

Abstract
Neonatal rats exposed to (+)-methamphetamine (MA) display spatial learning and reference memory deficits in the Morris water maze. In separate experiments the emergence and permanence of these effects were determined. Twenty litters were used in each experiment, and two male/female pairs/litter received saline or MA (5 mg/kg four times a day) on postnatal days (P) 11–20. In experiment 1, one MA and one saline pair from each litter began testing on either P30 or P40, whereas in experiment 2, testing began on P180 or P360. Animals received trials in a straight swimming channel and then in the Morris maze (acquisition, reversal, and reduced platform phases). In both experiments, MA-treated groups showed impaired learning in the platform trials and impaired reference memory in the probe trials, which were largely independent of age. The P30 and P40 MA impairments were seen on acquisition and reduced platform trials but not on reversal. In the probe trials, MA effects were seen during all phases. The P180 and P360 MA-induced deficits were seen in all phases of the platform trials. In probe trials, deficits were only seen during the reversal and reduced platform phases. The results demonstrate that neonatal MA treatment induces spatial learning and reference memory deficits that emerge early and persist until at least 1 year of age, suggesting permanence.
Keywords: cognition, development, developmental effects, methamphetamine, Morris water maze, rats, reference memory, spatial learning
Introduction
(+)-Methamphetamine (MA) is a widely abused drug, especially among adolescents and young adults. Annual US prevalence rates from 1999 to 2005 have varied from 2.5 to 4.7% for high school seniors (Johnston et al., 2006a), and from 2.4 to 2.8% for those between 19 and 28 years of age (Johnston et al., 2006b). By comparison, annual prevalence rates for cocaine are 4.8–6.2% for high school seniors and 5.4–7.1% for 19–28 year olds. For women, rates of drug use are highest among those of reproductive age (Day et al., 1993). Such widespread use inevitably leads to significant numbers of children being exposed prenatally. The prenatal effects of cocaine have been well documented (e.g. Richardson et al., 1996a, b; Lester et al., 1998). By contrast, little information is available on the effects of prenatal MA exposure.
Considerable information is available about the effects of MA on the mature brain (e.g. Wilson et al., 1996; Moszczynska et al., 2004; Meredith et al., 2005; Barr et al., 2006), but much less is known about its effects on the developing brain. Early studies of infants exposed to MA (Little et al., 1988) or MA and cocaine found reduced birth weight, increased rates of prematurity, and intra-ventricular hemorrhage (Oro and Dixon, 1987; Dixon and Bejar, 1989). MA–cocaine exposed infants were also found to have increased signs of withdrawal, using the Finnegan rating scale (sleep disturbances, tremors, poor feeding, hypertonia, vomiting, increased sneezing, high-pitched cry, increased fist sucking, tachypnea, loose stool, fever, hyperreflexia, increased yawning, and excoriation) (Dixon, 1989). In addition, 78% of flash-evoked visual potentials were abnormal in MA-cocaine exposed infants. A later study found reduced visual recognition memory on the Fagan test of Infant Intelligence among retrospectively ascertained MA-exposed infants of 6–12 months of age (Struthers and Hansen, 1992).
More recently, reduced creatine and glutamate/glutamine ratios in prenatally MA-exposed children were found by magnetic resonance spectroscopy (Smith et al., 2001). Magnetic resonance imaging identified volumetric reductions in the hippocampus and other forebrain areas in prenatally MA-exposed children (Chang et al., 2004). In a large study, 13 808 pregnant women were screened. Of these, 7119 were eligible for the study, 1618 consented to participate, and 84 were identified as abusing MA. The MA-exposed infants had reduced birth weight and increased rates of being small for gestational age (Smith et al., 2006). Fifty-six percent of the MA-abusing women used it during the first and second trimesters, whereas 44% used it throughout pregnancy (Smith et al., 2006). This indicates that nearly half of all women who use MA during gestation have third-trimester exposure. In a study from Thailand, 47 prenatally MA-exposed and 49 MA-unexposed infants were examined; it was found that MA-exposed infants had reduced birth weight and head circumference, and higher rates of being small for gestational age. They also reported that MA-exposed infants showed increased agitation, vomiting, and tachypnea (Chomchai et al., 2004).
