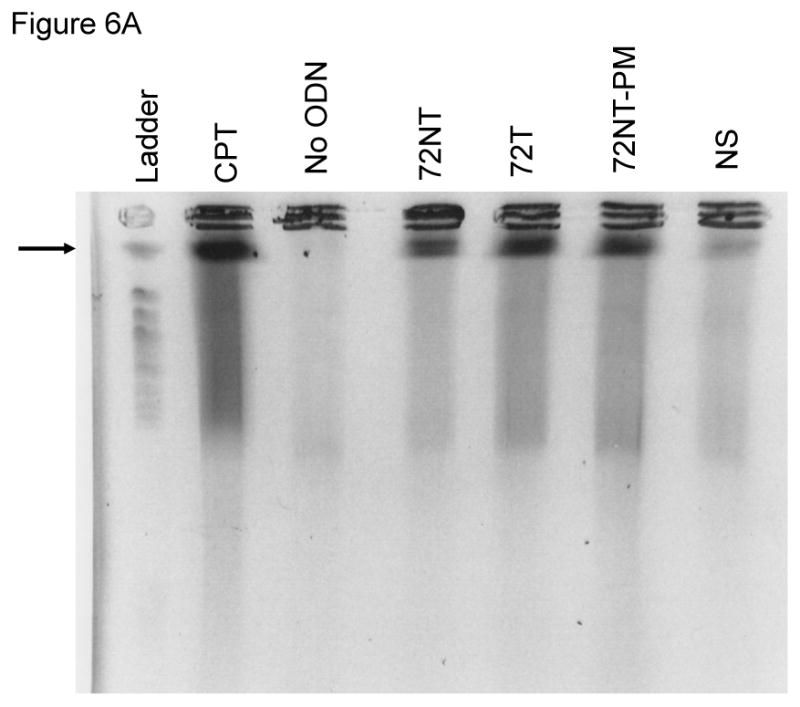
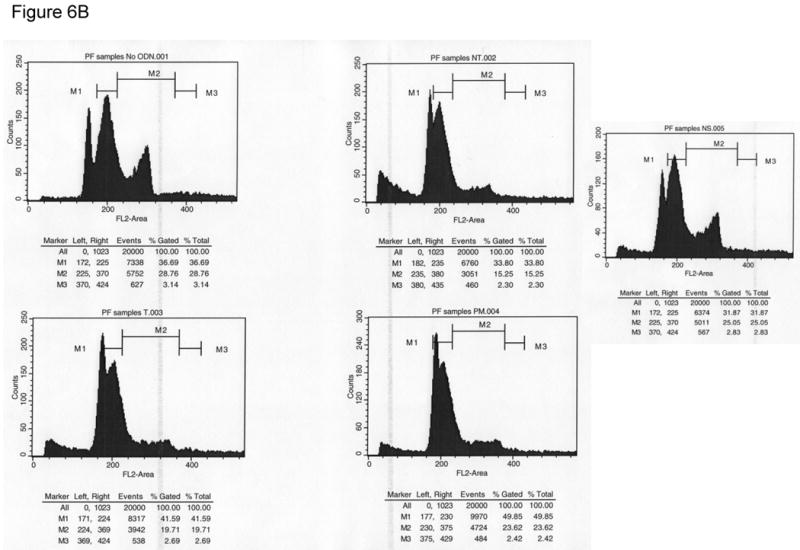
(A) PFGE of HCT116-19 pre-treated with 2 μM aphidicolin for 24 hours, electroporated with 8 μM ODN, and then allowed to recover for 24 hours in full growth media supplemented with 2 μM aphidicolin to block S-phase progression. (B) Cell cycle profiles of HCT116-19 cells after 24 hours recovery in the presence of 2 μM aphidicolin show the majority of cells are still in early S-phase.
4. Discussion
Targeted gene alteration remains a viable approach for creating single base changes in genomic DNA [6]. The mechanism of the nucleotide exchange reaction is being studied in several model systems including yeast, worms, bacteria and cultured human cells. In many cases, the target gene is a reporter construct that enables confirmation of genetic alteration by both phenotypic and genotypic readout. Recently, the regulation of TGA has been a particular focus of work in several labs and the consensus view is that S phase is the part of the cell cycle when TGA activity is maximized [11-13, 39]. Slowing down the progression of a cell through S phase, by reducing the rate of replication, enhances the frequency with which TGA takes place [5]. This is due, in all likelihood, to maintenance of an open chromatin configuration created by stalled replication forks. But, little is known about metabolic or nucleolytic events that take place either at the target site or as a function of base exchange itself. While it is clear that D-loops are created during the initiation or pairing phase of the reaction [3, 40, 41], the resolution phase of TGA, in which the ODN strand is incorporated into the genome, remains to be elucidated [9, 42].
