

Abstract
Na,K-ATPase is composed of two essential α- and β-subunits, both of which have multiple isoforms. Evidence indicates that the Na,K-ATPase enzymatic activity as well as its α1, α3 and β1 isoforms are reduced in the failing human heart. The catalytic α-subunit is the receptor for cardiac glycosides such as digitalis, used for the treatment of congestive heart failure. The role of the Na,K-ATPase β1-subunit (Na,K-β1) in cardiac function is not known. We used Cre/loxP technology to inactivate the Na,K-β1 gene exclusively in the ventricular cardiomyocytes. Animals with homozygous Na,K-β1 gene excision were born at the expected Mendelian ratio, grew into adulthood, and appeared to be healthy until 10 months of age. At 13–14 months, these mice had 13% higher heart/body weight ratios, and reduced contractility as revealed by echocardiography compared to their wild-type (WT) littermates. Pressure overload by transverse aortic constriction (TAC) in younger mice, resulted in compensated hypertrophy in WT mice, but decompensation in the Na,K-β1 KO mice. The young KO survivors of TAC exhibited decreased contractile function and mimicked the effects of the Na,K-β1 KO in older mice. Further, we show that intact hearts of Na,K-β1 KO anesthetized mice as well as isolated cardiomyocytes were insensitive to ouabain-induced positive inotropy. This insensitivity was associated with a reduction in NCX1, one of the proteins involved in regulating cardiac contractility. In conclusion, our results demonstrate that Na,K-β1 plays an essential role in regulating cardiac contractility and that its loss is associated with significant pathophysiology of the heart.
Keywords: Na,K-ATPase; Na,K-ATPase β1-subunit; hypertrophy; contractility; TAC; ouabain; cardiomyocytes; NCX1
INTRODUCTION
Na,K-ATPase, also called sodium pump, is a ubiquitous plasma-membrane bound oligomeric enzyme consisting of two essential non-covalently linked subunits, the α-subunit (Na,K-α), which contains the binding sites for ATP, Na+ and K+; and the β-subunit (Na,K-β), required for the proper membrane insertion of Na,K-α. Four Na,K-α (α1–α4) and three Na,K-β (β1-α3) isoforms expressed in a tissue-specific manner, have been described so far. Mouse heart expresses α1, α2, β1, and a small amount of β2. The human heart has α3 in addition [1]. Recently, the FXYD family of proteins has been shown to associate with the Na,K-ATPase [2, 3]. FXYD-1, also known as phospholemman (PLM), is mainly expressed in the heart and regulates the transport properties of Na,K-ATPase [4].
Na,K-ATPase catalyzes the transport of two K+ in and three Na+ out at the expense of one molecule of ATP. Besides maintaining cellular ion homeostasis, Na,K-ATPase performs specialized functions in specific tissues. The role of Na,K-ATPase in the cardiac muscle is very well documented. The excitation-contraction coupling (ECC) of the ventricular myocytes is greatly dependent on the transport of Ca2+ and Na+ into and out of the cytoplasm. The decline in intracellular Ca2+ concentration ([Ca2+]i) is a prerequisite for myocyte relaxation. This is in part mediated by the sarcolemmal Na+/Ca2+ exchanger 1 (NCX1), which physically interacts with the Na,K-ATPase in the heart [5, 6]. NCX1 transports one Ca2+ out and three Na+ in, thus Ca2+ extrusion results in an increase in intracellular Na+ concentration ([Na+]i). The major route for the extrusion of this excess Na+ is via the Na,K-ATPase. Thus, Na,K-ATPase functions in close partnership with the NCX1 to maintain the balance of [Ca2+]i and [Na+]i. Na,K-α1 is also the pharmacological receptor for cardiac glycosides, such as digitalis, which have been used for centuries for the treatment of congestive heart failure. The mechanism of digitalis mediated positive inotropy involves moderate inhibition of the enzymatic activity of Na,K-ATPase, leading to an elevation in the [Na+]i. This reduces the transport activity of NCX1 resulting in elevated [Ca2+]i and subsequent increased contractility of the heart.
Genetic knockout (KO) studies have provided valuable information regarding the role of Na,K-ATPase in the heart. Na,K-α1 heterozygous KO mice were hypocontractile, whereas Na,K-α2 heterozygous KO mice were hypercontractile [7]. The Na,K-β2 homozygous KO mice had enlarged ventricles, without any reduction in the enzymatic activity of Na,K-ATPase [8]. PLM KO mice showed depressed left ventricular function and increased Na,K-ATPase activity [9], suggesting that PLM regulates cardiac contractility [10]. However, the role of Na,K-β1 in heart function has not been described so far.
It is well known that Na,K-β1 functions as a chaperone for the Na,K-α and is required for efficient translation, transport and stability of the Na,K-α at the plasma membrane. Studies from our laboratory and others have demonstrated additional functions for Na,K-β1 in epithelial cells [11]. Independent of the ion transport activity, Na,K-β1 functions as a cell adhesion molecule [12]. Another isoform Na,K-β2 was initially identified as a cell adhesion molecule that mediates neuron- astrocyte interaction in the brain and was referred to as AMOG (adhesion molecule on glia) [13]. Furthermore, in cardiac myocytes, it was shown that Na,K-β1 determines the concentration of the functional Na,K-ATPase enzyme in the caveolae [14], which are hot-spots for signal transduction and involved in the regulation of Ca2+-dependent signaling. In order to define the role of Na,K-β1 in cardiac function, we generated mice with cardiomyocyte-specific KO of Na,K-β1 by using Cre/loxP technology. Homozygous KO mice lacking Na,K-β1 in the heart were born as expected by Mendelian inheritance and grew into healthy adults. With aging however, the KO hearts showed modest hypertrophy and reduced contractility. A similar phenotype was observed in younger KO mice when challenged with pressure overload by transverse aortic constriction. Furthermore, we show that the hearts of Na,K-β1 KO mice as well as isolated cardiomyocytes were insensitive to ouabain-induced positive inotropy with reduced expression of NCX1 in Na,K-β1 KO hearts. Thus, this study reveals a novel role for Na,K-β1 in heart function and ouabain-induced cardiac contractility.
MATERIALS AND METHODS
Generation of Na,K-β1 conditional knockout mice
A targeting vector was constructed such that loxP sites flanked the exon 2 of the Na,K-β1 gene, and electro-porated into 129SvEv embryonic stem (ES) cells to generate germ-line chimeras. Southern analysis was carried out to identify the clones in which a single homologous recombination event had taken place at the Na,K-β1 locus (Targeted allele). These clones were transiently transfected with a Cre recombinase gene under the control of the cytomegalovirus (CMV) promoter. Clones from each parental ES line that showed successful recombination to remove the selection marker cassette and still retained the floxed second exon (Floxed allele) were identified by Southern hybridization screening and confirmed by PCR. These clones were microinjected into C57Bl/6 blastocysts, to generate chimeras. All animal protocols were approved by the Animal Care and Use Committee at University of California, Los Angeles, and Nemours Biomedical Research of the Alfred I. duPont Hospital for Children.
