

Abstract
Molecular chaperones and heat shock proteins (Hsp) have emerged as critical regulators of proteins associated with neurodegenerative disease pathologies. The very nature of the chaperone system, which is to maintain protein quality control, means that most nascent proteins come in contact with chaperone proteins. Thus, amyloid precursor protein (APP), members of the gamma-secretase complex (presenilin 1 [PS1] collectively), the microtubule-associated protein tau (MAPT) as well as a number of neuroinflammatory components are all in contact with chaperones from the moment of their production. Chaperones are often grouped together as one machine presenting abnormal or mutant proteins to the proteasome for degradation, but this is not at all the case. In fact, the chaperone family consists of more than 100 proteins in mammalian cells, and the primary role for most of these proteins is to protect clients following synthesis and during stress; only as a last resort do they facilitate protein degradation. To the best of our current knowledge, the chaperone system in eukaryotic cells revolves around the ATPase activities of Hsp70 and Hsp90, the two primary chaperone scaffolds. Other chaperones and co-chaperones manipulate the ATPase activities of Hsp70 and Hsp90, facilitating either folding of the client or its degradation. In the case of Alzheimer's disease (AD), a number of studies have recently emerged describing the impact that these chaperones have on the proteotoxic effects of tau and amyloid-β accumulation. Here, we present the current understandings of chaperone biology and examine the literature investigating these proteins in the context of AD.
Keywords: Alzheimer's disease, chaperones, heat shock proteins, tau, degradation, protein misfolding
Introduction
Genetic analyses of families presenting with Alzheimer's dementia revealed that mutations in the amyloid precursor protein (APP) and presenilin 1 (PS1) protein were the cause of the disease for the affected kindred [1–7]. Overexpression of these mutant genes in transgenic mice showed that they enhance production of a 42-amino acid N-terminal peptide from APP, which enters the extracellular milieu [8–13]. This Aβ peptide has amyloidogenic properties, forming multimers in a β-sheet structure [14–17]. As enough of this material is produced, amyloid plaques form spontaneously and remain quite stable over time [18, 19]. Amyloidosis around the vasculature is also found in Alzheimer's disease (AD), and these plaques have been shown to grow more gradually with time [20]. This plaque production facilitates gliosis, and the role of microglia and astrocytes in AD is an area of intense investigation [21–25]. Microglia or macrophages become activated and can phagocytose Aβ[26, 27]; however, more current work suggests that multiple activation states are found in the brain, and it may be possible to manipulate these states to promote Aβ clearance [28–33]. Although amyloid plaques are the pathological end-point of aberrant Aβ production, a large number of studies have emerged suggesting that oligomeric forms of Aβ are the most pathogenic species [34–38]. A recent study demonstrated that dimeric Aβ species isolated from AD brain tissue impair synaptic plasticity and memory function when injected into normal rats [39].
Although sporadic AD follows the same pathological roadmap as the familial cases, a definitive genetic cause has not been established [40, 41]. However, polymorphisms in the APOE (apolipoprotein E) gene have been identified as significant risk factors for late-onset AD (LOAD) [42]. The function of ApoE in AD has been extensively reviewed [43]. Briefly, this protein is produced by microglia and astrocytes in the brain and has a major impact on scavenging of the Aβ peptide from the extracellular space, fostering the clearance of amyloid [44, 45]. The mutations in APOE linked to LOAD reduce the efficiency with which ApoE clears the Aβ peptide, facilitating amyloid accumulation and disease progression [46, 47].
Unfortunately, despite all of these data describing how Aβ is produced and which forms of Aβ are most toxic, recent work using therapies to reduce Aβ burden have met with limited success in the clinic. The immunotherapeutic strategy spearheaded by Elan/Wyeth Pharmaceuticals (Athlone, Ireland) has revealed that reducing amyloid burden does not halt the disease process, but rather slows the decline, and it carries with it a significant risk of cerebral haemorrhagic events [48–52]. In addition, clinical trials carried out by Myriad Pharmaceuticals (Salt Lake City, UT, USA) using the gamma-secretase modulator, flurizan, a drug which potently inhibited the PS1 complex to prevent Aβ 1–42 production in pre-clinical testing, was entirely ineffective at preventing cognitive decline in AD patients enrolled in a phase III clinical trial [53]. The optimistic and hopefully accurate interpretation of these results is that the pharmacological properties of these drugs were insufficient to achieve relevant concentrations at the target site. These trials may also suggest that perhaps the mild-to-moderate patient subsets selected for these studies were already at a stage in the disease process where anti-amyloid therapies may be less effective. Regardless, alternative therapeutic strategies to stop disease progression and even reverse the clinical phenotype should be explored.
Another major pathological component of AD is intracellular aggregation of the tau protein into tangles. Several recent studies have suggested that although Aβ may trigger this dysfunction of the tau protein, ultimately it becomes self-perpetuating [54, 55]. Tau pathology is found in a number of neurodegenerative diseases [56–59], and this generality decreased the enthusiasm for researching modifiers of tau, since it was perceived that tau accumulation was simply a result of any neurotoxic insult. This perspective changed when mutations in the microtubule-associated protein tau (MAPT)/tau gene were found to cause disease in affected individuals from families with a history of fronto-temporal dementia (FTD) [60–62]. The identification of these genetic variants proved that tau was capable of causing neurotoxicity; that it was not simply a resulting pathology of neurodegeneration. Since then, tangle pathology has been found to most closely correlate with neuron loss and cognitive deficits. Transgenic mice expressing these tau mutants have shown significant neuron loss [55, 63]. Very recently, the first clinical trial with a putative tau aggregation inhibitor significantly slowed cognitive decline in AD patients and tau pathology was reduced [64]. These data suggest that targeting the tau protein in patients already presenting with clinical symptoms of AD may be a more effective strategy than anti-Aβ therapies.
Tau mRNA can be alternatively spliced into six distinct isoforms and undergoes multiple post-translational modifications [65–71]. Since it was recently shown that mice transgenic for mutant APP lacked cognitive deficits when the tau protein was deleted [72], pinning down the interface where Aβ triggers tau dysfunction has become a major focus of current research efforts. The most commonly studied tau modification is phosphorylation, and a number of drug-screening efforts have focused on inhibiting the kinases most often associated with this process, namely GSK3β and CdK5 [73, 74]. But in addition to phosphorylation, other aspects of tau biology include folding [75], nitration [76], O-linked glycosylation, ubiquitination [77] and protease cleavage [78]. These modifications can alter tau's normal function of microtubule (MT) stabilisation; however, they likely contribute to its pathobiology as well. Recent efforts by our group and others have focused on trying to remove those particular tau species that are thought to be disease-related, preserving the normal tau to allow proper MT stabilisation. We have found that facilitating the clearance of these species may be achieved by manipulating the chaperone complex, the cell's protein quality control system [77, 79–81]. Since then, chaperone biology has emerged as an area of intense investigation with regard to AD research. Therefore, the remainder of this review will focus on the role that chaperones have in the biology and pathobiology of AD-related proteins.
Chaperones: the basics
The term ‘chaperone’ is defined as a guide whose purpose is to ensure propriety or restrict activity. In all cells, there are a group of proteins that have been dubbed chaperones because they carry out a very similar function; they make sure that other proteins arrive safely and are functional at their destination within the cell; however, these cellular chaperones also ensure that proteins deemed to be ‘ill-behaved’ or abnormal are destroyed. These unique properties of cellular chaperones bring them into intimate contact with many ‘client’ proteins within cells, including those associated with AD, such as APP, PS1 and tau (for a description of all abbreviations related to chaperones, see Table 1).
1.
Abbreviations
| Acronym | Protein name |
|---|---|
| AD | Alzheimer's disease |
| ApoE | Apolipoprotein E |
| APP | Amyloid-beta precursor protein |
| Aβ | Amyloid-beta |
| Bag1 | BCL2-associated athanogene |
| BiP/Grp78 | ER isoform of heat shock protein 70 |
| CDC37 | Cell division cycle 37 kD protein |
| CDK5 | Cyclin-dependent kinase 5 |
| CHIP | Carboxy-terminus of Hsc70-interacting protein |
| DNAJ | see Hsp40 |
| FTD | Frontal temporal dementia |
| GAPDH | Glyceraldehyde phosphate dehydrogenase |
| Grp94 | ER isoform of heat shock protein 90 |
| GSK3β | Glycogen synthase kinase 3 beta |
| Hsc70 | Heat shock cognate 70 kD protein 8 |
| HSE | Heat shock element |
| HSF1 | Heat shock factor 1 |
| Hsp | Heat shock protein |
| Hsp22 | Heat shock protein 22 |
| Hsp27 | Heat shock protein 27 |
| Hsp40 | Heat shock protein 40 |
| Hsp70 | Heat shock protein 70 |
| Hsp90 | Heat shock protein 90 |
| HtrA2 | Putative serine peptidase |
| MAPK | Mitogen-activated protein kinase |
| MAPT | Microtubule-associated protein tau |
| MARK2 (PAR1) | Microtubule affinity-regulating kinase 2 |
| MT | Microtubule |
| NFTs | Neurofibrillary tangles |
| P38 | See SAPK |
| PS1 | Presenilin 1 |
| SAPK | Stress-activated protein kinase |
| TPR | Tetratricopeptide repeat |
| UPR | Unfolded protein response |
Hsp90, Hsp70 and CHIP
Different chaperones work together to form cellular machines to differentially regulate the function of these client proteins by manipulating the ATPase activity of the two main chaperone scaffolds, Hsp70 and Hsp90. These interactions are mainly mediated by a tetratricopeptide domain that tightly binds with the C-terminal EEVD domains present on Hsp70 and Hsp90. A critical TPR-containing chaperone is carboxy-terminus of Hsc70-interacting protein (CHIP), a highly conserved ubiquitin ligase that is critical for quality control and stress recovery systems in most cell types. CHIP mitigates stress-related proteins after a cellular stress response [77, 82] and has anti-apoptotic properties.