We report here that DNA breakage is associated with TGA. There is an increase in the level/number of single-stranded breaks and the appearance of double-stranded breaks in a population of cells that are being targeted for correction. In our experiments, we targeted a single copy of the mutant eGFP gene integrated into HCT116 cells and monitored DNA breakage as a function of the TGA reaction. On a purely mechanistic level, our results validate previous observations made by several labs, using a variety of different gene and cell systems [11, 26 and references therein]. Transfection of increasing levels of ODN results in higher levels of gene repair [6] and targeting the non-transcribed strand of the mutant gene leads to a significantly higher level of gene repair than targeting the transcribed strand [7, 9, 24, 28, 39, 43]. An ODN bearing complete complementarity does not catalyze any TGA reaction even though it is known to pair stably at the target site. This last observation is at least consistent with the belief that TGA activity acts with some degree of specificity.
The activation of H2AX, as seen in Fig. 3, and related proteins [5, 12, 38] have led us to suspect that some DNA breakage might take place during TGA. Our results are consistent with the notion that strand interruptions result from TGA activity and that sequence homology enhances the level of single-strand breaks found in treated cells. This conclusion is based, in part, on data generated in the COMET assay by taking into consideration the decrease in replication rates of cells transfected with the ODN. Clearly, a higher number of breaks is observed in cells transfected with target-specific ODNs (see Fig. 4D).
Double-stranded DNA breakage is also observed as a result of ODN-promoted TGA activity. The response is dilutable and dose-dependent as breakage can be eliminated when the specific targeting ODN is replaced with nonspecific ODNs (Fig. 5). The unexpected result is that ODNs targeting either the non-transcribed or the transcribed strand induce DSBs at equal levels. Furthermore, an ODN designed to hybridize to the target site with perfect complementarity also induces dsDNA cleavage, yet only the 72NT ODN supports TGA activity in the HCT116 cell system. These data suggest that initial pairing of the ODN to the target site provides a substrate for DNA cleavage in a reaction mediated perhaps by enzymes involved in DNA replication or recombinational repair. We have shown that an ODN paired at the target site creates a three-stranded complex, which may evolve into a preferred substrate for nuclease cleavage [3, 40, 44]. In yeast, nucleolytic activity in the TGA reaction is promoted by Mre11 [8]. Striking similarities exist between the mechanism of TGA in yeast and mammalian cells as they are currently defined. Both require DNA pairing activities in the cell to form a common D-loop structure; both missense and frameshift mutations can be repaired with comparable efficiencies; and, TGA is regulated by both suppressor and enhancer protein activities in yeast that have functional homologs in mammalian cells [5, 7, 12, 45, 46]. So, it is likely that an Mre11-like function in HCT116 cells drives the DNA breakage, and we are currently determining the identity of the nuclease(s) catalyzing cleavage in mammalian cells.
The appearance of DNA breaks in reactions involved in nucleotide exchange, are not without precedent. Wang et al. [47] showed that ssODNs can produce specific deletions in plasmid and genomic DNA in mammalian cells. This activity was precise in nature since the readout was the generation of functional eGFP, but strand breaks undoubtedly took place. These results align with our data although the frequency of correction is low.
Chromosomal patch repair directed by ODNs relies on the creation of a DNA lesion at the site prior to the addition of the ODN [48-52]. ODNs then act to bridge the break site, essentially patching the lesion and providing a template for repair; RNA may also function in this type of repair reaction. Our data suggest that pre-existing DSBs at the site of correction are not a prerequisite for TGA and thus, in this case, breakage may follow or be coincident with the nucleotide exchange reaction.