Reverse transcription and quantitative real-time PCR
Total RNA extracted from ventricles using Trizol reagent (Invitrogen, Carlsbad, CA) was purified by on-column DNase digestion (Qiagen, Valencia, CA) to remove any contaminating genomic DNA. First-strand cDNA was generated from total RNA by using iScript cDNA synthesis kit (Bio-Rad, Hercules, CA) according to manufacturer’s protocol. Quantitative PCR was done by using Bio-Rad Syber Green on Bio-Rad iCycler iQ5 Real Time PCR instrument. The “comparative threshold” method was used to calculate relative gene expression. Values were normalized against GAPDH as endogenous control.
Echocardiography
Ultrasound echocardiographs were performed as described previously [15, 16]. Mice were sedated with isoflurane vaporized in O2 for ultrasound echocardiographic evaluation using the Acuson Sequoia C256 equipped with a 15L8, 15 MHz probe. The mice were positioned in the left lateral decubitus position for 2-D, M-mode and Doppler imaging, and ECG needle electrodes were attached to the extremities of the mice. Acuson and AccessPoint software (FreelandSystems, LLC, Santa Fe, NM) were used for data analysis. Heart rates were maintained at resting physiological levels of 500±50 beats per minute for all these procedures.
Transverse aortic constriction (TAC)
Transverse aortic constriction surgeries were carried out as previously described [17]. Age-matched, WT and Na,K-β1 knockout mice (10–11 wks old, weighing 20–25 g) were anesthetized with a mixture of Ketamine (100 mg/kg), xylazine (2 mg/kg) and Buprenorphine (0.1 mg/kg). A blunt dissection was performed at the second intercostals space to expose the aortic arch. A 7-0 silk suture was placed around the arch of the aorta between the left common carotid artery and the brachiocephalic trunk. A 26 or 27-gauge needle was placed on top of the exposed aorta, the suture tied and the needle subsequently removed to constrict the aorta. The chest and the skin incisions were closed with 5-0 silk and 5-0 Vycril respectively. All the mice were monitored for 8 weeks. Just prior to euthanasia, the right carotid and femoral arteries were catheterized in anesthetized mice to obtain the pressure gradient from the TAC. Only data from mice whose pressure gradients exceeded 35 mmHg were included in the analyses. Hemodynamic measurements were acquired, digitized, displayed and analyzed with HEM V3.3 software (Notocord Systems, Croissy sur Seine, France). All hemodynamic data were recorded continuously for at least 30 minutes to ensure physiological levels of pressures and heart rates.
Contractility
Ten to eleven month old mice were anesthetized with Pentobarbital (50 mg/Kg body mass), intubated and hemodynamic data obtained as previous described [17]. For assessment of contractility, the right carotid artery was catheterized with a 1.4Fr catheter (Millar Instruments, Houston, TX) advanced into the left ventricle. After stabilization of baseline LV pressure and ±dP/dT recordings, sequential 1 and 2 nmol/g body weight doses (0.1 ml bolus, IV) infusions of ouabain were administered at 20 minute intervals as described [18]. Hemodynamic measurements were acquired and analyzed as for the TAC gradient.
Histology and biochemistry
At the conclusion of physiological assessment studies, the mice were euthanized; their hearts were excised and weighed. The left tibia was also removed, cleaned and the length measured with calipers. Some hearts were fixed overnight in 4% paraformaldehyde, dehydrated, cleared with xylenes and embedded in paraffin for immunohistochemistry and Elastin-Verhoef/trichrome staining. Some hearts were used for the isolation of protein or total RNA for the biochemical and molecular analyses.
Heart lysates were prepared by homogenizing left ventricles in a buffer containing (in mmol/L): 25 Tris-HCl (pH 7.4), 10 EDTA, 10 EGTA, 100 NaF, 50 NaPPi, 10 Na3VO4, 1 dithiothreitol, 2 phenylmethylsulfonyl fluoride, 1% NP-40, 8 µg/mL each of leupeptin, pepstatin and aprotinin. The lysate was clarified by centrifugation at 15,000 rpm for 15 min. Immunoblot analyses were performed as described [19]. Antibodies against Na,K-β1 (for immunoblots), Na,K-α2 were obtained from Millipore, Billerica, MA. Na,K-α1 antibody was kindly provided by Dr. William James Ball Jr, University of Cincinnati, Cincinnati. Na,K-β1 (for IHC) and Na,K-β2 antibodies were kind gift from Dr. Pablo Martin-Vasallo, Universidad de La Laguna, Tenerife, Spain. Antibodies against Ca2+-handling proteins were generously provided by Dr. Kenneth Philipson, University of California, Los Angeles. The bands from three independent experiments were quantified using GeneTools software from PerkinElmer, Waltham, MA and the ratio of the band intensity to the intensity of the tubulin band was determined.
Na,K-ATPase activity
Na,K-ATPase activity assay was performed as described previously with some modifications [20]. A crude sarcolemma fraction isolated from mouse hearts was used for the assay. Heart was minced and homogenized using a ground glass homogenizer in a buffer containing 20 mM Tris, pH 7.5, 1 mM EDTA, and 0.315 mM sucrose. The homogenates were centrifuged at 186,000 g in Beckman Coulter SW41 Ti rotor for 1 h at 4°C. The pellet was collected, rehomogenized in the same buffer and centrifuged again. The new pellets were resuspended and layered on a sucrose-step gradient (0.75, 0.9, 1.2, 1.4 mM sucrose in 20 mM Tris, pH 7.5 and 1 mM EDTA) and centrifuged at 87,000 g in Beckman Coulter SW41 Ti rotor for 16 h at 4°C. The sarcolemma-enriched membrane fraction was collected from the interface of 0.75 and 0.9 mM sucrose, diluted with buffer lacking sucrose and centrifuged for 1 h at 165,000 g in the same rotor. Na,K-ATPase activity was assayed as the ouabain-sensitive ATP hydrolysis observed in a reaction mixture containing 25 mM histidine, pH 7.5, 130 mM NaCl, 20 mM KCl, 3 mM MgCl2, 3 mM ATP. Assays were performed in the presence or absence of 1 mM ouabain. The hydrolysis of ATP after 5 min at 37°C was quantified by Baginski method to detect inorganic phosphate using colorimetry.