In rats, we developed models of the effects of MA exposure during the early (Acuff-Smith et al., 1995) and later (Vorhees et al., 1994a, b) stages of brain development, analogous to the first and second half of human intrauterine brain development (West and Pierce, 1987; Bayer et al., 1993; Rice and Barone, 2000; Clancy et al., 2001, 2006, 2007; Herlenius and Lagercrantz, 2004). We found that (male and female) rats exposed to MA from P11 to P20 exhibit long-term spatial learning and memory deficits in the Morris water maze (MWM), whereas cued MWM performance and multiple T-maze learning in the Cincinnati water maze (CWM) were spared. Moreover, no learning impairments were seen after exposure on P1–10 (Vorhees et al., 1994a, b, 2000). The effects of P11–20 MA exposure on spatial learning and reference memory in the MWM are also seen in ACI (Vorhees et al., 1998) and Dark Agouti rats (Vorhees et al., 1999), and have also been reported recently, after comparable exposure in mice (Acevedo et al., 2007). In rats, these exposures caused no changes in MWM matching-to-sample performance, in which the platform was moved to new locations daily (Williams et al., 2003b). Using the same exposure, that is from P11–20, with half of each litter receiving pretraining to a randomly placed platform and half receiving no pretraining, it was revealed that the MA-induced MWM deficits represent a compound impairment; pretrained rats no longer showed acquisition deficits, but still showed reversal deficits, whereas non-pretrained MA-exposed animals showed both acquisition and reversal deficits (Williams et al., 2002).
Follow-up experiments showed that doses as low as 0.625 mg/kg × 4 doses/day on P11–20 induce MWM performance deficits, although higher doses are more effective (Williams et al., 2004b). In addition, treatment of offspring on P11–15 or P16–20 caused MWM deficits only in the P11–15 exposed MA group, but not in the P16–20 MA group (Williams et al., 2003a).
The mechanism by which P11–20 MA treatment affects brain development is not yet known. P11–20 MA treatment causes significant increases in the release of corticosterone (Williams et al., 2000) and this effect is both age-dependent and previous drug experience-dependent. For example, we found a U-shaped response function, in which corticosterone release was higher 30 min after MA treatment on P1 and 3 than on P5, 7, 9, 11, or 13, and was higher again on P15, 17, and 19. When examined 105 min after dosing, corticosterone release showed a similar U-shaped response with highest responses on P1, followed by P3 and 5, attenuated responses on P7, 9, 11, and 13, and larger responses on P15, 17, and 19. If the drug was, however, given chronically (4 doses/day) for 4 days with one final dose on the 5th day, the 30-min drug × age corticosterone response was no longer U-shaped but increased progressively with age. This was evident as the 30-min corticosterone levels showed smaller increases on P5 or 7 than after an acute dose, but were dramatically higher on P13, 15, 17, and 19 than after an acute dose. By contrast, 105-min corticosterone levels were lower on all these days than after an acute dose. This suggests that the attenuated corticosterone response on P7–15 seen after acute MA (Williams et al., 2006) follows a pattern similar to that observed during the stress hyporesponsive period (Sapolsky and Meaney, 1986), that is, on P2–14 when stressors do not elicit an adult-like release of corticosterone. In contrast, chronic MA treatment modifies this pattern by inhibiting the early corticosterone response and amplifying the later response (after P15). Therefore, one effect of MA exposure during development is to induce an exaggerated stress-like corticosterone release, especially as levels are still elevated 18 h after the fourth dose (Schaefer et al., 2006). What causes the prolonged rise in corticosterone or how it relates to the later cognitive deficits is not known.
MA treatment on P11–20 also results in long-term changes to dopaminergic systems. Rats treated with 10 mg/kg × 4/day MA on P11–20 and assessed at P90 show 10–15% reductions in neostriatal 3H-spiperone binding to D2-like receptors with no changes in affinity, 20% reductions in neostriatal protein kinase activity, and 15% reductions in neostriatal dopamine and DOPAC tissue concentrations (Crawford et al., 2003). When MA is administered on P11, and dopamine and DOPAC are examined on P12, no differences in monoamine levels are observed compared with saline-treated animals (Schaefer et al., 2006), indicating that chronic dosing is required to induce long-term neurochemical changes in young animals. These effects were found in both male and female animals.
Another aspect of modeling the long-term effects of early MA exposure is determining how soon the deficits emerge and how long they last. The latter is particularly important: if the effects are transient or dissipate with age, they might be of less significance than if they are permanent. In contrast, if the effects are long lasting, they may have more serious implications. Accordingly, the purpose of the present experiments was to ascertain whether the spatial learning deficits observed previously, after P11–20 MA exposure, emerge early (animals that began testing on P30 or P40) and whether they persist (animals that began testing on P180 or P360).
Methods
Subjects
Sprague–Dawley CD IGS rats weighing 251–275 g (males) and 151–175 g (females) were obtained from Charles River (Raleigh, North Carolina, USA) and acclimatized for a minimum of 2 weeks before breeding. Detection of a sperm plug was designated embryonic day 0 (E0). Before parturition, gravid female rats were transferred to polycarbonate cages. Starting on E21, female rats were checked for the presence of a litter; birth was counted as P0 (postnatal day 0). On P1, litters were randomly culled to four male and four female rats. On P11, offspring were uniquely identified (ear-punched). Food and water were provided ad libitum in rooms where ambient temperature was controlled at 21 ± 1°C and humidity at 50 ± 10%. The vivarium is accredited by the Association for the Assessment and Accreditation of Laboratory Animal Care (AAALAC), and the protocol was approved by the Institutional Animal Care and Use Committee of the Cincinnati Children’s Research Foundation. Dams were separated from their litters on P28. Thereafter, same-sex pairs from the same litters were housed together, with one treated and one control animal in the same cage.