Two issues remain to be addressed; is the break within or at the target site and why is ds breakage, stimulated by 72T and 72PM even though these two ODNs do not support detectable TGA in HCT116 cells? With regard to the site(s) of breakage, we are beginning a detailed study to map the location of any and all DSBs in genomic DNA. In response to the question of why breaks are seen in the absence of correction, our first hypothesis is based on the fact that the target site is transcriptionally active, and thus the D-loop structure could be viewed as a helix distortion by the cell's metabolic machinery. This structural aberration would thus engage TCR-related repair activities that involve nucleolytic cleavage or excision. If the transcribed strand is targeted, then the repair may be biased toward the strand in the helix rather than the invading strand (ODN). As such, 72T would be removed prior to directing a TGA event. In contrast, since 72NT is not directly impeding the movement of RNA polymerase on the transcribed strand, TGA may be take place prior to scission and resolution, assuming that TGA is not reliant on scission for nucleotide exchange. In yeast, ODNs designed to hybridize to the transcribed strand of the target site can be displaced by transcriptional activity and movement of RNA polymerase [29]. This may also account for the widely observed bias toward correction on the non-transcribed strand.
The data reported herein also enable us to develop a second hypothesis to explain our results. The creation of strand breaks may be a byproduct of the processing and/or resolution of a reaction intermediate, the stabilized D-loop. In one proposed mechanism of TGA, a sequence complementary ODN is aligned in homologous register with the target site [26]. Once bound stably, the ODN is incorporated into an actively growing replication module providing a primer for extension, akin to an Okazaki fragment [9]. As described above, the incorporation of an ODN into a replication bubble, specific for a particular complementary sequence of a replicating genome may induce fork stalling. This action could be a response to the presence of the D-loop or may be the result of an untethered double-stranded region near the fork. In either case, mechanical stress or nucleolytic attack leads to DNA cleavage at such chromosomal sites [19]. It is pertinent to point out that the lagging strand of the target site has been shown to be more amenable to TGA [53]. These observations are predictable if we can assume that ODN incorporation plays a major role in generating a repaired gene. Thus, the non-transcribed strand may also be the lagging strand; both T and NT ODNs pair at the site to enable breakage, but only the NT ODN incorporates producing a “corrected” sequence. Incorporation itself likely requires some strand breakage during strand assimilation, both of which can leave the “footprints” we observe. We cannot conclude that such breaks are irrefutably associated with successful correction but the evidence certainly points to what appears to be a link between genomic cleavage and TGA.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference List
- 1.Gamper HB, Parekh H, Rice MC, Bruner M, Youkey H, Kmiec EB. The DNA strand of chimeric RNA/DNA oligonucleotides can direct gene repair/conversion activity in mammalian and plant cell-free extracts. Nucleic Acids Res. 2000;28:4332–4339. doi: 10.1093/nar/28.21.4332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gamper HB, Cole-Strauss A, Metz R, Parekh H, Kumar R, Kmiec EB. A plausible mechanism for gene correction by chimeric oligonucleotides. Biochemistry. 2000;39:5808–5816. doi: 10.1021/bi9921891. [DOI] [PubMed] [Google Scholar]