Isolation of cardiac myocytes, and Ca2+ transient recordings
Ten to eleven month old mice were anesthetized and their hearts quickly removed by thoractomy. Single ventricular myocytes were isolated enzymatically using collagenase (2 mg/ml Type II collagenase, Invitrogen) and protease (0.166 mg/ml, Type XIV protease, Sigma Chemical Company, St. Louis, MO) digestion as described earlier [15, 21]. The dissociated cells were stored at room temperature for up to 6 hrs in modified Tyrode solution containing 136 mM NaCl, 5.4 mM KCl, 10 mM HEPES, 1 mM MgCl2, 0.33 mM NaH2PO4, 1 mM CaCl2, 10 mM glucose, pH 7.4. Electrophysiology on isolated cardiac myocytes was performed in a flow-through chamber mounted on the stage of a Zeiss Axiovert 100 inverted microscope (Carl Zeiss Microimaging, Thornwood, NY). Cells were paced externally using a Grass S9 stimulator (Grass-Telefactor, West Warwick, RI) via bipolar platinum electrodes in the bath. For measurement of Ca2+ transients, cardiomyocytes were loaded with the Ca2+ indicator fluo-3 AM (10 µM) for 10 minutes, followed by two washes in standard bath solution for 10 minutes each. Fluo-3 fluorescence was recorded using the line scan mode of a Zeiss Pascal 5 laser scanning confocal microscope (15 ms/line), with an excitation wavelength of 488 nm and an emission wavelength of 505 nm. Fluorescence transients were normalized to diastolic fluorescence and are expressed as F/Fo. Ouabain at a concentration of 0.1 to 1 µM was applied to resting cells in the bath solution 10 minutes prior to electrical stimulation.
Statistics
Data was compiled and expressed as means ± standard errors of the mean. Data was evaluated using unpaired, two-tailed t-tests (95% confidence interval). Data with a p < 0.05 was considered statistically significant.
RESULTS
Cardiac-specific knockout of Na,K-β1
To conditionally ablate the Na,K-β1, we generated floxed mice with a loxP site inserted into each intron flanking exon 2. The targeting strategy is outlined in Figure 1A. The presence of the floxed allele was confirmed by Southern hybridization (Figure 1B) and polymerase chain reaction (PCR) (Figure 1C). The floxed mice were indistinguishable from the WT mice and were used as control. Heart-specific ablation of Na,K-β1 was achieved by crossing mice homozygous for the Na,K-β1 floxed allele with transgenic mice expressing Cre recombinase under the control of the ventricular cardiomyocyte-specific myosin light chain-2 (MLC-2v) promoter [22, 23], kindly provided by Dr. Yibin Wang, University of California, Los Angeles. The KO mice were born at the expected Mendelian ratio with no detectable defects and seemed to grow healthy into adulthood. The heterozygous KO mice did not show any phenotype (data not shown), and only homozygous KO mice were included for these studies.
Figure 1.

Generation of Na,K-β1 conditional knockout mice. (A) A schematic representation of the targeting strategy. The wild-type ‘Na,K-β1 locus’ showing exons 1–4 (E1–E4) and the NdeI restriction sites (N) within the sequence is depicted. Exon 2 targeted for deletion is shown in yellow. The ‘Targeting Construct’ shows the selection marker cassette in orange flanked by the loxP sites (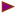 ) and the gene sequence used for homologous recombination (in blue, between the two dotted lines). The sizes of the DNA fragments expected from NdeI digestion and Southern hybridization using the probe (
) and the gene sequence used for homologous recombination (in blue, between the two dotted lines). The sizes of the DNA fragments expected from NdeI digestion and Southern hybridization using the probe ( ) are indicated on the green lines. (B) Southern blot analysis of NdeI digested genomic DNAs isolated from positive ES clones before (Targeted) or after (Floxed) transient transfection with CMV-Cre. (C) PCR analysis of genomic DNAs from positive ES clones. The floxed allele yields a 900 bp PCR product using the indicated primers (
) are indicated on the green lines. (B) Southern blot analysis of NdeI digested genomic DNAs isolated from positive ES clones before (Targeted) or after (Floxed) transient transfection with CMV-Cre. (C) PCR analysis of genomic DNAs from positive ES clones. The floxed allele yields a 900 bp PCR product using the indicated primers ( ), whereas the targeted allele fails to generate any product under the PCR conditions used.
), whereas the targeted allele fails to generate any product under the PCR conditions used.
Real-time RT-PCR showed 80% reduction specifically in the Na,K-β1 mRNA level and no significant change in the mRNA levels of Na,K-β2, Na,K-α1 or Na,K-α2 in homozygous Na,K-β1 KO hearts when compared to WT hearts (Figure 2A). The protein levels of Na,K-β1, Na,K-β2, Na,K-α1 and Na,K-α2 in these hearts were assessed using immunoblots. The Na,K-β1 protein level was greatly reduced in homozygous KO hearts (81%), whereas the level of Na,K-β2 remained the same as in WT hearts (Figure 2B). The Na,K-α1 protein level was reduced by 42%, although the mRNA level was unchanged in homozygous KO hearts. This is in accordance with the knowledge that Na,K-β1 is the major β isoform in the rodent heart [1], and that Na,K-β1 protein is required for the membrane insertion and stabilization of the Na,K-α1 [24]. Thus, in Na,K-β1 KO hearts, Na,K-α1 protein may be targeted for degradation. The level of Na,K-α2 protein however remained unchanged.
Figure 2.

Characterization of cardiac-specific Na,K-β1 knockout mice. Quantitative RT-PCR (A) and representative western blots (B) from heart lysates of wild-type (WT), heterozygous (Het), and homozygous KO (Homo) mice (n=4 each). The staining of Na,K-β1 as a narrow doublet has also been observed by other investigators in heart lysates. For immunoblots, the densitometric quantitation of the band intensities normalized to the loading control, tubulin are placed below each panel. (C) Comparison of the heart tissues of WT and KO mice by immunohistochemistry (IHC) and trichrome staining. Arrows represent staining of Na,K-β1 in the intercalated discs. Representative images from at least 5 mice are shown. (D) Na,K-ATPase pump activity measured as the amount of ouabain-sensitive inorganic phosphate from sarcolemmal membrane preparations from WT and KO hearts. Values are represented as Mean ± SE (n=3 each, * p < 0.05) and plotted as a percentage of the WT.
It has been reported earlier that cardiac-specific knockouts using Cre recombinase under the control of tissue-specific promoters do not occur with 100% efficiency [15]. Thus, a reduction of ~80% at both mRNA and protein levels is acceptable. Consistent with the immunoblot data, immunohistochemical analysis showed absence of Na,K-β1 in most cardiomyocytes in the KO heart (Figure 2C). Thus, these results revealed that the Na,K-β1 gene is efficiently deleted in majority of the homozygous KO cardiomyocytes.
Cardiac hypertrophy and dysfunction in aging Na,K-β1 knockout mice
Immunohistochemistry confirmed the localization of Na,K-β1 at the sarcolemma, and intercalated discs in WT mice as reported earlier (Figure 2C, arrows) [25]. The Elastin Verhoef/trichrome stain showed deposition of collagen in the spaces between the muscle fibers in 4 month old KO hearts but not in WT hearts suggesting interstitial fibrosis of the myocardium (Figure 2C), and heart dysfunction. Therefore, we assessed cardiac function and the degree of ventricular hypertrophy in Na,K-β1 KO mice and WT littermates by echocardiography (Table 1). At 4 months of age, there was no appreciable difference in the ventricular function in KO mice as compared to the WT mice. The echocardiography data and post mortem morphometry indicated a slight, but not significant increase in heart mass from wall thickening. At 10 months, contractility and ventricular function increased marginally. However, 13–14 month old homozygous KO mice exhibited significant cardiac hypertrophy and reduced ventricular function. The post-mortem heart mass, the heart weight/body weight (HW/BW), and the heart weight/tibia length (HW/TL) ratios showed a modest, but statistically significant increase (16%, 13%, and 14%, respectively) in KO mice as compared to their WT littermates. All three indices of ventricular function, i.e. fractional shortening (LVFS), velocity of circumferential fiber shortening (Vcf), and ejection fraction (LVEF), showed appreciable reduction, suggesting compromised heart function (Table 1). LVEF was reduced to 56.7% in KO mice as compared to 66.4% in WT littermates (n = 7 each, p<0.01). Thus, these physiological and histological data revealed that Na,K-β1 KO mice over a year old showed cardiac hypertrophy and reduced heart function.