Substrate processing
The basic pathway established for most client proteins upon synthesis is recognition by an Hsp40 variant, which then associates with Hsp70. The client is then passed to Hsp70 and either enters an Hsp70-exclusive cycle or, in coordination with Hsp70/Hsp90-organising protein (Hop), passes the client to Hsp90. Hsp90 has the unique ability to maintain mutated proteins in a folded and partially active state at the expense of ATP, permitting proteins that would otherwise be immediately targeted for degradation to persist within the cell.
Client degradation versus folding
Small molecules aptly named Hsp90 inhibitors have been generated and functionally force these clients to be degraded through the proteasome [83–85]. CHIP is a critical component for this degradation, and since it binds with both Hsp70 and Hsp90, it has an opportunity to interact with and ubiquitinate a number of proteins via their scaffold. Conversely, other chaperones and/or co-chaperones that are part of the complex at the time of the client–Hsp90 interaction can facilitate the ATPase activity of Hsp90, protecting these clients from degradation [79, 80]. For example, tau degradation was prevented by suppressing CHIP or Hsp90, but was enhanced by suppressing co-chaperones that promote re-folding or maintenance of Hsp90 clients, such as prostaglandin E synthase 3 (P23) or Cdc37 [80]. Hsp90 inhibitors also lead to increased activation of heat shock factor (HSF).
Chaperone expression
Hsp90 normally inhibits HSF1 by tethering it to the cytosol, preventing it from translocating to the nucleus. When Hsp90 levels are depleted by Hsp90 inhibition, HSF1 is released. Once it becomes phosphorylated, it trimerises and is able to enter the nucleus and begin transcribing genes containing heat shock elements (HSE), such as Hsp70 and Hsp27. In humans, there are 4 HSF genes, 6 Hsp90 genes, 13 Hsp70 genes and 41 Hs40/DNAJ genes, along with 11 small Hsps (for an extensive review on this topic, see Reference 86). In addition to this, there are a number of other known interacting proteins, such as P23, CHIP and Cdc37.
Small Hsps
Small Hsps such as Hsp22 and Hsp27 participate in the chaperone network in a unique way. These proteins are composed of two domains: a phosphorylation domain and a crystallin domain. Under normal conditions, cellular depots of these proteins are preserved as aggregates; however, upon stress, these aggregates break down into dimers and tetramers following phosphorylation by stress-activated protein kinases (SAPK/P38 MAPK) [87]. These smaller oligomers show enhanced chaperoning activity and can process proteins to the proteasome in an ATP-independent manner. Small Hsps are also newly transcribed upon stress, perhaps in anticipation of restoring the dormant stockpile following the stress event. Their chaperoning function relies on assistance from other chaperones, the complement of which is unknown. There is also evidence for extracellular localisation of Hsps, which may play an important part in immune signalling and epineuronal protein aggregation, that is, amyloid plaques [88]. Thus, this growing complexity of the chaperone network introduces an entirely new class of potential therapeutic targets, making the necessity for drawing their connection to AD pathogenesis of utmost importance.
Chaperone regulation in AD
Involvement of chaperones in the pathogenesis of AD was first proposed following promoter analysis of the APP gene, where an HSE was found within its promoter [89]. Immunohistochemical studies and expression analyses in AD brain tissue showed that expression levels of a number of Hsps, particularly Hsp27 and Hsp70, were elevated in affected regions from AD brain tissue, and this elevation appears to be a hybridisation of activated glia and dysregulated/stressed neurons (Fig. 1A and B) [90–96]. Since then, pathological structures typical of AD have been shown to harbour other chaperone proteins, such as CHIP [97] and Parkin [98]. These findings have served as the foundation for a number of mechanistic studies regarding the impact of chaperones and stress on the contributors to AD pathogenesis.
1.
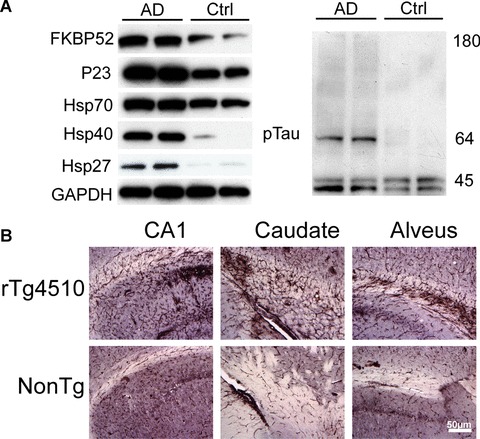
Heat shock proteins and chaperones are elevated in Alzheimer's brain. (A) Brain tissue from medial temporal gyrus of two Alzheimer's disease (AD) patients and two age/gender-matched controls were homogenised and analysed by Western blot. Chaperone proteins were dramatically elevated in AD brain compared with control. ptau antibody recognising tau phosphorylated at pS212 was used to confirm pathology. GAPDH levels were unchanged. (B) Brain sections from rTg4510 transgenic mice that have inducible P301L human tau expression in the forebrain and non-transgenic littermates were stained with an anti-Hsp27 antibody. Dramatic gliosis was evident throughout the CA1 and caudate putamen and along the alveus in transgenic mice compared with control.
Chaperone involvement in APP, presenilins and amyloid processing
APP is a membrane-associated protein and, as such, is processed through the endoplasmic reticulum (ER) and Golgi complex [99]. The ER chaperone BiP/Grp78 (the ER isoform of Hsp70) associates with APP, likely indicating that Grp94 (the ER isoform of Hsp90) can regulate APP as well [100]. Grp78 is now known to reduce amyloid production, further belying the importance of the ER chaperone system in AD [101]. CHIP also interacts with APP, and this is restricted to the ER and Golgi complex [102]. Another report suggests that the stress-activated protease, HtrA2, can regulate APP degradation at the ER, and when this protease is deleted, APP and consequently Aβ levels are elevated [103].
Although the ER chaperone family regulates APP processing, the cytosolic complement of chaperones, which includes the stress-inducible family, has also been linked to APP and amyloid biology [104]. Several reports demonstrate that small Hsps such as Hsp22 and Hsp27 bind to fibrillar amyloid plaques and actually inhibit their fibrillarisation [105]. Overexpression of the Caenorhabditis elegans small Hsp16.2 protected these organisms from Aβ-induced toxicity [106]. Interestingly, the critical pro-inflammatory cytokines, interleukin-1 and tumour necrosis factor-α, facilitate the phosphorylation of small Hsps, perhaps suggesting that inflammation increases the Hsp27 chaperoning function [107].
In addition to small Hsps, cytosolic Hsp70 and Hsp90 were shown to inhibit early stages of amyloid aggregation [108]. Hsp70 was able to protect against intracellular Aβ as well [109]. More recently, administration of Hsp90 inhibitors to primary neurons prevented Aβ-induced neurotoxicity, perhaps by increasing levels of Hsp70 and Hsp90 [110]; however, it is also possible that this improved toxicity profile is due to reductions in aberrant tau by Hsp90 inhibition [80], particularly in light of evidence demonstrating that tau knockout primary neurons are less susceptible to Aβ-induced toxicity [111]. Another critical regulator for all of these proteins is HSF1. While HSF1 transcribes de novo Hsp22, Hsp27 and variants of Hsp40, Hsp70 and Hsp90, it also induces the APP gene during stress [112–114]. In a C. elegans model of accumulation, HSF1 was shown to mediate the disaggregation of Aβ fibrils [115]. Thus, there appears to be multiple outcomes from HSF activation with regard to Aβ accumulation that may ultimately lead to a higher availability of toxic oligomeric intermediates; APP expression is elevated, leading to increases in both intra- and extracellular Aβ, amyloid plaques disaggregate in response to inductions in Hsps, and APP trafficking is enhanced due to Hsp expression (Fig. 2).
2.
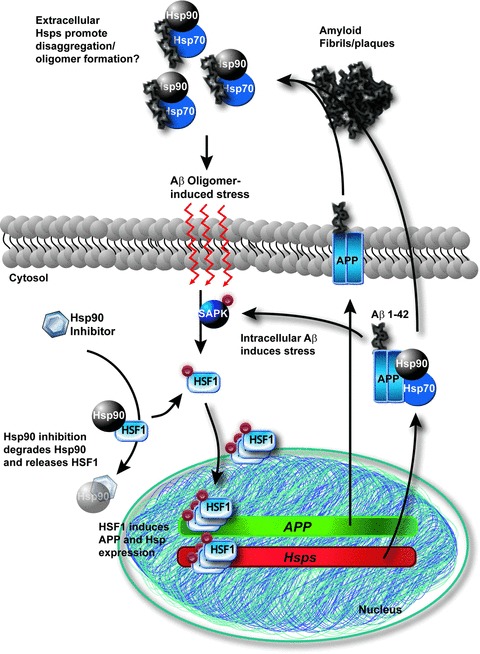
Role of chaperones and HSF1 in the processing of Aβ: a cycle of toxic soluble intermediates? Following a stress stimulus, HSF1 is phosphorylated by the stress-activated protein kinase cascade in the cytosol. HSF1 free of Hsp90 binding then trimerises and enters the nucleus, producing de novo copies of heat shock protein and APP mRNA. The HSPS shepherd APP to its destination and more Aβ is released. Extracellular chaperones prevent amyloid plaque formation and preserve soluble toxic amyloid species 90 (oligomers), leading to a stress loop. Intracellular Aβ may facilitate this stress and even further it. Hsp90 inhibition may compound the problem by indirectly allowing more HSF1 to become active.