Methamphetamine and treatments
Two experiments were performed. For each experiment, 20 litters were used. In each experiment, half of the male and half of the female animals in each litter received MA and the remaining half received saline (Sal): hence two male and two female rats per litter received MA and two male and two female rats per litter received Sal (split-litter design). (+)-Methamphetamine HCl (MA) was obtained from the National Institute on Drug Abuse (purity > 95%). The dose of MA (calculated as the free base) was 5 mg/kg body weight and was given subcutaneously four times a day at 2-h intervals on P11–20. Control animals were administered an equivalent volume of saline (3 ml/kg).
The two experiments were identical in the treatment aspects and differed only in the test ages of the offspring. In each experiment, half of the MA-treated and half of the Sal-treated offspring in each litter were tested at each of two separate ages. In experiment 1, half of each litter (i.e. one male and one female MA-treated pair and one male and one female Sal-treated pair) was tested beginning on either P30 or P40. In experiment 2, half of each litter was tested beginning on either P180 or P360.
Body weights
Body weights were obtained before each dose on P11–20; however, only two weights from each day were used in the analysis of body weights during dosing (before the first and last doses). Preweaning and adult weights were obtained once weekly beginning on P21 and continued through P91 in the first experiment, and through P238 (for the P180 test group) or P392 (for the P360 test group) in the second experiment.
Behavioral assessments
Straight channel swimming (latency to reach an escape ladder) was assessed in a 15 × 244 cm channel with 20 cm high sides filled to a depth of 10 cm with water on four trials (2-min limit/trial). This procedure was used to assess swimming ability and motivation to escape from water and to teach the animals that escape was possible.
MWM hidden platform trials were conducted in three phases, each consisting of 4 trials/day on each of 5 consecutive days, as described previously (Vorhees and Williams, 2006). The apparatus was a 210-cm (diameter) black circular stainless-steel water tank with 51-cm high walls filled to a depth of 30 cm with water (temperature 21 ± 1°C). Phase I (acquisition) used a platform of area 10 × 10 cm submerged 2 cm below the water. Phase II (reversal) used the same platform, but it was positioned diagonally in the opposite quadrant. Phase III (reduced) used a smaller 5 × 5 cm platform placed back in the original quadrant location. For all trials, rats were given a 2-min trial limit and 15-s intertrial interval (ITI) on the platform. If an animal did not find the platform within 2 min, it was placed on the platform. Platform positions within each phase were counterbalanced (e.g. for acquisition, half of the animals had the platform in the SE and half in the NW quadrant). Start positions were arranged such that those adjacent to the quadrant containing the platform were excluded. Rats were placed in the water facing the wall. Performance was recorded automatically using a video-tracking system (Polytrack; San Diego Instruments, San Diego, California, USA). Dependent variables were path length, cumulative distance, latency, and angle of first bearing to the goal.
MWM probe trials were conducted on the 6th day of each phase. Each probe trial was 30 s with the platform removed. Start positions on probe trials were from a location 180° to the goal, which was not one of the four learning trial start locations. Dependent variables on probe trials were angle of first bearing, average distance to the target site, and percent time spent in the target quadrant.
Statistics
Body-weight data were analyzed by analysis of variance (ANOVA) for repeated measures using the general linear model (SAS Proc GLM, SAS Institute, Cary, North Carolina, USA) with treatment Group, Age of testing, and Sex as within-litter factors; therefore litter was the statistical unit in these analyses. Variance–covariance matrices were tested for sphericity. In cases where matrices were significantly nonspherical, the Greenhouse–Geisser adjusted degrees of freedom and F ratios were used. Significant interactions with treatment were further analyzed using simple-effect ANOVAs on each day. As litter effects are an important source of variance in developmental studies, behavioral data were analyzed using a mixed general linear-model ANOVA (SAS Proc Mixed, SAS Institute). In this model, litter was a block factor. With litter accounted for in the block factor, treatment Group, Sex, platform Position, and test Age were between-group factors. Day was a repeated-measure factor. Variance–covariance models were selected based on best-fit statistics for each dependent variable. The models were either compound symmetry or first-order autoregressive. Significant interactions were further analyzed on each day using the slice-ANOVA option within Proc Mixed, which, as there were only two groups at each test age, was functionally equivalent to a t-test. For clarity of presentation, only significant Group (MA or Sal) main effects or interactions are provided with F ratios. Significance was set at P ≤ 0.05.
Results
Experiment 1
Mortality
No animal died in experiment 1.