Table 1.
Cardiac function in heart-specific Na,K-β1 knockout mice
| 4 months | 10 months | 13–14 months | ||||
|---|---|---|---|---|---|---|
| WT | KO | WT | KO | WT | KO | |
| (n=3) | (n=5) | (n=4) | (n=5) | (n=7) | (n=7) | |
| Body weight, g | 30.3 ± 4.2 | 29.4 ± 2.2 | 31.7 ± 2.5 | 36.4 ± 1.2 | 37.0 ± 2.9 | 37.5 ± 1.7 |
| Heart weight, mg | 143.7 ± 16.0 | 180.8 ± 23.7 | 159.2 ± 12.7 | 188.3 ± 7.3 | 162.3 ± 8.5 | 188.7 ± 7.7* |
| Tibia length, mm | 17.9 ± 0.1 | 18.0 ± 0.1 | 19.2 ± 0.6 | 19.5 ± 0.5 | 18.8 ± 0.1 | 19.3 ± 0.2 |
| HW/BW, mg/g | 4.8 ± 0.2 | 5.7 ± 0.4 | 5.0 ± 0.1 | 5.2 ± 0.2 | 4.5 ± 0.3 | 5.1 ± 0.2* |
| HW/TL, mg/mm | 8.0 ± 0.8 | 8.8 ± 1.2 | 8.3 ± 0.4 | 9.7 ± 0.4* | 8.6 ± 0.5 | 9.8 ± 0.4* |
| Left ventricular mass | 81 ± 7 | 102 ± 16 | 118 ± 7 | 121 ± 11 | 113 ± 9 | 117 ± 14 |
| PWT, mm | 0.63 ± 0.04 | 0.73 ± 0.06 | 0.68 ± 0.01 | 0.75 ± 0.04 | 0.71 ± 0.02 | 0.68 ± 0.03 |
| VST, mm | 0.59 ± 0.02 | 0.71 ± 0.03 | 0.71 ± 0.03 | 0.79 ± 0.04 | 0.73 ± 0.02 | 0.72 ± 0.05 |
| EDD, mm | 3.97 ± 0.20 | 3.94 ± 0.15 | 4.44 ± 0.15 | 4.16 ± 0.10 | 4.20 ± 0.14 | 4.34 ± 0.17 |
| ESD, mm | 2.73 ± 0.12 | 2.68 ± 0.12 | 3.00 ± 0.09 | 2.70 ± 0.11 | 2.81 ± 0.13 | 3.24 ± 0.13* |
| LVFS, % | 31.0 ± 0.8 | 32.0 ± 1.6 | 32.4 ± 1.4 | 35.1 ± 2.3 | 33.0 ± 1.7 | 25.3 ± 0.8# |
| Vcf, mm/s | 5.27 ± 0.26 | 5.53 ± 0.45 | 6.54 ± 0.30 | 7.29 ± 0.40 | 6.61 ± 0.48 | 4.59 ± 0.17# |
| LVEF, % | 65.5 ± 1.3 | 66.5 ± 2.4 | 68.5 ± 2.3 | 71.0 ± 2.3 | 66.4 ± 2.5 | 56.7 ± 1.8# |
Values are SE of the Mean.
p<0.01
p<0.05
Heart function in younger mice following pressure overload
The younger homozygous KO mice appeared relatively healthy and did not exhibit any noticeable symptoms of heart dysfunction as determined by echocardiography measurements. We decided to challenge 10–11 week old mice with pressure overload by transverse aortic constriction (TAC) to study the effect of stress on cardiac function in KO mice. Cardiac hypertrophy was observed in both the WT and KO hearts; two weeks post TAC, as expected. There was an increase in the left ventricular mass, the posterior wall thickness (PWT), ventricular septal wall thickness (VST), and the end diastolic dimension (EDD) based on echocardiography measurements before and after TAC (Table 2, Figure 3). There was a tendency for KO hearts to exhibit a greater degree of hypertrophy than their WT littermates in response to TAC. The hypertrophied WT hearts exhibited a compensatory increase in the LVEF, Vcf, and LVFS in response to pressure overload. The KO hearts on the other hand showed visible signs of heart dysfunction. Although the surgery was successful, the KO mice were less tolerant to the aortic constriction. The survivors exhibited significantly decreased ventricular function two weeks post TAC, as seen from the reduction in the LVFS, Vcf and LVEF (Table 2). The differential response of WT and KO hearts to pressure overload is summarized in Figure 3. The Vcf was increased by 39% post-TAC in WT hearts, while it suffered a 40% reduction in KO hearts post-TAC as compared to their pre-TAC values respectively. A similar trend was observed for LVEF and LVFS. Thus, the pressure overload study strongly indicated that Na,K-β1 plays an important role in sustaining heart function under conditions of stress.
Table 2.
Cardiac function following pressure overload induced hypertrophy in heart-specific Na,K-β1 knockout mice
| pre-TAC | 14 d post-TAC | p | ||||||
|---|---|---|---|---|---|---|---|---|
| WT (n=7) |
KO (n=10) |
WT (n=4) |
KO (n=4) |
|||||
| Group Number | 1 | 2 | 3 | 4 | 1&2 | 1&3 | 2&4 | 3&4 |
| Body weight, g | 22.8 ± 1.7 | 21.0 ± 1.4 | 22.4 ± 1.2 | 22.0 ± 1.0 | ||||
| Heart weight, mg | ND | ND | 155.5 ± 7.7 | 185.7 ± 28.4 | ||||
| Tibia length, mm | ND | ND | 17.5 ± 0.2 | 17.5 ± 0.3 | ||||
| HW/BW, mg/g | ND | ND | 7.0 ± 0.6 | 8.5 ± 1.4 | ||||
| HW/TL, mg/mm | ND | ND | 8.9 ± 0.5 | 10.6 ± 1.6 | ||||
| Left ventricular mass | 73 ± 4 | 78 ± 5 | 120 ± 10 | 161 ± 21 | <0.001 | <0.001 | ||
| PWT, mm | 0.57 ± 0.03 | 0.56 ± 0.02 | 0.78 ± 0.04 | 0.89 ± 0.07 | <0.01 | <0.001 | ||
| VST, mm | 0.61 ± 0.03 | 0.62 ± 0.03 | 0.73 ± 0.03 | 0.80 ± 0.01 | <0.05 | <0.001 | <0.05 | |
| EDD, mm | 3.81 ± 0.12 | 3.95 ± 0.10 | 4.28 ± 0.16 | 4.63 ± 0.23 | <0.01 | |||
| ESD, mm | 2.61 ± 0.14 | 2.71 ± 0.12 | 2.58 ± 0.10 | 3.73 ± 0.29 | <0.05 | <0.01 | <0.01 | |
| LVFS, % | 31.7 ± 2.2 | 31.5 ± 2.1 | 39.4 ± 3.8 | 19.8 ± 2.8 | <0.01 | <0.01 | ||
| Vcf, mm/s | 5.63 ± 0.38 | 5.66 ± 0.40 | 7.83 ± 0.77 | 3.41 ± 0.55 | <0.05 | <0.05 | <0.01 | |
| LVEF, % | 66.0 ± 3.2 | 65.8 ± 2.6 | 70.9 ± 1.8 | 46.6 ± 5.6 | <0.01 | <0.01 | ||
Values are SE of the Mean. The respective p values are indicated for different groups if significant. TAC = transverse aortic constriction, ND = not determined.