PS1 resides within the cellular membrane and is a central component of the gamma-secretase complex that cleaves the Aβ peptide from APP. Although there is no pathologic accumulation of the PS1 protein in AD, the mutations found in this gene clearly implicate it in the disease process. PS1 has been predominantly found in the ER and can be phosphorylated. PS1 was shown to associate with an unknown protein containing TPR domains, suggesting for the first time that the PS1 may be regulated by the chaperone network. There have been several conflicting reports suggesting that presenilins actually trigger the unfolded protein response (UPR) in the ER, a process that leads to chaperone induction. The UPR is a process that is activated when unfolded or misfolded proteins begin accumulating in the ER lumen. Initially, chaperone protein transcription is increased in an effort to restore homeostasis. When this fails, translation is suppressed and general transcription is decreased. When all of these processes fail to rescue the cell, apoptosis is initiated. Intially, mutant PS1 was shown to down-regulate the UPR in the ER [116]; however, later reports suggested that presenilins were not tied to the UPR process [117]. Although this conflict is yet to be resolved, it is now known that PS1 forms aggresomes within the ER upon heat shock stress [118]. The functional consequence of these aggresomes remains unclear.
Chaperone regulation of the MAPT
The majority of strides that have been taken with regard to the role of chaperones in AD have revolved around the tau protein and its aberrant intracellular accumulation. Tau is thought to be an inherently unfolded protein that promotes MT polymerisation and stability. Once tau aggregation is induced by hyperphosphorylation, folding and cleavage are thought to follow [75, 119, 120]. These post-translational modifications can impact the interaction of tau with MTs, and thus there may be specific forms of tau that are preferred chaperone substrates relative to others. It was recently reported that chaperone proteins, including Hsp27, HSP70 and CHIP, can recognise abnormal tau and reduce its concentration by facilitating its degradation and dephosphorylation [97, 121, 122]. Hsp27 preferentially binds to hyperphosphorylated tau as well as paired helical filamentous tau but not to non-phosphorylated tau [122]. The expression of another small Hsp, alphaB-crystallin has been found in glial inclusions of tauopathies [123]. Hsp27 also increases tau phosphorylation at Ser262, whereas alphaB-crystallin decreases phosphorylated tau and GSK-3β levels [124]. Interestingly, Hsp27 is cross-linked with tau in NFTs from AD brain [98]. Moreover, positive correlations have been found in the soluble protein levels from AD brain tissue between tau and molecular chaperones including Hsp27, Hsp40, Hsp90, alphaB-crystallin and CHIP [125]. Conversely, the levels of HSPs were inversely correlated with the levels of granulated tau oligomers, an intermediate of tau filaments. In a separate study, increased levels of Hsp70 and Hsp90 were found to promote tau solubility and MT binding in various cellular models [121]. Subsequent studies show that tau binds directly to Hsp70, and Bag1 has a role in this interaction [126, 127]. Taken together, these findings suggest that chaperones are necessary to maintain tau in a non-aggregated state, a consequence that may ultimately be deleterious for the brain.
Other studies have provided new insights into the mechanisms employed by the chaperone network to handle abnormally accumulating tau. Initial studies from our group showed that pharmacologic inhibition of Hsp90 significantly reduced intracellular levels of the disease-associated phosphorylated tau species pS202/T205 and pS396/S404 [128]. These Hsp90 inhibitors primarily facilitated the clearance of phospho-tau via proteasomal degradation, although lysosomal clearance also seemed to play a more minor role [79]. We also found that these inhibitors could reduce tau in transgenic tau mice. However, perhaps, more importantly, we identified a novel mechanism demonstrating that there are two pathways that can lead to opposing outcomes for tau biology with regard to Hsp90; some chaperones preserve tau, whereas others promote its degradation [80]. Furthermore, mutant, but not wild-type (WT) tau, is maintained in tauopathies by Hsp90; and the inhibition of Hsp90 leads to reductions in the pathogenicity of these mutant species [129].
CHIP has also been found to be critical for tau degradation. CHIP is found in tau lesions from AD and Pick's disease by immunohistochemistry [130]. CHIP also binds to and ubiquitinates tau directly, and this association is facilitated by Hsp70 [97]. This ubiquitination process preferentially occurs on exon 10 (+) tau, the material that abnormally accumulates in a number of tauopathies [131]. Conversely, mice deficient in CHIP had significant accumulation of hyperphosphorylated tau species that did not aggregate and were ubiquitin-negative [77]. Biochemical analyses showed increased levels of CHIP and Hsp70 in AD, and CHIP levels were inversely proportional to sarkosyl-insoluble tau [132]. We also demonstrated that CHIP is the critical mediator of Hsp90 inhibitor-mediated tau reductions [80]. In aggregate, these studies suggest that ubiquitination may be critical for insoluble tau filament formation, and any clearance mechanisms of tau may be dependent on CHIP.
As these chaperone machines survey the cell, they come in contact with many different proteins, and we speculated as to whether these scaffolds might serve as a nexus for modifying proteins to act on tau. Akt, an oncogenic master kinase that can phosphorylate tau, is also elevated in AD [133]. We found that Akt is also a client of CHIP, and that this interaction is highly dependent on Hsp90 [81]. Akt prevented tau ubiquitination and subsequent degradation by regulating the Hsp90/CHIP complex directly, by competing with tau as a client, or by modifying tau in such a way as to make it a less favourable substrate for the Hsp90–CHIP complex. We also found that Akt can regulate CHIP expression levels and that it has a functional interaction with the MT affinity-regulating kinase 2 (PAR1/MARK2). Akt enhances the activity of PAR1, to promote phosphorylation of tau at S262/S356, a phospho-tau species that is not recognised by the Hsp90/CHIP complex [79]. These data suggest that kinases must be considered as part of the chaperone machinery, perhaps using the Hsp90 scaffold as a tether to interact with potential substrates. This could help to explain the promiscuity of kinases that may lack specific binding domains for each substrate but use Hsp90 or Hsp70 to facilitate the interaction. A model demonstrating our current understanding of tau processing through the Hsp70/Hsp90 network is shown in Fig. 3.
3.
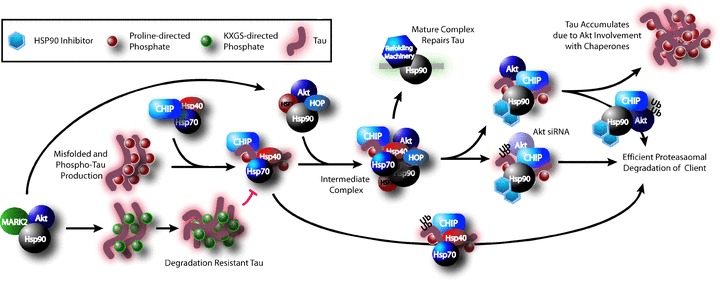
Current model of tau processing through the chaperone network. Kinases such as MARK2 and Akt appear to operate in concert with Hsp90 to affect phosphorylation. MARK2 and Akt synergise to phosphorylate tau at KXGS motifs, which prevents tau degradation by the chaperone system. Other aberrant or misfolded tau is recognised by the Hsp40/Hsp70 complex. From here, tau can be either directly degraded by this complex or passed to Hsp90 to form an intermediate Hsp90 complex. Akt is associated with this complex. Here several courses can be taken based on the context: The Hsp90 complex can mature into a refolding machine and repair tau, or tau degradation can be facilitated, either out of necessity or Hsp90 inhibition. When Akt is present, tau degradation is impaired and thus it accumulates; however, when Akt is reduced by siRNA, tau degradation is enhanced. Akt is also a client of Hsp90 and thus is degraded in a fashion similar to tau.
Conclusions
The field of chaperone biology continues to gain momentum because of the critical role that these proteins appear to be playing in a number of diseases. The intricacies of these proteinaceous machines are just beginning to be imagined. For AD, chaperones represent an entirely new aspect of disease biology, an aspect that may provide a common link to a great number of neurodegenerative disorders. However, within this complex system likely lies at least one specific therapeutic target that is yet to be identified for each of these diseases. By gaining insights into the mechanisms of chaperone biology, there is no doubt that these targets will come to light and a new generation of AD therapeutics will emerge.