Body weight
Body weights obtained during treatment showed that there were significant effects of MA treatment. There was a significant main effect of Group [F(1,19) = 462.21, P < 0.001], and significant interactions of Group × Day [F(9,171) = 436.20, P < 0.001], Group × Time of Day [F(1,19) = 6.60, P < 0.02], and Group × Day × Time of Day [F(9,171) = 11.43, P < 0.001]. Further analyses of the interactions on each day and at each time of day revealed that the groups did not differ on P11 before the first morning injection, but were different in the afternoon before the second injection. Group differences were found on all other days and times from P12 through P20. In all cases, MA treatment significantly reduced body weight compared with Sal treatment. The body weight taken closest to the beginning of testing is shown in Table 1. No interactions were observed between Sex and Group.
Table 1.
Body weights (mean ± SEM) in grams (N)
| Group | Sal | MA | Sal | MA | ||
|---|---|---|---|---|---|---|
| Test age | P30 | P40 | ||||
| Nearest weighing age | P28 | P35 | ||||
| Female | 95.0 ± 2.1 (20) | 83.7 ± 1.9* (20) | 140.2 ± 2.8 (20) | 131.0 ± 2.0* (20) | ||
| Male | 103.7 ± 2.0 (20) | 88.7 ± 2.3* (20) | 163.9 ± 3.1 (20) | 150.4 ± 2.8 (20) | ||
| Test age | P180 | P360 | ||||
| Nearest weighing age | P175 | P357 | ||||
| Female | 342.3 ± 10.8 (20) | 365.5 ± 10.7 (19) | 498.7 ± 26.4 (19)a | 503.5 ± 23.0 (19) | ||
| Male | 692.6 ± 16.6 (20) | 711.8 ± 19.7 (20) | 841.1 ± 32.1 (19) | 874.3 ± 27.3 (19) |
P<0.05 vs. Sal. MA, methamphetamine; Sal, saline.
One additional animal in this group died before completing all phases of the Morris water maze and its data were excluded from the phase it did not complete.
Animals were weighed one day after the end of treatment (P21) and on the day they were separated from their dams (P28). An ANOVA on these body weights showed a significant Group main effect [F(1,19) = 264.84, P < 0.001]; no other main effects or interactions were significant. After weaning, animals continued to be weighed weekly until the end of the experiment (P35-91). An ANOVA of these adolescent through adult body weights showed no treatment main effect. The main effects of Age and Sex were significant as expected (P < 0.001). The Group × Age interaction showed a nonsignificant trend (P < 0.07).
Straight channel
Straight channel swimming times were unaffected by MA treatment regardless of test age group (P30 or P40), indicating no differences in swimming ability between the groups. There were no interactions with Sex.
Morris water maze
Four primary measures of performance were used to assess learning: latency, path length, cumulative distance from the platform, and first bearing (i.e. the angle of deviation from a direct line to the platform center, based on average directional heading during the first 13 cm of movement at the beginning of each trial). We have previously demonstrated that latency, path length, and cumulative distance are highly correlated with each other (r > 0.93), whereas first bearing, because it reflects initial performance rather than the entire trial, is less correlated with the three other measures (r = 0.41–0.57) (Vorhees and Williams, 2006). Accordingly, we use path length for illustrative purposes and describe the latency and cumulative distance effects. We use first bearing to reflect the animal’s initial sense of where to go. A third measure we use to clarify certain trends in data is peripheral swim time (thigmotaxis). This has been shown to be useful in dissecting the effects of other treatments (Cain and Saucier, 1996; Cain et al., 1996) and we have previously shown its value in rats neonatally treated with MA (Williams et al., 2002). On probe trials, we use average distance from the platform, number of platform site crossings, first bearing, and percent time and distance in the target quadrant. Average distance and first bearing are used for illustrative purposes.
Phase 1, acquisition The Group × Day interaction on path length was significant [F(4,408) = 2.78, P < 0.05], but the main effect of Group and Group × Age interaction was not; there were no interactions with Sex. Predicted pairwise group comparisons at each age, based on the Group × Age interaction on average distance, showed that the MA-treated P40 group was farther from the platform site than the Sal-treated P40 controls (P < 0.05). The learning curves at each age and combined across ages for path length are illustrated in Fig. 1. For latency, the Group main effect was significant [F(1,126) = 7.47, P < 0.01], as was the Group × Day interaction [F(4,401) = 2.56, P < 0.05]. For cumulative distance, the main effect of Group fell short of significance, as did the Group × Day or Group × Age interactions (both P < 0.06). The Group main effect was significant for first bearing [F(1,119) = 23.18, P < 0.001], and is illustrated in Fig. 2a. As can be seen, when the data are combined across test ages, the MA-treated group had larger average first bearings than Sal-treated controls. A follow-up analysis of time in the periphery confirmed that MA-treated offspring spent more time around the edge of the pool than Sal-treated controls [F(1,138) = 12.77, P < 0.001]; Fig. 2b).