- 3.Drury MD, Kmiec EB. DNA pairing is an important step in the process of targeted nucleotide exchange. Nucleic Acids Res. 2003;31:899–910. doi: 10.1093/nar/gkg171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Igoucheva O, Alexeev V, Pryce M, Yoon K. Transcription affects formation and processing of intermediates in oligonucleotide-mediated gene alteration. Nucleic Acids Res. 2003;31:2659–2670. doi: 10.1093/nar/gkg360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ferrara L, Parekh-Olmedo H, Kmiec E. Enhanced oligonucleotide-directed gene targeting in mammalian cells following treatment with DNA damaging agents. Exp Cell Res. 2004;300:170–179. doi: 10.1016/j.yexcr.2004.06.021. [DOI] [PubMed] [Google Scholar]
- 6.Parekh-Olmedo H, Ferrara L, Brachman E, Kmiec EB. Gene therapy progress and prospects: targeted gene repair. Gene Ther. 2005;12:639–646. doi: 10.1038/sj.gt.3302511. [DOI] [PubMed] [Google Scholar]
- 7.Liu L, Cheng S, van Brabant AJ, Kmiec EB. Rad51p and Rad54p, but not Rad52p, elevate gene repair in Saccharomyces cerevisiae directed by modified single-stranded oligonucleotide vectors. Nucleic Acids Res. 2002;30:2742–2750. doi: 10.1093/nar/gkf397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Liu L, Usher M, Hu Y, Kmiec EB. Nuclease activity of Saccharomyces cerevisiae Mre11 functions in targeted nucleotide alteration. Appl Environ Microbiol. 2003;69:6216–6224. doi: 10.1128/AEM.69.10.6216-6224.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Radecke S, Radecke F, Peter I, Schwarz K. Physical incorporation of a single-stranded oligodeoxynucleotide during targeted repair of a human chromosomal locus. J Gene Med. 2006;8:217–228. doi: 10.1002/jgm.828. [DOI] [PubMed] [Google Scholar]
- 10.Huen MS, Li XT, Lu LY, Watt RM, Liu DP, Huang JD. The involvement of replication in single stranded oligonucleotide-mediated gene repair. Nucleic Acids Res. 2006;34:6183–6194. doi: 10.1093/nar/gkl852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Engstrom JU, Kmiec EB. DNA Replication, cell cycle progression and the targeted gene repair reaction. Cell Cycle. 2008;7 doi: 10.4161/cc.7.10.5826. [DOI] [PubMed] [Google Scholar]
- 12.Ferrara L, Kmiec EB. Camptothecin enhances the frequency of oligonucleotide-directed gene repair in mammalian cells by inducing DNA damage and activating homologous recombination. Nucleic Acids Res. 2004;32:5239–5248. doi: 10.1093/nar/gkh822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Olsen PA, Randol M, Krauss S. Implications of cell cycle progression on functional sequence correction by short single-stranded DNA oligonucleotides. Gene Ther. 2005;12:546–551. doi: 10.1038/sj.gt.3302454. [DOI] [PubMed] [Google Scholar]
- 14.Olsen PA, Randol M, Luna L, Brown T, Krauss S. Genomic sequence correction by single-stranded DNA oligonucleotides: role of DNA synthesis and chemical modifications of the oligonucleotide ends. J Gene Med. 2005;7:1534–1544. doi: 10.1002/jgm.804. [DOI] [PubMed] [Google Scholar]
- 15.Wu XS, Xin L, Yin WX, Shang XY, Lu L, Watt RM, Cheah KS, Huang JD, Liu DP, Liang CC. Increased efficiency of oligonucleotide-mediated gene repair through slowing replication fork progression. Proc Natl Acad Sci U S A. 2005;102:2508–2513. doi: 10.1073/pnas.0406991102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Engstrom JU, Kmiec EB. Manipulation of cell cycle progression can counteract the apparent loss of correction frequency following oligonucleotide-directed gene repair. BMC Mol Biol. 2007;8:9. doi: 10.1186/1471-2199-8-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Brachman EE, Kmiec EB. Gene repair in mammalian cells is stimulated by the elongation of S phase and transient stalling of replication forks. DNA Repair (Amst) 2005;4:445–457. doi: 10.1016/j.dnarep.2004.11.007. [DOI] [PubMed] [Google Scholar]
- 18.Ferrara L, Kmiec EB. Targeted gene repair activates Chk1 and Chk2 and stalls replication in corrected cells. DNA Repair (Amst) 2006;5:422–431. doi: 10.1016/j.dnarep.2005.11.009. [DOI] [PubMed] [Google Scholar]
- 19.Lobachev KS, Rattray A, Narayanan V. Hairpin- and cruciform-mediated chromosome breakage: causes and consequences in eukaryotic cells. Front Biosci. 2007;12:4208–4220. doi: 10.2741/2381. [DOI] [PubMed] [Google Scholar]