References
- 1.Mullan M, Houlden H, Windelspecht M, Fidani L, Lombardi C, Diaz P, Rossor M, Crook R, Hardy J, Duff K, Crawford F. A locus for familial early-onset Alzheimer's disease on the long arm of chromosome 14, proximal to the alpha 1-antichy-motrypsin gene. Nat Genet. 1992;2:340–2. doi: 10.1038/ng1292-340. [DOI] [PubMed] [Google Scholar]
- 2.Van Broeckhoven C, Haan J, Bakker E, Hardy JA, Van Hul W, Wehnert A, Vegter-Van der Vlis M, Roos RA. Amyloid beta protein precursor gene and hereditary cerebral hemorrhage with amyloidosis (Dutch) Science. 1990;248:1120–2. doi: 10.1126/science.1971458. [DOI] [PubMed] [Google Scholar]
- 3.Goate A, Chartier-Harlin MC, Mullan M, Brown J, Crawford F, Fidani L, Giuffra L, Haynes A, Irving N, James L, Mant R, Newton P, Rooke K, Roques P, Talbot C, Pericak-Vance M, Roses A, Williamson R, Rossor M, Owen M, Hardy J. Segregation of a missense mutation in the amyloid precursor protein gene with familial Alzheimer's disease. Nature. 1991;349:704–6. doi: 10.1038/349704a0. [DOI] [PubMed] [Google Scholar]
- 4.Chartier-Harlin MC, Crawford F, Houlden H, Warren A, Hughes D, Fidani L, Goate A, Rossor M, Roques P, Hardy J, Mullan M. Early-onset Alzheimer's disease caused by mutations at codon 717 of the beta-amyloid precursor protein gene. Nature. 1991;353:844–6. doi: 10.1038/353844a0. [DOI] [PubMed] [Google Scholar]
- 5.Campion D, Flaman JM, Brice A, Hannequin D, Dubois B, Martin C, Moreau V, Charbonnier F, Didierjean O, Tardieu S, Penet C, Puel M, Pasquier F, Le Doze F, Bellis G, Calenda A, Heiolig R, Martinez M, Mallet J, Bellis M, Clerget-Darpoux F, Agid Y, Trebourg T. Mutations of the presenilin I gene in families with early-onset Alzheimer's disease. Hum Mol Genet. 1995;4:2373–7. doi: 10.1093/hmg/4.12.2373. [DOI] [PubMed] [Google Scholar]
- 6.Perez-Tur J, Croxton R, Wright K, Phillips H, Zehr C, Crook R, Hutton M, Hardy J, Karran E, Roberts GW, Lancaster S, Haltia T. A further presenilin 1 mutation in the exon 8 cluster in familial Alzheimer's disease. Neurodegeneration. 1996;5:207–12. doi: 10.1006/neur.1996.0028. [DOI] [PubMed] [Google Scholar]
- 7.Sherrington R, Rogaev EI, Liang Y, Rogaeva EA, Levesque G, Ikeda M, Chi H, Lin C, Li G, Holman K, Tsuda T, Mar L, Foncin J-F, Bruni AC, Montesi MP, Sorbi S, Rainero I, Pinessi L, Nee L, Chumakov I, Pollen D, Brookes A, Sanseau P, Polinsky RJ, Wasco W, Da Silva HAR, Haines JL, Pericak-Vance MA, Tanzi RE, Roses AD, Fraser PE, Rommens JM, St George-Hyslop PH. Cloning of a gene bearing missense mutations in early-onset familial Alzheimer's disease. Nature. 1995;375:754–60. doi: 10.1038/375754a0. [DOI] [PubMed] [Google Scholar]
- 8.Hsiao K, Chapman P, Nilsen S, Eckman C, Harigaya Y, Younkin S, Yang F, Cole G. Correlative memory deficits, Abeta elevation, and amyloid plaques in transgenic mice. Science. 1996;274:99–102. doi: 10.1126/science.274.5284.99. [DOI] [PubMed] [Google Scholar]
- 9.Duff K, Eckman C, Zehr C, Yu X, Prada CM, Perez-tur J, Hutton M, Buee L, Harigaya Y, Yager D, Morgan D, Gordon MN, Holcomb L, Refolo L, Zenk B, Hardy J, Younkin S. Increased amyloid-beta42(43) in brains of mice expressing mutant presenilin 1. Nature. 1996;383:710–3. doi: 10.1038/383710a0. [DOI] [PubMed] [Google Scholar]
- 10.Holcomb L, Gordon MN, McGowan E, Yu X, Benkovic S, Jantzen P, Wright K, Saad I, Mueller R, Morgan D, Sanders S, Zehr C, O’Campo K, Hardy J, Prada CM, Eckman C, Younkin S, Hsiao K, Duff K. Accelerated Alzheimer-type phenotype in transgenic mice carrying both mutant amyloid precursor protein and presenilin 1 transgenes. Nat Med. 1998;4:97–100. doi: 10.1038/nm0198-097. [DOI] [PubMed] [Google Scholar]
- 11.Holcomb LA, Gordon MN, Jantzen P, Hsiao K, Duff K, Morgan D. Behavioral changes in transgenic mice expressing both amyloid precursor protein and presenilin-1 mutations: lack of association with amyloid deposits. Behav Genet. 1999;29:177–85. doi: 10.1023/a:1021691918517. [DOI] [PubMed] [Google Scholar]
- 12.Takeuchi A, Irizarry MC, Duff K, Saido TC, Hsiao Ashe K, Hasegawa M, Mann DM, Hyman BT, Iwatsubo T. Age-related amyloid beta deposition in transgenic mice overexpressing both Alzheimer mutant presenilin 1 and amyloid beta precursor protein Swedish mutant is not associated with global neuronal loss. Am J Pathol. 2000;157:331–9. doi: 10.1016/s0002-9440(10)64544-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Borchelt DR, Ratovitski T, Van Lare J, Lee MK, Gonzales V, Jenkins NA, Copeland NG, Price DL, Sisodia SS. Accelerated amyloid deposition in the brains of transgenic mice coexpressing mutant presenilin 1 and amyloid precursor proteins. Neuron. 1997;19:939–45. doi: 10.1016/s0896-6273(00)80974-5. [DOI] [PubMed] [Google Scholar]
- 14.Golde TE, Estus S, Younkin LH, Selkoe DJ, Younkin SG. Processing of the amyloid protein precursor to potentially amyloidogenic derivatives. Science. 1992;255:728–30. doi: 10.1126/science.1738847. [DOI] [PubMed] [Google Scholar]
- 15.Estus S, Golde TE, Kunishita T, Blades D, Lowery D, Eisen M, Usiak M, Qu XM, Tabira T, Greenberg BD, Youn Kin SG. Potentially amyloidogenic, carboxyl-terminal derivatives of the amyloid protein precursor. Science. 1992;255:726–8. doi: 10.1126/science.1738846. [DOI] [PubMed] [Google Scholar]
- 16.Shoji M, Golde TE, Ghiso J, Cheung TT, Estus S, Shaffer LM, Cai XD, McKay DM, Tintner R, Frangione B, Youn Kin SG. Production of the Alzheimer amyloid beta protein by normal proteolytic processing. Science. 1992;258:126–9. doi: 10.1126/science.1439760. [DOI] [PubMed] [Google Scholar]
- 17.Halverson K, Fraser PE, Kirschner DA, Lansbury PT., Jr Molecular determinants of amyloid deposition in Alzheimer's disease: conformational studies of synthetic beta-protein fragments. Biochemistry. 1990;29:2639–44. doi: 10.1021/bi00463a003. [DOI] [PubMed] [Google Scholar]
- 18.Meyer-Luehmann M, Spires-Jones TL, Prada C, Garcia-Alloza M, De Calignon A, Rozkalne A, Koenigsknecht-Talboo J, Holtzman DM, Bacskai BJ, Hyman BT. Rapid appearance and local toxicity of amyloid-beta plaques in a mouse model of Alzheimer's disease. Nature. 2008;451:720–4. doi: 10.1038/nature06616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Spires-Jones TL, De Calignon A, Matsui T, Zehr C, Pitstick R, Wu HY, Osetek JD, Jones PB, Bacskai BJ, Feany MB, Carlson GA, Ashe KH, Lewis J, Hyman BT. In vivo imaging reveals dissociation between caspase activation and acute neuronal death in tangle-bearing neurons. J Neurosci. 2008;28:862–7. doi: 10.1523/JNEUROSCI.3072-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Robbins EM, Betensky RA, Domnitz SB, Purcell SM, Garcia-Alloza M, Greenberg C, Rebeck GW, Hyman BT, Greenberg SM, Frosch MP, Bacskai BJ. Kinetics of cerebral amyloid angiopathy progression in a transgenic mouse model of Alzheimer disease. J Neurosci. 2006;26:365–71. doi: 10.1523/JNEUROSCI.3854-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Frautschy SA, Yang F, Irrizarry M, Hyman B, Saido TC, Hsiao K, Cole GM. Microglial response to amyloid plaques in APPsw transgenic mice. Am J Pathol. 1998;152:307–17. [PMC free article] [PubMed] [Google Scholar]
- 22.Blume AJ, Vitek MP. Focusing on IL-1-promotion of beta-amyloid precursor protein synthesis as an early event in Alzheimer's disease. Neurobiol Aging. 1989;10:406–8. doi: 10.1016/0197-4580(89)90077-8. [DOI] [PubMed] [Google Scholar]
- 23.Gordon MN, Holcomb LA, Jantzen PT, DiCarlo G, Wilcock D, Boyett KW, Connor K, Melachrino J, O’Callaghan JP, Morgan D. Time course of the development of Alzheimer-like pathology in the doubly transgenic PS1 + APP mouse. Exp Neurol. 2002;173:183–95. doi: 10.1006/exnr.2001.7754. [DOI] [PubMed] [Google Scholar]