Fig. 1.

Experiment 1. (P30, P40) Morris water maze, acquisition phase, platform trials for path length: (a), path length (cm) averaged across 4 trials/day and across sexes for the P30 groups. (b) Same as in panel (a) except that data are for the P40 groups. (c) Same as in (a) except that data are the combined averages of the P30 and P40 groups. Data are means ± SEM. Rats received 5 mg/kg methamphetamine (MA) four times a day on P11–20. **P<0.01 vs. saline.
Fig. 2.
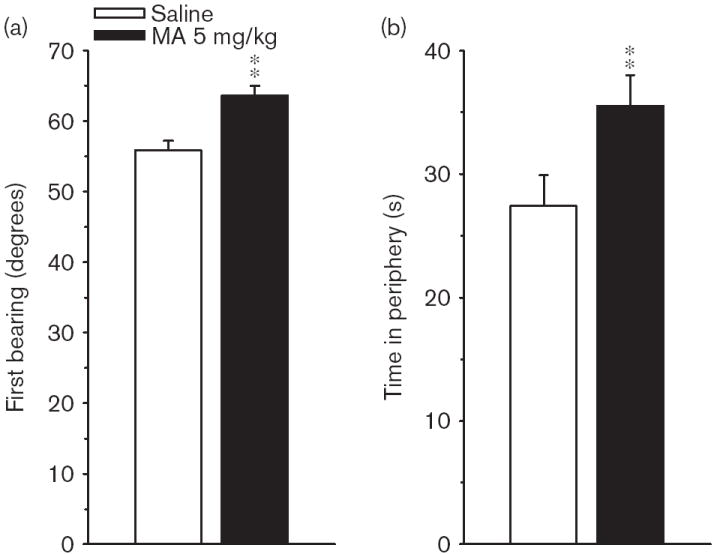
Experiment 1. (P30, P40) Morris water maze, acquisition phase, platform trials. (a) Angle of first bearing to the platform site measured in the first 13 cm at the start of each trial averaged across 4 trials/day for 5 days for male and female rats. (b) Time in the periphery of the maze (outer annulus) on platform trials averaged as in (a). Data are means ± SEM. Rats received 5 mg/kg methamphetamine (MA) four times per day on P11–20. **P<0.01.
On the probe trial, there was a significant Group × Age interaction on average distance [F(1,119) = 3.91, P < 0.05], a significant Group main effect on first bearing [F(1,119) = 10.87, P < 0.002], and a significant Group × Age interaction on percent distance in the target quadrant [F(1,119) = 3.99, P < 0.05]. No significant interactions were observed between Sex and Group. Group comparisons on percent distance in the target quadrant showed a deficit in the MA-treated P30 group compared with the P30 Sal-treated group (P < 0.05; not shown). ANOVAs on platform site crossings and percent time in the target quadrant were not significant.
Phase 2, reversal No significant main effects or interactions were observed on reversal trials for path length (Fig. 3), latency, or cumulative distance, and no interactions with Sex. A significant interaction was observed of Group × Age × Day [F(4,462) = 2.80, P < 0.05; Fig. 4a and b] on first bearing and a significant Group main effect, [F(1,133) = 13.02, P < 0.001; Fig. 4c]. There were no significant Group effects on time in the periphery. On the probe trial, there were no significant Group main effects on average distance, site crossings, percent time, or distance in the target quadrant, but there was a significant Group × Sex effect on first bearing [F(1,133) = 6.38, P < 0.02]. This was attributable to a significantly increased first bearing among MA-treated male rats averaged across test ages (Fig. 5).
Fig. 3.
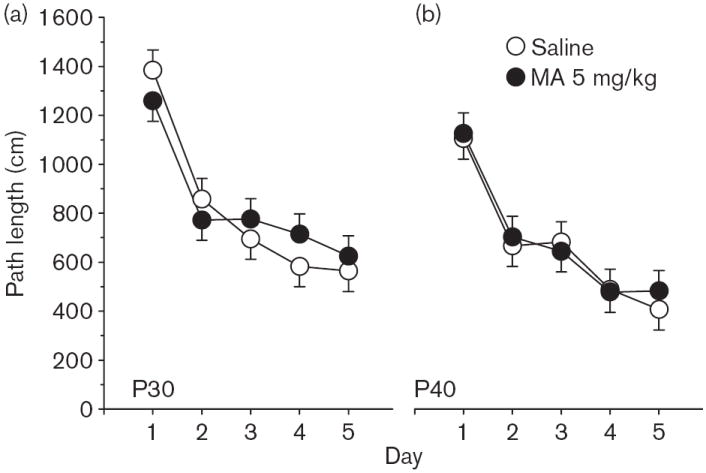
Experiment 1. (P30, P40) Morris water maze, reversal phase, platform trials for path length: (a) Path length (cm) averaged across 4 trials/day and across sexes for the P30 groups. (b) Same as in (a) except as applicable to the P40 groups. Data are means ± SEM. Rats received 5 mg/kg methamphetamine (MA) four times a day on P11–20.