Figure 3.

Differential response of WT and knockout hearts to TAC. The graph depicts the response to TAC procedure and is based on data from Table 2. The values plotted are the % change in WT and KO hearts following TAC. The pre-TAC values for WT and KO were considered 100%. Error bars denote SE of the mean.
Na,K-β1 knockout hearts are insensitive to ouabain-induced contractility
The Na,K-β1 KO hearts had reduced levels of Na,K-α1 protein. Whether there was a reduction in the pump activity in KO hearts was tested by the quantification of the ouabain-sensitive inorganic phosphate released in a sarcolemma-enriched membrane fraction. Sarcolemmal membrane preparations from KO hearts had 30% reduced Na,K-ATPase activity as compared to the WT (Figure 2D). To evaluate the effect of further inhibition of the pump in KO hearts, the maximal (dP/dtMax) and minimal (dP/dtMin) rate of contraction and relaxation under basal conditions and following infusion with ouabain was measured in 10–11 month old anesthetized mice. The maximum rate of heart contraction (+dP/dt) and relaxation (−dP/dt), measured by Millar catheterization, under basal conditions in Na,K-β1 KO mice was higher as compared to the WT mice (Figure 4, A and C). Administration of ouabain increased the dP/dtMax (p = 0.004) and dP/dtMin (p = 0.009) in WT mice in a dose-dependent manner consistent with previous reports [18]. Surprisingly, no significant increase in the dP/dtMax or dP/dtMin was observed in KO mice even at the higher ouabain dose (p = 0.324 and 0.549 respectively, n = 6). In order to account for animal to animal variability, the change in contractility following ouabain infusion was plotted as ΔdP/dtMax or ΔdP/dtMin (Figure 4, B and D). This allowed a more accurate analysis of the effect of ouabain on the cardiac contractility. At 1 nmol/g body weight of ouabain, the WT mice showed an increase of over 2600 mm of Hg/sec in dP/dtMax, whereas there was a slight reduction in KO mice at the same ouabain concentration. Also, at the higher ouabain concentration, the ΔdP/dtMax was over 3700 mm of Hg/sec higher in WT mice compared to the KO mice (Figure 4B). Similarly, there was a ~5000 mm of Hg/sec difference in the ΔdP/dtMin in WT mice as compared to the KO mice at each ouabain dosage (Figure 4D). These results suggest that the hearts of KO mice are insensitive to alterations of contractility induced by ouabain.
Figure 4.

Effect of ouabain on cardiac contractility in WT and knockout mice. The effect of 0, 1 or 2 nmol/g body weight ouabain on the maximum rate of contraction dP/dtMax (A), and the maximum rate of relaxation dP/dtMin (C) was determined in WT and KO mice (n=6 each). Values are represented as Mean ± SE. * p < 0.01, # p < 0.05 versus WT baseline (0 nmol/g body weight of ouabain). The differences in pressure values compared to the baseline were plotted as ΔdP/dtMax (B) and ΔdP/dtMin (D). Values are represented as Mean ± SE. * p < 0.01, # p < 0.05 between WT and KO.
Ca2+-handling in isolated cardiomyocytes from knockout hearts
Since [Ca2+]i is an important determinant of cardiac contractility, we examined whether there were any alterations in the Ca2+-handling properties of cardiac myocytes isolated from KO mice. Intracellular Ca2+ transients were recorded in enzymatically-isolated contracting ventricular myocytes from 10–11 month old mice loaded with fluo-3 AM. We found a 1.6-fold increase in the amplitude of Ca2+ release (measured as the amplitude of the fluorescence transient normalized to resting fluorescence, ΔF/Fo) in KO myocytes compared to WT controls (Table 3, p < 0.01 and Figure 5A). Also, the time to peak (TTP) showed a modest, but statistically significant 1.25-fold increase in KO compared to WT myocytes (p < 0.01). These results were consistent with the slight increase in contractility measured by echocardiography (Table 1) and the small increase in dP/dt at baseline in KO anaesthetized mice (Figure 4) in the same age group.
Table 3.
Ca2+ transient characteristics in isolated WT and Na,K-β1 knockout cardiomyocytes
| ΔF/Fo | TTP (s) | Half decay (s) | |
|---|---|---|---|
| WT (n=17) | 0.840 ± 0.085 | 0.034 ± 0.002 | 0.180 ± 0.010 |
| KO (n=10) | 1.410 ± 0.163* | 0.043 ± 0.002* | 0.195 ± 0.008 |
Data are Mean ± SEM.
p = 0.01, ΔF/Fo = amplitude of Ca2+ transient normalized to resting fluorescence, TTP = time to peak, Half decay = time to 50% relaxation.
Figure 5.

Analysis of Ca2+ transients in isolated WT and knockout cardiomyocytes. Cardiomyocytes were paced at 0.5 Hz in the presence or absence of ouabain. (A) Representative plots of Ca2+ transients are shown. Ouabain had no effect on Ca2+ transients in KO myocytes. (B) Summary of the effects of ouabain on the kinetics of intracellular Ca2+ transient. Data are represented as Mean ± SE and expressed as a percentage of the respective pre-ouabain values. n=10 for WT, n=7 for KO. * p < 0.01, ΔF/Fo corresponds to the change in the amplitude of the Ca2+ transient normalized to the resting fluorescence, TTP indicates time to peak, Half decay is the time to 50% relaxation.
Since the hearts of KO mice failed to show increased contractility in response to ouabain, we examined the effect of ouabain on Ca2+ transients in isolated cardiomyocytes. In WT cells, low ouabain concentrations (0.1 µmol/L), increased the amplitude of the Ca2+ transient (Figure 5B). Higher ouabain concentrations (1 µmol/L) caused Ca2+ overload with a reduction in the amplitude of the transient (ΔF/Fo), and a delay in the TTP consistent with previous reports [26]. There was an increase in resting fluorescence to account for the decreased amplitude in WT myocytes treated with 1 µmol/L ouabain. On the other hand, Ca2+ transients in KO myocytes did not respond to ouabain, even at the higher concentration, consistent with the lack of response to ouabain in intact animals.