- 24.Apelt J, Schliebs R. Beta-amyloid-induced glial expression of both pro- and anti-inflammatory cytokines in cerebral cortex of aged transgenic Tg2576 mice with Alzheimer plaque pathology. Brain Res. 2001;894:21–30. doi: 10.1016/s0006-8993(00)03176-0. [DOI] [PubMed] [Google Scholar]
- 25.Dickson DW, Farlo J, Davies P, Crystal H, Fuld P, Yen SH. Alzheimer's disease. A double-labeling immunohistochemical study of senile plaques. Am J Pathol. 1988;132:86–101. [PMC free article] [PubMed] [Google Scholar]
- 26.Stalder AK, Ermini F, Bondolfi L, Krenger W, Burbach GJ, Deller T, Coomaraswamy J, Staufenbiel M, Landmann R, Jucker M. Invasion of hematopoietic cells into the brain of amyloid precursor protein transgenic mice. J Neurosci. 2005;25:11125–32. doi: 10.1523/JNEUROSCI.2545-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Simard AR, Soulet D, Gowing G, Julien JP, Rivest S. Bone marrow-derived microglia play a critical role in restricting senile plaque formation in Alzheimer's disease. Neuron. 2006;49:489–502. doi: 10.1016/j.neuron.2006.01.022. [DOI] [PubMed] [Google Scholar]
- 28.Morgan D, Gordon MN, Tan J, Wilcock D, Rojiani AM. Dynamic complexity of the microglial activation response in transgenic models of amyloid deposition: implications for Alzheimer therapeutics. J Neuropathol Exp Neurol. 2005;64:743–53. doi: 10.1097/01.jnen.0000178444.33972.e0. [DOI] [PubMed] [Google Scholar]
- 29.Town T, Vendrame M, Patel A, Poetter D, DelleDonne A, Mori T, Smeed R, Crawford F, Klein T, Tan J, Mullan M. Reduced Th1 and enhanced Th2 immunity after immunization with Alzheimer's beta-amyloid(1–42) J Neuroimmunol. 2002;132:49–59. doi: 10.1016/s0165-5728(02)00307-7. [DOI] [PubMed] [Google Scholar]
- 30.Colton CA, Mott RT, Sharpe H, Xu Q, Van Nostrand WE, Vitek MP. Expression profiles for macrophage alternative activation genes in AD and in mouse models of AD. J Neuroinflammation. 2006;3:27. doi: 10.1186/1742-2094-3-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Frautschy SA, Cole GM, Baird A. Phagocytosis and deposition of vascular beta-amyloid in rat brains injected with Alzheimer beta-amyloid. Am J Pathol. 1992;140:1389–99. [PMC free article] [PubMed] [Google Scholar]
- 32.Maier M, Peng Y, Jiang L, Seabrook TJ, Carroll MC, Lemere CA. Complement C3 deficiency leads to accelerated amyloid beta plaque deposition and neurodegeneration and modulation of the microglia/macrophage phenotype in amyloid precursor protein transgenic mice. J Neurosci. 2008;28:6333–41. doi: 10.1523/JNEUROSCI.0829-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Town T, Laouar Y, Pittenger C, Mori T, Szekely CA, Tan J, Duman RS, Flavell RA. Blocking TGF-beta-Smad2/3 innate immune signaling mitigates Alzheimer-like pathology. Nat Med. 2008;14:681–7. doi: 10.1038/nm1781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shankar GM, Bloodgood BL, Townsend M, Walsh DM, Selkoe DJ, Sabatini BL. Natural oligomers of the Alzheimer amyloid-beta protein induce reversible synapse loss by modulating an NMDA-type glutamate receptor-dependent signaling pathway. J Neurosci. 2007;27:2866–75. doi: 10.1523/JNEUROSCI.4970-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Walsh DM, Klyubin I, Fadeeva JV, Cullen WK, Anwyl R, Wolfe MS, Rowan MJ, Selkoe DJ. Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo. Nature. 2002;416:535–9. doi: 10.1038/416535a. [DOI] [PubMed] [Google Scholar]
- 36.Cleary JP, Walsh DM, Hofmeister JJ, Shankar GM, Kuskowski MA, Selkoe DJ, Ashe KH. Natural oligomers of the amyloid-beta protein specifically disrupt cognitive function. Nat Neurosci. 2005;8:79–84. doi: 10.1038/nn1372. [DOI] [PubMed] [Google Scholar]
- 37.Gong Y, Chang L, Viola KL, Lacor PN, Lambert MP, Finch CE, Krafft GA, Klein WL. Alzheimer's disease-affected brain: presence of oligomeric A beta ligands (ADDLs) suggests a molecular basis for reversible memory loss. Proc Natl Acad Sci USA. 2003;100:10417–22. doi: 10.1073/pnas.1834302100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Caughey B, Lansbury PT. Protofibrils, pores, fibrils, and neurodegeneration: separating the responsible protein aggregates from the innocent bystanders. Annu Rev Neurosci. 2003;26:267–98. doi: 10.1146/annurev.neuro.26.010302.081142. [DOI] [PubMed] [Google Scholar]
- 39.Shankar GM, Li S, Mehta TH, Garcia-Munoz A, Shepardson NE, Smith I, Brett FM, Farrell MA, Rowan MJ, Lemere CA, Regan CM, Walsh DM, Sabatini BL, Selkoe DJ. Amyloid-beta protein dimers isolated directly from Alzheimer's brains impair synaptic plasticity and memory. Nat Med. 2008;14:837–42. doi: 10.1038/nm1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Clarimon J, Djaldetti R, Lleo A, Guerreiro RJ, Molinuevo JL, Paisan-Ruiz C, Gomez-Isla T, Blesa R, Singleton A, Hardy J. Whole genome analysis in a consanguineous family with early onset Alzheimer's disease. Neurobiol Aging. 2008 doi: 10.1016/j.neurobiolaging.2008.02.008. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Coon KD, Myers AJ, Craig DW, Webster JA, Pearson JV, Lince DH, Zismann VL, Beach TG, Leung D, Bryden L, Halperin RF, Marlowe L, Kaleem M, Walker DG, Ravid R, Heward CB, Rogers J, Papassotiropoulos A, Reiman EM, Hardy J, Stephan DA. A high-density whole-genome association study reveals that APOE is the major susceptibility gene for sporadic late-onset Alzheimer's disease. J Clin Psychiatry. 2007;68:613–8. doi: 10.4088/jcp.v68n0419. [DOI] [PubMed] [Google Scholar]
- 42.Corder EH, Saunders AM, Strittmatter WJ, Schmechel DE, Gaskell PC, Small GW, Roses AD, Haines JL, Pericak-Vance MA. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer's disease in late onset families. Science. 1993;261:921–3. doi: 10.1126/science.8346443. [DOI] [PubMed] [Google Scholar]
- 43.Cedazo-Minguez A. Apolipoprotein E and Alzheimer's disease: molecular mechanisms and therapeutic opportunities. J Cell Mol Med. 2007;11:1227–38. doi: 10.1111/j.1582-4934.2007.00130.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yang DS, Smith JD, Zhou Z, Gandy SE, Martins RN. Characterization of the binding of amyloid-beta peptide to cell culture-derived native apolipoprotein E2, E3, and E4 isoforms and to isoforms from human plasma. J Neurochem. 1997;68:721–5. doi: 10.1046/j.1471-4159.1997.68020721.x. [DOI] [PubMed] [Google Scholar]
- 45.Jiang Q, Lee CY, Mandrekar S, Wilkinson B, Cramer P, Zelcer N, Mann K, Lamb B, Willson TM, Collins JL, Richardson JC, Smith JD, Comery TA, Riddell D, Holtzman DM, Tontonoz P, Landreth GE. ApoE promotes the proteolytic degradation of Abeta. Neuron. 2008;58:681–93. doi: 10.1016/j.neuron.2008.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Van Duijn CM, De Knijff P, Cruts M, Wehnert A, Havekes LM, Hofman A, Van Broeckhoven C. Apolipoprotein E4 allele in a population-based study of early-onset Alzheimer's disease. Nat Genet. 1994;7:74–8. doi: 10.1038/ng0594-74. [DOI] [PubMed] [Google Scholar]
- 47.Hartman RE, Laurer H, Longhi L, Bales KR, Paul SM, McIntosh TK, Holtzman DM. Apolipoprotein E4 influences amyloid deposition but not cell loss after traumatic brain injury in a mouse model of Alzheimer's disease. J Neurosci. 2002;22:10083–7. doi: 10.1523/JNEUROSCI.22-23-10083.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Schenk DB, Seubert P, Grundman M, Black R. A beta immunotherapy: lessons learned for potential treatment of Alzheimer's disease. Neurodegener Dis. 2005;2:255–60. doi: 10.1159/000090365. [DOI] [PubMed] [Google Scholar]
- 49.Hock C, Konietzko U, Streffer JR, Tracy J, Signorell A, Muller-Tillmanns B, Lemke U, Henke K, Moritz E, Garcia E, Wollmer MA, Umbricht D, De Quervain DJ, Hofmann M, Maddalena A, Papassotiropoulos A, Nitsch RM. Antibodies against beta-amyloid slow cognitive decline in Alzheimer's disease. Neuron. 2003;38:547–54. doi: 10.1016/s0896-6273(03)00294-0. [DOI] [PubMed] [Google Scholar]