Fig. 4.

Experiment 1. (P30, P40) Morris water maze, reversal phase, platform trials for first bearing: (a) First bearing (degrees) for the P30 groups averaged across 4 trials/day and across sexes. (b) Same as in (a) except as applied to the P40 groups. (c) The average across trials, days, sexes, and test ages to show the main effect of Group. Data are means ± SEM. Rats received 5 mg/kg methamphetamine (MA) four times a day on P11–20. *P<0.05 **P<0.01 vs. saline.
Fig. 5.
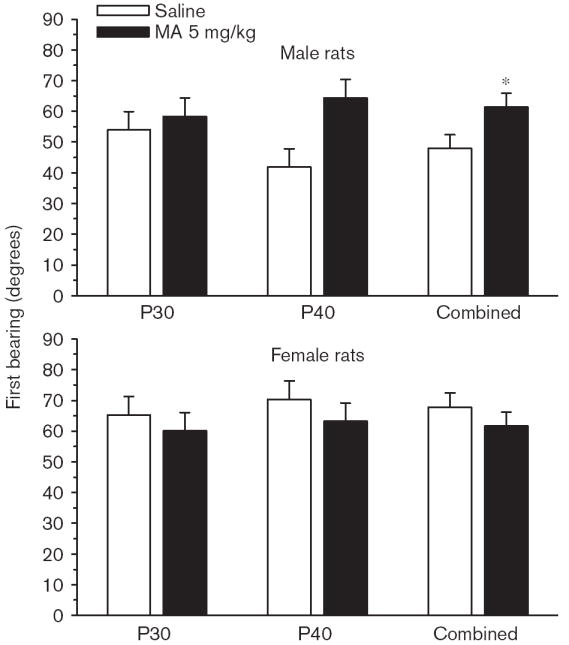
Experiment 1. (P30, P40) Morris water maze, reversal phase, first bearing probe trial: Data are shown separately for male and female rats to reflect Group × Sex interaction. Data are means ± SEM. Rats received 5 mg/kg methamphetamine (MA) four times a day on P11–20. *P<0.05 vs. saline.
Phase 3, reduced On the third phase of MWM, when a smaller, relocated platform was used, there were no significant Group main effects on path length, latency, or cumulative distance, but there were significant Group × Age × Day interactions on path length [F(4,424) = 3.60, P < 0.01], latency [F(4,424) = 2.85, P < 0.05], and cumulative distance [F(4,423) = 3.73, P < 0.01]. No significant interactions between Group and Sex were observed. For path length, the Group × Age × Day interaction is illustrated in Fig. 6a and b. As can be seen, the MA-treated P40 group had significantly longer path lengths on the first day of testing. A significant Group main effect was also seen on first bearing [F(1,126) = 5.27, P < 0.05; Fig. 6c], and a significant Group × Sex × Day × Age interaction [F(4,425) = 2.76, P < 0.05]. Converging evidence that the MA-treated animals at both P30 and P40 were impaired was provided by a follow-up ANOVA on time in the periphery, in which a significant Group main effect was observed [F(1,126) = 4.86, P < 0.05]. As can be seen in Fig. 6d, the MA-treated group spent approximately 30% more time in the periphery than Sal-treated controls. On the probe trial, average distance, site crossings, and quadrant analyses showed no significant Group main effects, but there was a significant Group main effect on first bearing [F(1,126) = 4.51 P < 0.05]. As can be seen in Fig. 7, the MA-treated animals were farther off course than the Sal-treated animals. There were no significant interactions between Group and Sex.
Fig. 6.

Experiment 1. (P30, P40) Morris water maze, phase 3: Phase 3 was with the platform position being moved to a different quadrant from that during acquisition or reversal and with a smaller (5 × 5 cm) platform. (a) Path lengths averaged across 4 trials/day and across sexes for the P30 groups. (b) Path lengths as in (a) but for the P40 age groups. (c) First bearing averaged across trials, days, sexes, and ages to show the main effect of Group. (d) Time in the periphery (s) averaged across trials, days, sexes, and ages to show the main effect of Group. Data are means ± SEM. Rats received 5 mg/kg methamphetamine (MA) four times a day on P11–20. *P<0.05 **P<0.01 vs. saline.
Fig. 7.