Na,K-β1 KO hearts have reduced NCX1 protein
The amplitude of the Ca2+ transients remained unaltered in Na,K-β1 KO cardiomyocytes treated with ouabain suggesting that the Na,K-β1 KO mice were defective in Ca2+-handling following ouabain treatment. In order to gain insights into the mechanism of altered Ca2+-handling in Na,K-β1 KO mice, we determined the levels of proteins involved in ECC; plasma membrane Ca2+-ATPase (PMCA), calsequestrin, sarco/endoplasmic reticulum Ca2+-ATPase (SERCA) and NCX1. Strikingly, the level of NCX1 protein was specifically reduced by 65%, whereas the expression of other proteins tested remained unaltered.
DISCUSSION
In this study, we show that the conditional KO of Na,K-β1 in cardiomyocytes resulted in mild hypertrophy and reduced ventricular function in aging mice. When younger KO mice with uncompromised heart function were challenged by TAC to induce pressure overload, they exhibited decreased contractility. We also show that Na,K-β1 KO mice were insensitive to ouabain-induced increase in contractility, and that this response appears to be at least partially due to an inability to raise Ca2+ following ouabain treatment. Consistent with this observation, a specific reduction of NCX1 protein in Na,K-β1 KO hearts was observed revealing a previously undescribed potential link between Na,K-β1 and NCX1. Thus, our results demonstrate an essential role for Na,K-β1 in cardiac contractility and function.
The conventional KO strategy deleting the gene product from the entire organism has been utilized for the generation of knockouts of Na,K-ATPase isoforms α1, α2, and β2. Homozygous KO mice lacking Na,K-α1 were embryonic lethal, while those lacking Na,K-α2 exhibited peri-natal lethality [7]. The Na,K-β2 homozygous KO mice did not survive past 17–18 days of age [8]. In addition, since cardiac function is intimately associated with renal function, complete KO may not define the specific role of a given Na,K-ATPase isoform in the heart. Therefore, we utilized the Cre-loxP technology to specifically ablate Na,K-β1 in the heart. The Na,K-β1 gene consists of six exons. The second exon encodes the single trans-membrane domain of Na,K-β1, which is essential for the function of this integral membrane protein. We have shown recently that the trans-membrane domain of Na,K-β1 not only serves to oligomerize the β-β homo-dimers but is also required for the coupling of Na,K-β1 with its functional partner Na,K-α1, and that these interactions are required for Na,K-β1 function [27]. Deletion of the second exon led to a shift of the reading frame and introduced a premature stop codon, resulting in the formation of a truncated protein. Thus, by targeting the exon 2, we were able to successfully generate mice with conditional ablation of Na,K-β1.
Mouse hearts express Na,K-ATPase α1, α2, β1 and β2, with α1 and β1 as the major isoforms. It has been observed in studies using KO mice for proteins with multiple isoforms that the absence of one isoform may result in the upregulated expression of the other in an attempt to compensate for the loss. For example, Na,K-α1 heterozygous mice showed an increase in Na,K-α2 levels [7]. On the other hand, Na,K-α2 heterozygous and Na,K-β2 homozygous KO mice had no compensatory increase in the levels of Na,K-α1 or Na,K-β1, respectively [28]. We found that Na,K-β1 KO hearts did not show increased expression of other isoforms of Na,K-β (Figure 2, A and B). However, there was a 42% reduction in Na,K-α1 at the protein level. This reduction may be due to the requirement of Na,K-β1 for the translation, stability and membrane insertion of Na,K-α1 [24]. Even with low levels of Na,K-α1 the young KO mice did not show any visible abnormalities. This suggests that there may be other compensatory mechanisms activated in Na,K-β1 KO cardiomyocytes, which are sufficient to maintain essential heart function for survival in young mice under non-stress conditions.
Studies using heterozygous KO mice of Na,K-α isoforms showed that the reduction in Na,K-α1 or Na,K-α2 led to completely contrasting phenotypes [7]. Mice with genetically reduced levels of Na,K-α1 were hypocontractile, whereas those with reduced levels of Na,K-α2 were hypercontractile, as determined in ex vivo working heart studies. Interestingly, our heart-specific homozygous Na,K-β1 KO mice, with 42% reduction in the level of Na,K-α1, and 30% reduction in the Na,K-ATPase activity, also showed significant hypocontractility as measured by echocardiography in 13–14 month old mice. Although the contractility data were similar, there were distinct differences between Na,K-β1 KO and Na,K-α1 heterozygous KO mice: 1) Considering that both these mice had a similar reduction in the levels of Na,K-α1, they showed distinct response to ouabain. Remarkably, ouabain increased the contractility in Na,K-α1 heterozygous KO mice, [7], whereas the Na,K-β1 KO mice were ouabain-insensitive. This contrasting phenotype indicates that Na,K-β1 has a role in ouabain-induced positive inotropy, which has not been described so far. 2) Consistent with the lack of response to ouabain in anesthetized mice and isolated cardiomyocytes, the NCX1 protein level was reduced by 65% in Na,K-β1 KO mice. However, in Na,K-α1 heterozygous KO mice there was only a small reduction in the levels of NCX1 and these hearts retained ouabain sensitivity. 3) The amplitude of the Ca2+ transients in isolated cardiomyocytes, in the absence of ouabain was identical to the WT in Na,K-α1 heterozygous KO mice, probably due to the compensatory increase in Na,K-α2 protein expression [7]. By contrast, the amplitude of the Ca2+ transients was increased in Na,K-β1 KO mice which did not show any change in Na,K-α2 levels. Thus, we conclude that Na,K-β1 KO mouse model specifically revealed a novel role for Na,K-β1 in cardiac function.
The classic mechanism of cardiac glycoside action is dependent on reduced Ca2+ efflux by the NCX1 when [Na+]i is increased by Na,K-ATPase inhibition [26]. Thus, a reduction in NCX1 levels may lead to reduced NCX1 activity resulting in ouabain insensitivity. Furthermore, the striking similarities in Ca2+ transients following ouabain treatment in Na,K-β1 KO and NCX1 KO mice [26] suggest that the alteration in NCX1 protein levels in Na,K-β1 KO mice is involved in the observed ouabain insensitivity. Heart-specific NCX1 KO mice also showed reduced contractility with age [15], suggesting that the reduction in NCX1 levels in Na,K-β1 KO mice may also be involved in the compromised contractile function. How Na,K-β1 regulates NCX1 protein level will be the subject of future investigation.