- 50.Nitsch RM, Hock C. Targeting beta-amyloid pathology in Alzheimer's Disease with Abeta immunotherapy. Neurotherapeutics. 2008;5:415–20. doi: 10.1016/j.nurt.2008.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wilcock DM, Rojiani A, Rosenthal A, Subbarao S, Freeman MJ, Gordon MN, Morgan D. Passive immunotherapy against Abeta in aged APP-transgenic mice reverses cognitive deficits and depletes parenchymal amyloid deposits in spite of increased vascular amyloid and microhemorrhage. J Neuroinflammation. 2004;1:24. doi: 10.1186/1742-2094-1-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wilcock DM, Alamed J, Gottschall PE, Grimm J, Rosenthal A, Pons J, Ronan V, Symmonds K, Gordon MN, Morgan D. Deglycosylated anti-amyloid-beta antibodies eliminate cognitive deficits and reduce parenchymal amyloid with minimal vascular consequences in aged amyloid precursor protein transgenic mice. J Neurosci. 2006;26:5340–6. doi: 10.1523/JNEUROSCI.0695-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Osborne R. Myriad stumbles, Wyeth closes on Alzheimer's. Nat Biotechnol. 2008;26:841–3. doi: 10.1038/nbt0808-841. [DOI] [PubMed] [Google Scholar]
- 54.Oddo S, Billings L, Kesslak JP, Cribbs DH, LaFerla FM. Abeta immunotherapy leads to clearance of early, but not late, hyperphosphorylated tau aggregates via the proteasome. Neuron. 2004;43:321–32. doi: 10.1016/j.neuron.2004.07.003. [DOI] [PubMed] [Google Scholar]
- 55.Santacruz K, Lewis J, Spires T, Paulson J, Kotilinek L, Ingelsson M, Guimaraes A, DeTure M, Ramsden M, McGowan E, Forster C, Yue M, Orne J, Janus C, Mariash A, Kuskowski M, Hyman B, Hutton M, Ashe KH. Tau suppression in a neurodegenerative mouse model improves memory function. Science. 2005;309:476–81. doi: 10.1126/science.1113694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Feany MB, Ksiezak-Reding H, Liu WK, Vincent I, Yen SH, Dickson DW. Epitope expression and hyperphosphorylation of tau protein in corticobasal degeneration: differentiation from progressive supranuclear palsy. Acta Neuropathol. 1995;90:37–43. doi: 10.1007/BF00294457. [DOI] [PubMed] [Google Scholar]
- 57.Feany MB, Dickson DW. Neurodegenerative disorders with extensive tau pathology: a comparative study and review. Ann Neurol. 1996;40:139–48. doi: 10.1002/ana.410400204. [DOI] [PubMed] [Google Scholar]
- 58.Goedert M, Spillantini MG, Crowther RA, Chen SG, Parchi P, Tabaton M, Lanska DJ, Markesbery WR, Wilhelmsen KC, Dickson DW, Petersen RB, Gambetti P. Tau gene mutation in familial progressive subcortical gliosis. Nat Med. 1999;5:454–7. doi: 10.1038/7454. [DOI] [PubMed] [Google Scholar]
- 59.Mattila P, Togo T, Dickson DW. The subthalamic nucleus has neurofibrillary tangles in argyrophilic grain disease and advanced Alzheimer's disease. Neurosci Lett. 2002;320:81–5. doi: 10.1016/s0304-3940(02)00006-x. [DOI] [PubMed] [Google Scholar]
- 60.Hutton M, Lendon CL, Rizzu P, Baker M, Froelich S, Houlden H, Pickering-Brown S, Chakraverty S, Isaacs A, Grover A, Hackett J, Adamson J, Lincoln S, Dickson D, Davies P, Petersen RC, Stevens M, De Graaff E, Wauters E, Van Baren J, Hillebrand M, Joosse M, Kwon JM, Nowotny P, Heutink P. Association of missense and 5’-splice-site mutations in tau with the inherited dementia FTDP-17. Nature. 1998;393:702–5. doi: 10.1038/31508. [DOI] [PubMed] [Google Scholar]
- 61.Spillantini MG, Murrell JR, Goedert M, Farlow MR, Klug A, Ghetti B. Mutation in the tau gene in familial multiple system tauopathy with presenile dementia. Proc Natl Acad Sci USA. 1998;95:7737–41. doi: 10.1073/pnas.95.13.7737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Rizzini C, Goedert M, Hodges JR, Smith MJ, Jakes R, Hills R, Xuereb JH, Crowther RA, Spillantini MG. Tau gene mutation K257T causes a tauopathy similar to Pick's disease. J Neuropathol Exp Neurol. 2000;59:990–1001. doi: 10.1093/jnen/59.11.990. [DOI] [PubMed] [Google Scholar]
- 63.Ramsden M, Kotilinek L, Forster C, Paulson J, McGowan E, SantaCruz K, Guimaraes A, Yue M, Lewis J, Carlson G, Hutton M, Ashe KH. Age-dependent neurofibrillary tangle formation, neuron loss, and memory impairment in a mouse model of human tauopathy (P301L) J Neurosci. 2005;25:10637–47. doi: 10.1523/JNEUROSCI.3279-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Opar A. Mixed results for disease-modification strategies for Alzheimer's disease. Nat Rev Drug Discov. 2008;7:717–8. doi: 10.1038/nrd2676. [DOI] [PubMed] [Google Scholar]
- 65.Kosik KS, Shimura H. Phosphorylated tau and the neurodegenerative foldopathies. Biochim Biophys Acta. 2005;1739:298–310. doi: 10.1016/j.bbadis.2004.10.011. [DOI] [PubMed] [Google Scholar]
- 66.Drewes G, Ebneth A, Preuss U, Mandelkow EM, Mandelkow E. MARK, a novel family of protein kinases that phos-phorylate microtubule-associated proteins and trigger microtubule disruption. Cell. 1997;89:297–308. doi: 10.1016/s0092-8674(00)80208-1. [DOI] [PubMed] [Google Scholar]
- 67.Sontag E, Nunbhakdi-Craig V, Lee G, Bloom GS, Mumby MC. Regulation of the phosphorylation state and microtubule-binding activity of Tau by protein phosphatase 2A. Neuron. 1996;17:1201–7. doi: 10.1016/s0896-6273(00)80250-0. [DOI] [PubMed] [Google Scholar]
- 68.Nishimura I, Yang Y, Lu B. PAR-1 kinase plays an initiator role in a temporally ordered phosphorylation process that confers tau toxicity in Drosophila. Cell. 2004;116:671–82. doi: 10.1016/s0092-8674(04)00170-9. [DOI] [PubMed] [Google Scholar]
- 69.Sperber BR, Leight S, Goedert M, Lee VM. Glycogen synthase kinase-3 beta phosphorylates tau protein at multiple sites in intact cells. Neurosci Lett. 1995;197:149–53. doi: 10.1016/0304-3940(95)11902-9. [DOI] [PubMed] [Google Scholar]
- 70.Baumann K, Mandelkow EM, Biernat J, Piwnica-Worms H, Mandelkow E. Abnormal Alzheimer-like phosphorylation of tau-protein by cyclin-dependent kinases cdk2 and cdk5. FEBS Lett. 1993;336:417–24. doi: 10.1016/0014-5793(93)80849-p. [DOI] [PubMed] [Google Scholar]
- 71.Roder HM, Eden PA, Ingram VM. Brain protein kinase PK40erk converts tau into a PHF-like form as found in Alzheimer's disease. Biochem Biophys Res Commun. 1993;193:639–47. doi: 10.1006/bbrc.1993.1672. [DOI] [PubMed] [Google Scholar]
- 72.Roberson ED, Scearce-Levie K, Palop JJ, Yan F, Cheng IH, Wu T, Gerstein H, Yu GQ, Mucke L. Reducing endogenous tau ameliorates amyloid beta-induced deficits in an Alzheimer's disease mouse model. Science. 2007;316:750–4. doi: 10.1126/science.1141736. [DOI] [PubMed] [Google Scholar]
- 73.Noble W, Planel E, Zehr C, Olm V, Meyerson J, Suleman F, Gaynor K, Wang L, LaFrancois J, Feinstein B, Burns M, Krishnamurthy P, Wen Y, Bhat R, Lewis J, Dickson D, Duff K. Inhibition of glycogen synthase kinase-3 by lithium correlates with reduced tauopathy and degeneration in vivo. Proc Natl Acad Sci USA. 2005;102:6990–5. doi: 10.1073/pnas.0500466102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kosik KS, Ahn J, Stein R, Yeh LA. Discovery of compounds that will prevent tau pathology. J Mol Neurosci. 2002;19:261–6. doi: 10.1385/JMN:19:3:261. [DOI] [PubMed] [Google Scholar]
- 75.Weaver CL, Espinoza M, Kress Y, Davies P. Conformational change as one of the earliest alterations of tau in Alzheimer's disease. Neurobiol Aging. 2000;21:719–27. doi: 10.1016/s0197-4580(00)00157-3. [DOI] [PubMed] [Google Scholar]
- 76.Reynolds MR, Berry RW, Binder LI. Site-specific nitration differentially influences tau assembly in vitro. Biochemistry. 2005;44:13997–4009. doi: 10.1021/bi051028w. [DOI] [PubMed] [Google Scholar]