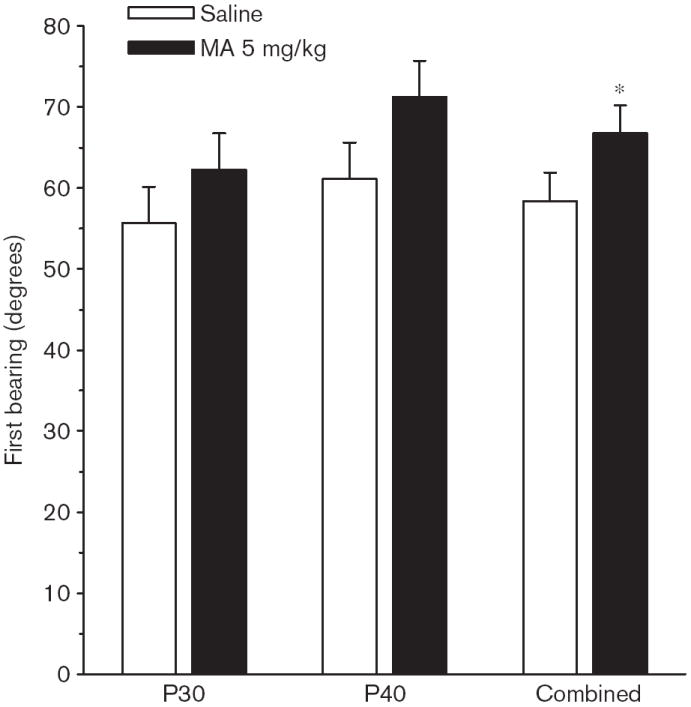
Experiment 1. (P30, P40) Morris water maze, phase 3, probe trial, first bearing: Data shown are for the P30 and P40 groups and for the combined averages of both test ages to show the main effect of Group. Data are averaged across sexes. Data are means ± SEM. Rats received 5 mg/kg methamphetamine (MA) four times a day on P11–20. *P<0.05 vs. saline.
Experiment 2
Mortality
No mortality occurred during dosing (P11–20) or during the week thereafter (P21–28). There were no deaths among the 40 Sal-treated female or the 40 Sal-treated male rats between P35 and P175. There was one death among the 40 MA-treated female rats during this time and no deaths among the 40 MA-treated male rats. Among the subgroups retained for testing beginning on P360, one death occurred in each of the four groups (Table 1) between P175 and P357; hence the N in each cell at this test age was 19. In addition, one of the 19 animals in the Sal female subgroup died before completing all three phases of the MWM and its data for the incomplete phase were excluded.
Body weight
An ANOVA on body weights recorded during dosing on P11–20 showed a significant Group main effect [F(1,19) = 464.91, P < 0.001], and significant interactions of Group × Day [F(9,171) = 423.89, P < 0.001], and Group × Day × Time of Day [F(9,171) = 10.44, P < 0.001]. Further analyses on each day and at each time of day showed that there were no body weight differences on P11 before the first injection. Differences were significant before the last injection of the afternoon on P11 (P < 0.05) and for all remaining days and times (P12–20). No significant interactions between Group and Sex were observed.
An ANOVA on P21 and P28 body weights showed a significant Group main effect [F(1,19) = 389.32, P < 0.001], and significant interactions of Group × Age [F(1,19) = 5.80, P < 0.05], Group × Week [F(1,19) = 5.25, P < 0.05], and Group × Sex ×Week [F(1,19) = 5.69, P < 0.05]. When further analyzed for the Group × Age interaction, Group was significant for both age groups. When the Group × Sex × Week interaction was further analyzed, there were group differences among male and female rats at both P21 and P28. The interaction arose because the magnitude of the MA effect was slightly different for each sex at these two ages, but the pattern of differences was the same.
After P28, all animals were weighed weekly until P238, which was after the subgroup that began testing on P180 completed their testing. At that point they were killed and only the subgroup scheduled for testing beginning on P360 were retained and weighed weekly until after the end of their testing on P392. An omnibus ANOVA on the P35–238 body weights showed no significant main effect of Group, but a significant Group × Week interaction was obtained [F(29,551) = 4.15, P < 0.02]. When the interaction was analyzed, there were significant group differences on P35, 42, 49, and 56, but from P63 onward the MA groups recovered such that no significant differences remained. In the subgroup that continued on to P360, no significant Group main effects or interactions were seen. Body weights at the age nearest to the age of testing are shown in Table 1. At no point were there any significant interactions between Group and Sex.
Morris water maze
Phase 1, acquisition The main effect of Group on path length (Fig. 8a and b) was not significant but there was a significant Group × Day interaction F(4,431) = 2.55, P < 0.05. As can be seen in Fig. 8c, the MA-treated animals had longer path lengths on day 2 of acquisition than did the Sal-treated animals. This same interaction (Group × Day) was also significant for cumulative distance, F(4,427) = 2.84, P < 0.05, but fell short of significance for latency (P < 0.09). There was a significant Group main effect on first bearing [F(1,126) = 10.16, P < 0.002], as well as a significant Group × Age interaction [F(1,126) = 4.53, P < 0.05]; both of these effects reflected the fact that the MA-treated animals were farther off course than the Sal-treated animals and that this was more pronounced for the P180 group compared with those tested at P360. No significant interactions of Group × Sex were obtained.