In Na,K-β1 KO mice, the hypertrophic process appears to have started around 4 months (as determined by collagen deposition in Figure 2C), and continues gradually until old age, leading to deterioration in myocardial function in aging KO mice. Although statistically insignificant, 4 month-old mice showed increased ventricular and septal wall thicknesses, which continued until 10 months of age. The small increase in the contractility evident from the echocardiography and the catheter measurements, and the increased Ca2+ transient at the age of 10 months might be an attempt to compensate for the underlying disease progression or may be related to the positive inotropic effect of reduced Na,K-ATPase activity. At 13–14 months of age, however the hypertrophic disease matured into dilated cardiomyopathy, as the chamber size was bigger, without any apparent gain in wall thicknesses. The reduction in contractility and diminished ventricular function was visible and statistically significant at this age. Similar decompensation in cardiac contractility has been observed in failing human hearts, which also exhibit reduced Na,K-ATPase activity [29]. Thus, our data suggests that the Na,K-β1 cardiac-specific KO mouse model mimics the progression of heart disease in human cardiomyopathies.
The direct link between altered Na,K-ATPase function and cardiomyopathy remains obscure, even though its physiological significance in the cardiac muscle is well established. In the failing human heart, the levels of Na,K-α1, Na,K-α3, Na,K-β1 and the Na,K-ATPase enzyme activity were found to be reduced [30]. Further, a close correlation was observed between decreased Na,K-ATPase concentration and reduced LVEF in two separate studies in patients with idiopathic dilated cardiomyopathy [31, 32]. It has been shown that a reduction of Na,K-ATPase expression occurs early in the development of heart failure, probably before the symptoms are evident [29]. Our studies now show that reduced expression of Na,K-β1 predisposes the hearts to a condition where they are incapable of enduring stress. This suggests that reduced expression of Na,K-β1 might prognosticate potential cardiac malfunction in the human heart and should be addressed in future studies.
Figure 6.

Immunoblot analysis of the levels of Ca2+-handling proteins from Na,K-β1 KO and WT hearts. Representative blots from three independent experiments are shown. The numbers below each panel represent the ratio of band intensities between the specific protein and the loading control tubulin normalized to the WT. For PMCA and NCX1, the major bands are pointed by arrows. Multiple bands representing either aggregation or degradation products have been reported for both PMCA and NCX1.
ACKNOWLEDGEMENTS
This work was supported by a grant from the National Institutes of Health (RO1-DK56216) and the Nemours Foundation. We thank Drs. Hong Wu and Xin Liu, University of California, Los Angeles, for providing us the DNA vector, pBS246 and helpful advice for generating the conditional knockout targeting construct. We thank Dr. Yibin Wang, University of California, Los Angeles, for making the MLC-2v Cre mice available to us. We also thank Dr. Pablo Martin-Vasallo, from Universidad de La Laguna, Tenerife, Spain, for the gift of Na,K-β1 and Na,K-α2 antibodies, and Dr. William James Ball Jr., University of Cincinnati, Cincinnati, for Na,K-α1 antibody. Technical assistance from Dr. Anilkumar Gopalakrishnapillai, Ryan McSpadden and Candice Krauthauser is gratefully acknowledged. We thank Dr. Kenneth Philipson, University of California, Los Angeles, for critical reading of the manuscript and for providing antibodies against Ca2+-handling proteins.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.McDonough AA, Velotta JB, Schwinger RH, Philipson KD, Farley RA. The cardiac sodium pump: structure and function. Basic Res Cardiol. 2002;97 Suppl 1:I19–I24. doi: 10.1007/s003950200024. [DOI] [PubMed] [Google Scholar]
- 2.Garty H, Karlish SJ. Role of FXYD proteins in ion transport. Annu Rev Physiol. 2006;68:431–459. doi: 10.1146/annurev.physiol.68.040104.131852. [DOI] [PubMed] [Google Scholar]
- 3.Geering K. FXYD proteins: new regulators of Na-K-ATPase. Am J Physiol Renal Physiol. 2006 Feb;290(2):F241–F250. doi: 10.1152/ajprenal.00126.2005. [DOI] [PubMed] [Google Scholar]
- 4.Crambert G, Fuzesi M, Garty H, Karlish S, Geering K. Phospholemman (FXYD1) associates with Na,K-ATPase and regulates its transport properties. Proc Natl Acad Sci U S A. 2002 Aug 20;99(17):11476–11481. doi: 10.1073/pnas.182267299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Juhaszova M, Shimizu H, Borin ML, Yip RK, Santiago EM, Lindenmayer GE, et al. Localization of the Na(+)-Ca2+ exchanger in vascular smooth muscle, and in neurons and astrocytes. Ann N Y Acad Sci. 1996 Apr 15;779:318–385. doi: 10.1111/j.1749-6632.1996.tb44804.x. [DOI] [PubMed] [Google Scholar]
- 6.Dostanic I, Schultz Jel J, Lorenz JN, Lingrel JB. The alpha 1 isoform of Na,K-ATPase regulates cardiac contractility and functionally interacts and co-localizes with the Na/Ca exchanger in heart. J Biol Chem. 2004 Dec 24;279(52):54053–54061. doi: 10.1074/jbc.M410737200. [DOI] [PubMed] [Google Scholar]
- 7.James PF, Grupp IL, Grupp G, Woo AL, Askew GR, Croyle ML, et al. Identification of a specific role for the Na,K-ATPase alpha 2 isoform as a regulator of calcium in the heart. Mol Cell. 1999 May;3(5):555–563. doi: 10.1016/s1097-2765(00)80349-4. [DOI] [PubMed] [Google Scholar]
- 8.Magyar JP, Bartsch U, Wang ZQ, Howells N, Aguzzi A, Wagner EF, et al. Degeneration of neural cells in the central nervous system of mice deficient in the gene for the adhesion molecule on Glia, the beta 2 subunit of murine Na,K-ATPase. J Cell Biol. 1994 Nov;127(3):835–845. doi: 10.1083/jcb.127.3.835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bell JR, Kennington E, Fuller W, Dighe K, Donoghue P, Clark JE, et al. Characterization of the phospholemman knockout mouse heart: depressed left ventricular function with increased Na-K-ATPase activity. Am J Physiol Heart Circ Physiol. 2008 Feb;294(2):H613–H621. doi: 10.1152/ajpheart.01332.2007. [DOI] [PubMed] [Google Scholar]