- 77.Dickey CA, Yue M, Lin WL, Dickson DW, Dunmore JH, Lee WC, Zehr C, West G, Cao S, Clark AM, Caldwell GA, Caldwell KA, Eckman C, Patterson C, Hutton M, Petrucelli L. Deletion of the ubiquitin ligase CHIP leads to the accumulation, but not the aggregation, of both endogenous phospho- and caspase-3-cleaved tau species. J Neurosci. 2006;26:6985–96. doi: 10.1523/JNEUROSCI.0746-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Ma J, Yee A, Brewer HB, Jr, Das S, Potter H. Amyloid-associated proteins alpha 1-antichymotrypsin and apolipoprotein E promote assembly of Alzheimer beta-protein into filaments. Nature. 1994;372:92–4. doi: 10.1038/372092a0. [DOI] [PubMed] [Google Scholar]
- 79.Dickey CA, Dunmore J, Lu B, Wang JW, Lee WC, Kamal A, Burrows F, Eckman C, Hutton M, Petrucelli L. HSP induction mediates selective clearance of tau phosphorylated at proline-directed Ser/Thr sites but not KXGS (MARK) sites. FASEB J. 2006;20:753–5. doi: 10.1096/fj.05-5343fje. [DOI] [PubMed] [Google Scholar]
- 80.Dickey CA, Kamal A, Lundgren K, Klosak N, Bailey RM, Dunmore J, Ash P, Shoraka S, Zlatkovic J, Eckman CB, Patterson C, Dickson DW, Nahman NS, Jr, Hutton M, Burrows F, Petrucelli L. The high-affinity HSP90-CHIP complex recognizes and selectively degrades phosphorylated tau client proteins. J Clin Invest. 2007;117:648–58. doi: 10.1172/JCI29715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Dickey CA, Koren J, Zhang YJ, Xu YF, Jinwal UK, Birnbaum MJ, Monks B, Sun M, Cheng JQ, Patterson C, Bailey RM, Dunmore J, Soresh S, Leon C, Morgan D, Petrucelli L. Akt and CHIP coregulate tau degradation through coordinated interactions. Proc Natl Acad Sci USA. 2008;105:3622–7. doi: 10.1073/pnas.0709180105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Qian SB, McDonough H, Boellmann F, Cyr DM, Patterson C. CHIP-mediated stress recovery by sequential ubiquitination of substrates and Hsp70. Nature. 2006;440:551–5. doi: 10.1038/nature04600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Basso AD, Solit DB, Chiosis G, Giri B, Tsichlis P, Rosen N. Akt forms an intracellular complex with heat shock protein 90 (Hsp90) and Cdc37 and is destabilized by inhibitors of Hsp90 function. J Biol Chem. 2002;277:39858–66. doi: 10.1074/jbc.M206322200. [DOI] [PubMed] [Google Scholar]
- 84.Neckers L. Hsp90 inhibitors as novel cancer chemotherapeutic agents. Trends Mol Med. 2002;8:S55–61. doi: 10.1016/s1471-4914(02)02316-x. [DOI] [PubMed] [Google Scholar]
- 85.Morano KA, Thiele DJ. The Sch9 protein kinase regulates Hsp90 chaperone complex signal transduction activity in vivo. EMBO J. 1999;18:5953–62. doi: 10.1093/emboj/18.21.5953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Vos MJ, Hageman J, Carra S, Kampinga HH. Structural and functional diversities between members of the human HSPB, HSPH, HSPA, and DNAJ chaperone families. Biochemistry. 2008;47:7001–11. doi: 10.1021/bi800639z. [DOI] [PubMed] [Google Scholar]
- 87.Lelj-Garolla B, Mauk AG. Self-association and chaperone activity of Hsp27 are thermally activated. J Biol Chem. 2006;281:8169–74. doi: 10.1074/jbc.M512553200. [DOI] [PubMed] [Google Scholar]
- 88.Calderwood SK, Mambula SS, Gray PJ, Jr, Theriault JR. Extracellular heat shock proteins in cell signaling. FEBS Lett. 2007;581:3689–94. doi: 10.1016/j.febslet.2007.04.044. [DOI] [PubMed] [Google Scholar]
- 89.Salbaum JM, Weidemann A, Lemaire HG, Masters CL, Beyreuther K. The promoter of Alzheimer's disease amyloid A4 precursor gene. EMBO J. 1988;7:2807–13. doi: 10.1002/j.1460-2075.1988.tb03136.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Yoo BC, Seidl R, Cairns N, Lubec G. Heat-shock protein 70 levels in brain of patients with Down syndrome and Alzheimer's disease. J Neural Transm Suppl. 1999;57:315–22. doi: 10.1007/978-3-7091-6380-1_22. [DOI] [PubMed] [Google Scholar]
- 91.Harrison PJ, Procter AW, Exworthy T, Roberts GW, Najlerahim A, Barton AJ, Pearson RC. Heat shock protein (hsx70) mRNA expression in human brain: effects of neurodegenerative disease and agonal state. Neuropathol Appl Neurobiol. 1993;19:10–21. doi: 10.1111/j.1365-2990.1993.tb00400.x. [DOI] [PubMed] [Google Scholar]
- 92.Hamos JE, Oblas B, Pulaski-Salo D, Welch WJ, Bole DG, Drachman DA. Expression of heat shock proteins in Alzheimer's disease. Neurology. 1991;41:345–50. doi: 10.1212/wnl.41.3.345. [DOI] [PubMed] [Google Scholar]
- 93.Perez N, Sugar J, Charya S, Johnson G, Merril C, Bierer L, Perl D, Haroutunian V, Wallace W. Increased synthesis and accumulation of heat shock 70 proteins in Alzheimer's disease. Brain Res Mol Brain Res. 1991;11:249–54. doi: 10.1016/0169-328x(91)90033-t. [DOI] [PubMed] [Google Scholar]
- 94.Cisse S, Perry G, Lacoste-Royal G, Cabana T, Gauvreau D. Immunochemical identification of ubiquitin and heat-shock proteins in corpora amylacea from normal aged and Alzheimer's disease brains. Acta Neuropathol. 1993;85:233–40. doi: 10.1007/BF00227716. [DOI] [PubMed] [Google Scholar]
- 95.Renkawek K, Bosman GJ, Gaestel M. Increased expression of heat-shock protein 27 kDa in Alzheimer disease: a preliminary study. Neuroreport. 1993;5:14–6. doi: 10.1097/00001756-199310000-00003. [DOI] [PubMed] [Google Scholar]
- 96.Renkawek K, Bosman GJ, De Jong WW. Expression of small heat-shock protein Hsp 27 in reactive gliosis in Alzheimer disease and other types of dementia. Acta Neuropathol. 1994;87:511–9. doi: 10.1007/BF00294178. [DOI] [PubMed] [Google Scholar]
- 97.Petrucelli L, Dickson D, Kehoe K, Taylor J, Snyder H, Grover A, De Lucia M, McGowan E, Lewis J, Prihar G, Kim J, Dillmann WH, Browne SE, Hall A, Voellmy R, Tsuboi Y, Dawson TM, Wolozin B, Hardy J, Hutton M. CHIP and Hsp70 regulate tau ubiquitination, degradation and aggregation. Hum Mol Genet. 2004;13:703–14. doi: 10.1093/hmg/ddh083. [DOI] [PubMed] [Google Scholar]
- 98.Nemes Z, Devreese B, Steinert PM, Van Beeumen J, Fesus L. Cross-linking of ubiquitin, HSP27, parkin, and alpha-synu-clein by gamma-glutamyl-epsilon-lysine bonds in Alzheimer's neurofibrillary tangles. FASEB J. 2004;18:1135–7. doi: 10.1096/fj.04-1493fje. [DOI] [PubMed] [Google Scholar]
- 99.Caporaso GL, Takei K, Gandy SE, Matteoli M, Mundigl O, Greengard P, De Camilli P. Morphologic and biochemical analysis of the intracellular trafficking of the Alzheimer beta/A4 amyloid precursor protein. J Neurosci. 1994;14:3122–38. doi: 10.1523/JNEUROSCI.14-05-03122.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Yang Y, Turner RS, Gaut JR. The chaperone BiP/GRP78 binds to amyloid precursor protein and decreases Abeta40 and Abeta42 secretion. J Biol Chem. 1998;273:25552–5. doi: 10.1074/jbc.273.40.25552. [DOI] [PubMed] [Google Scholar]
- 101.Hoshino T, Nakaya T, Araki W, Suzuki K, Suzuki T, Mizushima T. Endoplasmic reticulum chaperones inhibit the production of amyloid-beta peptides. Biochem J. 2007;402:581–9. doi: 10.1042/BJ20061318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Kumar P, Ambasta RK, Veereshwarayya V, Rosen KM, Kosik KS, Band H, Mestril R, Patterson C, Querfurth HW. CHIP and HSPs interact with beta-APP in a proteasome-dependent manner and influence Abeta metabolism. Hum Mol Genet. 2007;16:848–64. doi: 10.1093/hmg/ddm030. [DOI] [PubMed] [Google Scholar]
- 103.Huttunen HJ, Guenette SY, Peach C, Greco C, Xia W, Kim DY, Barren C, Tanzi RE, Kovacs DM. HtrA2 regulates beta-amyloid precursor protein (APP) metabolism through endoplasmic reticulum-associated degradation. J Biol Chem. 2007;282:28285–95. doi: 10.1074/jbc.M702951200. [DOI] [PubMed] [Google Scholar]
- 104.Cottrell BA, Galvan V, Banwait S, Gorostiza O, Lombardo CR, Williams T, Schilling B, Peel A, Gibson B, Koo EH, Link CD, Bredesen DE. A pilot proteomic study of amyloid precursor interactors in Alzheimer's disease. Ann Neurol. 2005;58:277–89. doi: 10.1002/ana.20554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Wilhelmus MM, Otte-Holler I, Wesseling P, De Waal RM, Boelens WC, Verbeek MM. Specific association of small heat shock proteins with the pathological hallmarks of Alzheimer's disease brains. Neuropathol Appl Neurobiol. 2006;32:119–30. doi: 10.1111/j.1365-2990.2006.00689.x. [DOI] [PubMed] [Google Scholar]