- 20.Michel B, Ehrlich SD, Uzest M. DNA double-strand breaks caused by replication arrest. EMBO J. 1997;16:430–438. doi: 10.1093/emboj/16.2.430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Michel B. Replication fork arrest and DNA recombination. Trends Biochem Sci. 2000;25:173–178. doi: 10.1016/s0968-0004(00)01560-7. [DOI] [PubMed] [Google Scholar]
- 22.Seigneur M, Ehrlich SD, Michel B. RuvABC-dependent double-strand breaks in dnaBts mutants require recA. Mol Microbiol. 2000;38:565–574. doi: 10.1046/j.1365-2958.2000.02152.x. [DOI] [PubMed] [Google Scholar]
- 23.Saintigny Y, Delacote F, Vares G, Petitot F, Lambert S, Averbeck D, Lopez BS. Characterization of homologous recombination induced by replication inhibition in mammalian cells. EMBO J. 2001;20:3861–3870. doi: 10.1093/emboj/20.14.3861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hu Y, Parekh-Olmedo H, Drury M, Skogen M, Kmiec EB. Reaction parameters of targeted gene repair in Mammalian cells. Mol Biotechnol. 2005;29:197–210. doi: 10.1385/MB:29:3:197. [DOI] [PubMed] [Google Scholar]
- 25.Santos JH, Meyer JN, Mandavilli BS, Van HB. Quantitative PCR-based measurement of nuclear and mitochondrial DNA damage and repair in mammalian cells. Methods Mol Biol. 2006;314:183–199. doi: 10.1385/1-59259-973-7:183. [DOI] [PubMed] [Google Scholar]
- 26.Parekh-Olmedo H, Kmiec EB. Progress and prospects: targeted gene alteration (TGA) Gene Ther. 2007;14:1675–1680. doi: 10.1038/sj.gt.3303053. [DOI] [PubMed] [Google Scholar]
- 27.Engstrom JU, Suzuki T, Kmiec EB. Regulation of targeted gene repair by intrinsic cellular processes. Bioessays. 2009;31:159–168. doi: 10.1002/bies.200800119. [DOI] [PubMed] [Google Scholar]
- 28.Bertoni C, Morris GE, Rando TA. Strand bias in oligonucleotide-mediated dystrophin gene editing. Hum Mol Genet. 2005;14:221–233. doi: 10.1093/hmg/ddi020. [DOI] [PubMed] [Google Scholar]
- 29.Liu L, Rice MC, Drury M, Cheng S, Gamper H, Kmiec EB. Strand bias in targeted gene repair is influenced by transcriptional activity. Mol Cell Biol. 2002;22:3852–3863. doi: 10.1128/MCB.22.11.3852-3863.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sorensen CB, Krogsdam AM, Andersen MS, Kristiansen K, Bolund L, Jensen TG. Site-specific strand bias in gene correction using single-stranded oligonucleotides. J Mol Med. 2005;83:39–49. doi: 10.1007/s00109-004-0592-6. [DOI] [PubMed] [Google Scholar]
- 31.Downey M, Durocher D. gammaH2AX as a checkpoint maintenance signal. Cell Cycle. 2006;5:1376–1381. doi: 10.4161/cc.5.13.2899. [DOI] [PubMed] [Google Scholar]
- 32.Burma S, Chen BP, Murphy M, Kurimasa A, Chen DJ. ATM phosphorylates histone H2AX in response to DNA double-strand breaks. J Biol Chem. 2001;276:42462–42467. doi: 10.1074/jbc.C100466200. [DOI] [PubMed] [Google Scholar]
- 33.Paull TT, Rogakou EP, Yamazaki V, Kirchgessner CU, Gellert M, Bonner WM. A critical role for histone H2AX in recruitment of repair factors to nuclear foci after DNA damage. Curr Biol. 2000;10:886–895. doi: 10.1016/s0960-9822(00)00610-2. [DOI] [PubMed] [Google Scholar]
- 34.Olsen PA, McKeen C, Krauss S. Branched oligonucleotides induce in vivo gene conversion of a mutated EGFP reporter. Gene Ther. 2003;10:1830–1840. doi: 10.1038/sj.gt.3302079. [DOI] [PubMed] [Google Scholar]
- 35.Ferrara L, Engstrom JU, Schwartz T, Parekh-Olmedo H, Kmiec EB. Recovery of cell cycle delay following targeted gene repair by oligonucleotides. DNA Repair (Amst) 2007;6:1529–1535. doi: 10.1016/j.dnarep.2007.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Maguire KK, Kmiec EB. Enhancement of in vivo targeted nucleotide exchange by nonspecific carrier DNA. Methods Mol Biol. 2004;262:209–219. doi: 10.1385/1-59259-761-0:209. [DOI] [PubMed] [Google Scholar]
- 37.Shang XY, Hao DL, Wu XS, Yin WX, Guo ZC, Liu DP, Liang CC. Improvement of SSO-mediated gene repair efficiency by nonspecific oligonucleotides. Biochem Biophys Res Commun. 2008;376:74–79. doi: 10.1016/j.bbrc.2008.08.119. [DOI] [PubMed] [Google Scholar]