- 10.Cheung JY. Regulation of cardiac contractility: High time for FXYD. Am J Physiol Heart Circ Physiol. 2008;294:584–585. doi: 10.1152/ajpheart.91430.2007. [DOI] [PubMed] [Google Scholar]
- 11.Rajasekaran SA, Barwe SP, Rajasekaran AK. Multiple functions of Na,K-ATPase in epithelial cells. Semin Nephrol. 2005 Sep;25(5):328–334. doi: 10.1016/j.semnephrol.2005.03.008. [DOI] [PubMed] [Google Scholar]
- 12.Rajasekaran SA, Rajasekaran AK. Na,K-ATPase and epithelial tight junctions. Front Biosci. 2009;14:2130–2148. doi: 10.2741/3367. [DOI] [PubMed] [Google Scholar]
- 13.Gloor S, Antonicek H, Sweadner KJ, Pagliusi S, Frank R, Moos M, et al. The adhesion molecule on glia (AMOG) is a homologue of the beta subunit of the Na,K-ATPase. J Cell Biol. 1990 Jan;110(1):165–174. doi: 10.1083/jcb.110.1.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu L, Askari A. Beta-subunit of cardiac Na+-K+-ATPase dictates the concentration of the functional enzyme in caveolae. Am J Physiol Cell Physiol. 2006 Oct;291(4):C569–C578. doi: 10.1152/ajpcell.00002.2006. [DOI] [PubMed] [Google Scholar]
- 15.Henderson SA, Goldhaber JI, So JM, Han T, Motter C, Ngo A, et al. Functional adult myocardium in the absence of Na+-Ca2+ exchange: cardiac-specific knockout of NCX1. Circ Res. 2004 Sep 17;95(6):604–611. doi: 10.1161/01.RES.0000142316.08250.68. [DOI] [PubMed] [Google Scholar]
- 16.Chu DK, Jordan MC, Kim JK, Couto MA, Roos KP. Comparing isoflurane with tribromoethanol anesthesia for echocardiographic phenotyping of transgenic mice. J Am Assoc Lab Anim Sci. 2006 Jul;45(4):8–13. [PubMed] [Google Scholar]
- 17.Roos KP, Jordan MC, Fishbein MC, Ritter MR, Friedlander M, Chang HC, et al. Hypertrophy and heart failure in mice overexpressing the cardiac sodium-calcium exchanger. J Card Fail. 2007 May;13(4):318–329. doi: 10.1016/j.cardfail.2007.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dostanic I, Lorenz JN, Schultz Jel J, Grupp IL, Neumann JC, Wani MA, et al. The alpha2 isoform of Na,K-ATPase mediates ouabain-induced cardiac inotropy in mice. J Biol Chem. 2003 Dec 26;278(52):53026–53034. doi: 10.1074/jbc.M308547200. [DOI] [PubMed] [Google Scholar]
- 19.Barwe SP, Anilkumar G, Moon SY, Zheng Y, Whitelegge JP, Rajasekaran SA, et al. Novel role for Na,K-ATPase in phosphatidylinositol 3-kinase signaling and suppression of cell motility. Mol Biol Cell. 2005 Mar;16(3):1082–1094. doi: 10.1091/mbc.E04-05-0427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jia LG, Donnet C, Bogaev RC, Blatt RJ, McKinney CE, Day KH, et al. Hypertrophy, increased ejection fraction, and reduced Na-K-ATPase activity in phospholemman-deficient mice. Am J Physiol Heart Circ Physiol. 2005 Apr;288(4):H1982–H1988. doi: 10.1152/ajpheart.00142.2004. [DOI] [PubMed] [Google Scholar]
- 21.Mitra R, Morad M. A uniform enzymatic method for dissociation of myocytes from hearts and stomachs of vertebrates. Am J Physiol. 1985 Nov;249(5 Pt 2):H1056–H1060. doi: 10.1152/ajpheart.1985.249.5.H1056. [DOI] [PubMed] [Google Scholar]
- 22.Chen J, Kubalak SW, Chien KR. Ventricular muscle-restricted targeting of the RXRalpha gene reveals a non-cell-autonomous requirement in cardiac chamber morphogenesis. Development. 1998 May;125(10):1943–1949. doi: 10.1242/dev.125.10.1943. [DOI] [PubMed] [Google Scholar]
- 23.Chen J, Kubalak SW, Minamisawa S, Price RL, Becker KD, Hickey R, et al. Selective requirement of myosin light chain 2v in embryonic heart function. J Biol Chem. 1998 Jan 9;273(2):1252–1256. doi: 10.1074/jbc.273.2.1252. [DOI] [PubMed] [Google Scholar]
- 24.Rajasekaran SA, Gopal J, Willis D, Espineda C, Twiss JL, Rajasekaran AK. Na,K-ATPase beta1-subunit increases the translation efficiency of the alpha1-subunit in MSV-MDCK cells. Mol Biol Cell. 2004 Jul;15(7):3224–3232. doi: 10.1091/mbc.E04-03-0222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McDonough AA, Zhang Y, Shin V, Frank JS. Subcellular distribution of sodium pump isoform subunits in mammalian cardiac myocytes. Am J Physiol. 1996 Apr;270(4 Pt 1):C1221–C1227. doi: 10.1152/ajpcell.1996.270.4.C1221. [DOI] [PubMed] [Google Scholar]
- 26.Reuter H, Henderson SA, Han T, Ross RS, Goldhaber JI, Philipson KD. The Na+-Ca2+ exchanger is essential for the action of cardiac glycosides. Circ Res. 2002 Feb 22;90(3):305–308. doi: 10.1161/hh0302.104562. [DOI] [PubMed] [Google Scholar]
- 27.Barwe SP, Kim S, Rajasekaran SA, Bowie JU, Rajasekaran AK. Janus model of the Na,K-ATPase beta-subunit transmembrane domain: distinct faces mediate alpha/beta assembly and beta-beta homo-oligomerization. J Mol Biol. 2007 Jan 19;365(3):706–714. doi: 10.1016/j.jmb.2006.10.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Weber P, Bartsch U, Schachner M, Montag D. Na,K-ATPase subunit beta1 knock-in prevents lethality of beta2 deficiency in mice. J Neurosci. 1998 Nov 15;18(22):9192–9203. doi: 10.1523/JNEUROSCI.18-22-09192.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schwinger RH, Bundgaard H, Muller-Ehmsen J, Kjeldsen K. The Na, K-ATPase in the failing human heart. Cardiovasc Res. 2003 Mar 15;57(4):913–920. doi: 10.1016/s0008-6363(02)00767-8. [DOI] [PubMed] [Google Scholar]
- 30.Schwinger RH, Wang J, Frank K, Muller-Ehmsen J, Brixius K, McDonough AA, et al. Reduced sodium pump alpha1, alpha3, and beta1-isoform protein levels and Na+,K+-ATPase activity but unchanged Na+-Ca2+ exchanger protein levels in human heart failure. Circulation. 1999 Apr 27;99(16):2105–2112. doi: 10.1161/01.cir.99.16.2105. [DOI] [PubMed] [Google Scholar]
- 31.Ishino K, Botker HE, Clausen T, Hetzer R, Sehested J. Myocardial adenine nucleotides, glycogen, and Na, K-ATPase in patients with idiopathic dilated cardiomyopathy requiring mechanical circulatory support. Am J Cardiol. 1999 Feb 1;83(3):396–399. doi: 10.1016/s0002-9149(98)00876-5. [DOI] [PubMed] [Google Scholar]
- 32.Norgaard A, Bagger JP, Bjerregaard P, Baandrup U, Kjeldsen K, Thomsen PE. Relation of left ventricular function and Na,K-pump concentration in suspected idiopathic dilated cardiomyopathy. Am J Cardiol. 1988 Jun 1;61(5):1312–1315. doi: 10.1016/0002-9149(88)91175-7. [DOI] [PubMed] [Google Scholar]