- 106.Fonte V, Kipp DR, Yerg J, III, Merin D, Forrestal M, Wagner E, Roberts CM, Link CD. Suppression of in vivo beta-amyloid peptide toxicity by overexpression of the HSP-16.2 small chaperone protein. J Biol Chem. 2008;283:784–91. doi: 10.1074/jbc.M703339200. [DOI] [PubMed] [Google Scholar]
- 107.Kaur P, Welch WJ, Saklatvala J. Interleukin 1 and tumour necrosis factor increase phosphorylation of the small heat shock protein. Effects in fibroblasts, Hep G2 and U937 cells. FEBS Lett. 1989;258:269–73. doi: 10.1016/0014-5793(89)81671-0. [DOI] [PubMed] [Google Scholar]
- 108.Evans CG, Wisen S, Gestwicki JE. Heat shock proteins 70 and 90 inhibit early stages of amyloid beta-(1–42) aggregation in vitro. J Biol Chem. 2006;281:33182–91. doi: 10.1074/jbc.M606192200. [DOI] [PubMed] [Google Scholar]
- 109.Magrane J, Smith RC, Walsh K, Querfurth HW. Heat shock protein 70 participates in the neuroprotective response to intracellularly expressed beta-amyloid in neurons. J Neurosci. 2004;24:1700–6. doi: 10.1523/JNEUROSCI.4330-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Ansar S, Burlison JA, Hadden MK, Yu XM, Desino KE, Bean J, Neckers L, Audus KL, Michaelis ML, Blagg BS. A non-toxic Hsp90 inhibitor protects neurons from Abeta-induced toxicity. Bioorg Med Chem Lett. 2007;17:1984–90. doi: 10.1016/j.bmcl.2007.01.017. [DOI] [PubMed] [Google Scholar]
- 111.Rapoport M, Dawson HN, Binder LI, Vitek MP, Ferreira A. Tau is essential to beta-amyloid-induced neurotoxicity. Proc Natl Acad Sci USA. 2002;99:6364–9. doi: 10.1073/pnas.092136199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Dewji NN. The structure and functions of the presenilins. Cell Mol Life Sci. 2005;62:1109–19. doi: 10.1007/s00018-005-4566-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Dewji NN. Presenilin structure in mechanisms leading to Alzheimer's disease. J Alzheimers Dis. 2006;10:277–90. doi: 10.3233/jad-2006-102-312. [DOI] [PubMed] [Google Scholar]
- 114.Dewji NN, Do C, Bayney RM. Transcriptional activation of Alzheimer's beta-amyloid precursor protein gene by stress. Brain Res Mol Brain Res. 1995;33:245–53. doi: 10.1016/0169-328x(95)00131-b. [DOI] [PubMed] [Google Scholar]
- 115.Cohen E, Bieschke J, Perciavalle RM, Kelly JW, Dillin A. Opposing activities protect against age-onset proteotoxicity. Science. 2006;313:1604–10. doi: 10.1126/science.1124646. [DOI] [PubMed] [Google Scholar]
- 116.Katayama T, Imaizumi K, Sato N, Miyoshi K, Kudo T, Hitomi J, Morihara T, Yoneda T, Gomi F, Mori Y, Nakano Y, Takeda J, Tsuda T, Itoyama Y, Murayama O, Takashima A, St George-Hyslop P, Takeda M, Tohyama M. Presenilin-1 mutations downregulate the signalling pathway of the unfolded-protein response. Nat Cell Biol. 1999;1:479–85. doi: 10.1038/70265. [DOI] [PubMed] [Google Scholar]
- 117.Sato N, Urano F, Yoon Leem J, Kim SH, Li M, Donoviel D, Bernstein A, Lee AS, Ron D, Veselits ML, Sisodia SS, Thinakaran G. Upregulation of BiP and CHOP by the unfolded-protein response is independent of presenilin expression. Nat Cell Biol. 2000;2:863–70. doi: 10.1038/35046500. [DOI] [PubMed] [Google Scholar]
- 118.Kovacs I, Lentini KM, Ingano LM, Kovacs DM. Presenilin 1 forms aggresomal deposits in response to heat shock. J Mol Neurosci. 2006;29:9–19. doi: 10.1385/JMN:29:1:29. [DOI] [PubMed] [Google Scholar]
- 119.Guillozet-Bongaarts AL, Garcia-Sierra F, Reynolds MR, Horowitz PM, Fu Y, Wang T, Cahill ME, Bigio EH, Berry RW, Binder LI. Tau truncation during neurofibrillary tangle evolution in Alzheimer's disease. Neurobiol Aging. 2005;26:1015–22. doi: 10.1016/j.neurobiolaging.2004.09.019. [DOI] [PubMed] [Google Scholar]
- 120.Pickering-Brown S, Baker M, Yen SH, Liu WK, Hasegawa M, Cairns N, Lantos PL, Rossor M, Iwatsubo T, Davies Y, Allsop D, Furlong R, Owen F, Hardy J, Mann D, Hutton M. Pick's disease is associated with mutations in the tau gene. Ann Neurol. 2000;48:859–67. [PubMed] [Google Scholar]
- 121.Dou F, Netzer WJ, Tanemura K, Li F, Hartl FU, Takashima A, Gouras GK, Greengard P, Xu H. Chaperones increase association of tau protein with microtubules. Proc Natl Acad Sci USA. 2003;100:721–6. doi: 10.1073/pnas.242720499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Shimura H, Miura-Shimura Y, Kosik KS. Binding of tau to heat shock protein 27 leads to decreased concentration of hyperphosphorylated tau and enhanced cell survival. J Biol Chem. 2004;279:17957–62. doi: 10.1074/jbc.M400351200. [DOI] [PubMed] [Google Scholar]
- 123.Dabir DV, Trojanowski JQ, Richter-Landsberg C, Lee VM, Forman MS. Expression of the small heat-shock protein alphaB-crystallin in tauopathies with glial pathology. Am J Pathol. 2004;164:155–66. doi: 10.1016/s0002-9440(10)63106-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Bjorkdahl C, Sjogren MJ, Zhou X, Concha H, Avila J, Winblad B, Pei JJ. Small heat shock proteins Hsp27 or alphaB-crystallin and the protein components of neurofibrillary tangles: tau and neurofilaments. J Neurosci Res. 2008;86:1343–52. doi: 10.1002/jnr.21589. [DOI] [PubMed] [Google Scholar]
- 125.Sahara N, Maeda S, Yoshiike Y, Mizoroki T, Yamashita S, Murayama M, Park JM, Saito Y, Murayama S, Takashima A. Molecular chaperone-mediated tau protein metabolism counteracts the formation of granular tau oligomers in human brain. J Neurosci Res. 2007;85:3098–108. doi: 10.1002/jnr.21417. [DOI] [PubMed] [Google Scholar]
- 126.Sarkar M, Kuret J, Lee G. Two motifs within the tau microtubule-binding domain mediate its association with the hsc70 molecular chaperone. J Neurosci Res. 2008;86(12):2763–73. doi: 10.1002/jnr.21721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Elliott E, Tsvetkov P, Ginzburg I. BAG-1 associates with Hsc70.Tau complex and regulates the proteasomal degradation of Tau protein. J Biol Chem. 2007;282:37276–84. doi: 10.1074/jbc.M706379200. [DOI] [PubMed] [Google Scholar]
- 128.Dickey CA, Eriksen J, Kamal A, Burrows F, Kasibhatla S, Eckman CB, Hutton M, Petrucelli L. Development of a high throughput drug screening assay for the detection of changes in tau levels—proof of concept with HSP90 inhibitors. Curr Alzheimer Res. 2005;2:231–8. doi: 10.2174/1567205053585927. [DOI] [PubMed] [Google Scholar]
- 129.Luo W, Dou F, Rodina A, Chip S, Kim J, Zhao Q, Moulick K, Aguirre J, Wu N, Greengard P, Chiosis G. Roles of heat-shock protein 90 in maintaining and facilitating the neurodegenerative phenotype in tauopathies. Proc Natl Acad Sci USA. 2007;104:9511–6. doi: 10.1073/pnas.0701055104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Dickey CA, Patterson C, Dickson D, Petrucelli L. Brain CHIP: removing the culprits in neurodegenerative disease. Trends Mol Med. 2007;13:32–8. doi: 10.1016/j.molmed.2006.11.003. [DOI] [PubMed] [Google Scholar]
- 131.Hatakeyama S, Matsumoto M, Kamura T, Murayama M, Chui DH, Planel E, Takahashi R, Nakayama KI, Takashima A. U-box protein carboxyl terminus of Hsc70-interacting protein (CHIP) mediates poly-ubiquitylation preferentially on four-repeat Tau and is involved in neurodegeneration of tauopathy. J Neurochem. 2004;91:299–307. doi: 10.1111/j.1471-4159.2004.02713.x. [DOI] [PubMed] [Google Scholar]
- 132.Sahara N, Murayama M, Mizoroki T, Urushitani M, Imai Y, Takahashi R, Murata S, Tanaka K, Takashima A. In vivo evidence of CHIP up-regulation attenuating tau aggregation. J Neurochem. 2005;94:1254–63. doi: 10.1111/j.1471-4159.2005.03272.x. [DOI] [PubMed] [Google Scholar]
- 133.Pei JJ, Khatoon S, An WL, Nordlinder M, Tanaka T, Braak H, Tsujio I, Takeda M, Alafuzoff I, Winblad B, Cowburn RF, Grundke-Iqbal I, Iqbal K. Role of protein kinase B in Alzheimer's neurofibrillary pathology. Acta Neuropathol. 2003;105:381–92. doi: 10.1007/s00401-002-0657-y. [DOI] [PubMed] [Google Scholar]