- 38.Schwartz T, Kmiec EB. Using methyl methanesulfonate (MMS) to stimulate targeting gene repair activity in mammalian cells. Gene Therapy and Molecular Biology. 2005;9:193–202. [Google Scholar]
- 39.Radecke F, Peter I, Radecke S, Gellhaus K, Schwarz K, Cathomen T. Targeted chromosomal gene modification in human cells by single-stranded oligodeoxynucleotides in the presence of a DNA double-strand break. Mol Ther. 2006;14:798–808. doi: 10.1016/j.ymthe.2006.06.008. [DOI] [PubMed] [Google Scholar]
- 40.Drury MD, Kmiec EB. Double displacement loops (double d-loops) are templates for oligonucleotide-directed mutagenesis and gene repair. Oligonucleotides. 2004;14:274–286. doi: 10.1089/oli.2004.14.274. [DOI] [PubMed] [Google Scholar]
- 41.Drury MD, Skogen MJ, Kmiec EB. A tolerance of DNA heterology in the mammalian targeted gene repair reaction. Oligonucleotides. 2005;15:155–171. doi: 10.1089/oli.2005.15.155. [DOI] [PubMed] [Google Scholar]
- 42.Maguire KK, Kmiec EB. Multiple roles for MSH2 in the repair of a deletion mutation directed by modified single-stranded oligonucleotides. Gene. 2007;386:107–114. doi: 10.1016/j.gene.2006.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Huen MS, Lu LY, Liu DP, Huang JD. Active transcription promotes single-stranded oligonucleotide mediated gene repair. Biochem Biophys Res Commun. 2007;353:33–39. doi: 10.1016/j.bbrc.2006.11.146. [DOI] [PubMed] [Google Scholar]
- 44.Parekh-Olmedo H, Drury M, Kmiec EB. Targeted nucleotide exchange in Saccharomyces cerevisiae directed by short oligonucleotides containing locked nucleic acids. Chem Biol. 2002;9:1073–1084. doi: 10.1016/s1074-5521(02)00236-3. [DOI] [PubMed] [Google Scholar]
- 45.Liu L, Kmiec EB. Engineered Saccharomyces cerevisiae Rad51 increases the frequency of targeted nucleotide exchange and gene repair in vivo. Proc Natl Acad Sci U S A. 2003 [Google Scholar]
- 46.Liu L, Maguire KK, Kmiec EB. Genetic re-engineering of Saccharomyces cerevisiae RAD51 leads to a significant increase in the frequency of gene repair in vivo. Nucleic Acids Res. 2004;32:2093–2101. doi: 10.1093/nar/gkh506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang Z, Zhou ZJ, Liu DP, Huang JD. Double-stranded break can be repaired by single-stranded oligonucleotides via the ATM/ATR pathway in mammalian cells. Oligonucleotides. 2008;18:21–32. doi: 10.1089/oli.2007.0093. [DOI] [PubMed] [Google Scholar]
- 48.Storici F, Durham CL, Gordenin DA, Resnick MA. Chromosomal site-specific double-strand breaks are efficiently targeted for repair by oligonucleotides in yeast. Proc Natl Acad Sci U S A. 2003;100:14994–14999. doi: 10.1073/pnas.2036296100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Storici F, Snipe JR, Chan GK, Gordenin DA, Resnick MA. Conservative repair of a chromosomal double-strand break by single-strand DNA through two steps of annealing. Mol Cell Biol. 2006;26:7645–7657. doi: 10.1128/MCB.00672-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Storici F, Resnick MA. The delitto perfetto approach to in vivo site-directed mutagenesis and chromosome rearrangements with synthetic oligonucleotides in yeast. Methods Enzymol. 2006;409:329–345. doi: 10.1016/S0076-6879(05)09019-1. [DOI] [PubMed] [Google Scholar]
- 51.Morrish TA, Gilbert N, Myers JS, Vincent BJ, Stamato TD, Taccioli GE, Batzer MA, Moran JV. DNA repair mediated by endonuclease-independent LINE-1 retrotransposition. Nat Genet. 2002;31:159–165. doi: 10.1038/ng898. [DOI] [PubMed] [Google Scholar]
- 52.Moore JK, Haber JE. Capture of retrotransposon DNA at the sites of chromosomal double-strand breaks. Nature. 1996;383:644–646. doi: 10.1038/383644a0. [DOI] [PubMed] [Google Scholar]
- 53.Brachman EE, Kmiec EB. DNA replication and transcription direct a DNA strand bias in the process of targeted gene repair in mammalian cells. J Cell Sci. 2004;117:3867–3874. doi: 10.1242/jcs.01250. [DOI] [PubMed] [Google Scholar]