Fig. 8.

Experiment 2. (P180, P360) Morris water maze, acquisition phase, platform trials, path length: (a) Path length (cm) averaged across 4 trials/day and across sexes for the P180 groups. (b) Same as in (a) except as applied to the P360 groups. (c) Same as in (a) except that data are the combined averages of the P180 and P360 groups. Data are means ± SEM. Rats received 5 mg/kg methamphetamine. (MA) four times a day on P11–20. *P<0.05 vs. saline.
On the probe trial there was a significant Group main effect on first bearing [F(1,114) = 6.00, P < 0.02], which is illustrated in Fig. 9. The first bearing data show that the MA-treated group was significantly more off course than the Sal-treated group across both ages combined. There were no significant Group effects on average distance, site crossings, or quadrant time or distance, and no Group × Sex interactions.
Fig. 9.
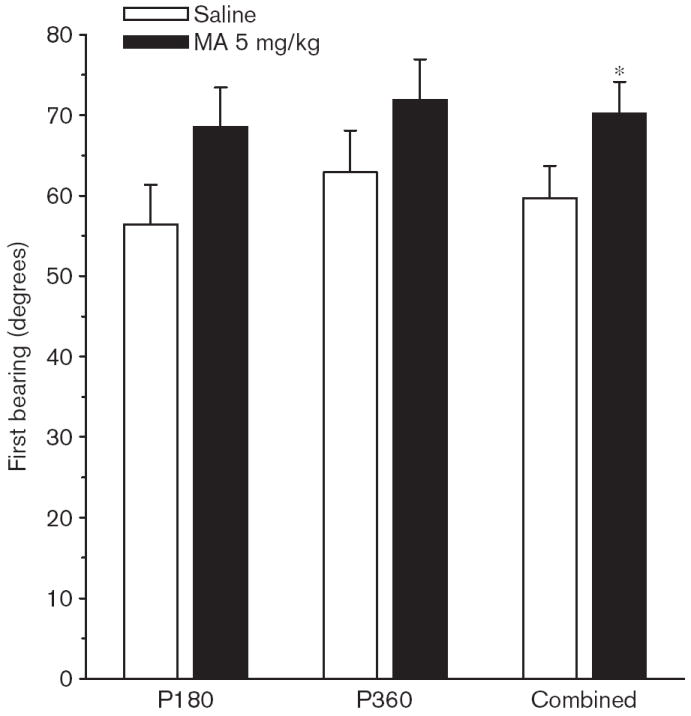
Experiment 2. (P180, P360) Morris water maze, acquisition phase, probe trial, first bearing: First bearing, averaged across male and female rats, for the groups tested at P180, P360 or the combined average of both. Data are means ± SEM. Rats received 5 mg/kg methamphetamine (MA) four times a day on P11-20. *P<0.05 vs. saline.
Phase 2, reversal The main effect of Group was significant for path length [F(1,119) = 5.87, P < 0.02]. None of the interactions was significant. As can been seen in Fig. 10, the MA-treated group had longer path lengths across ages. Although, visually, Fig. 10 suggests that the P360 group contributed more to this effect than the P180 age group, none of the interactions was significant. The main effect of Group was also significant for latency [F(1,118) = 9.72, P < 0.005], and cumulative distance [F(1,118) = 5.44, P < 0.05]. A significant Group main effect was also observed on first bearing [F(1,119) = 18.32, P < 0.001 (not shown)]. All of these effects confirmed what was found for path length.
Fig. 10.

Experiment 2. (P180, P360) Morris water maze, reversal phase, platform trials, path length: (a) Path length (cm) averaged across 4 trials/day and across sexes for the P180 groups. (b) Same as in (a) except as applied to the P360 groups. (c) Same as in (a) except that data are the combined averages of the P180 and P360 groups, which are also averaged across days, to show the main effect of Group. Data are means ± SEM. Rats received 5 mg/kg methamphetamine (MA) four times a day on P11–20. *P<0.05 vs. saline.
On the probe trial, there were significant main effects of Group on average distance [F(1,118) = 19.12, P < 0.001], site crossings [F(1,118) = 5.00, P < 0.05], and percent time [F(1,118) = 5.55, P < 0.02], and percent distance in the target quadrant [F(1,118) = 6.07, P < 0.02], but the effect on first bearing fell short of significance (P < 0.07). The effect on average distance is shown in Fig. 11, and as can be seen, the MA-treated group was farther from the platform site when averaged across test ages than the Sal-treated group. The interaction of Group × Sex was not significant.
Fig. 11.
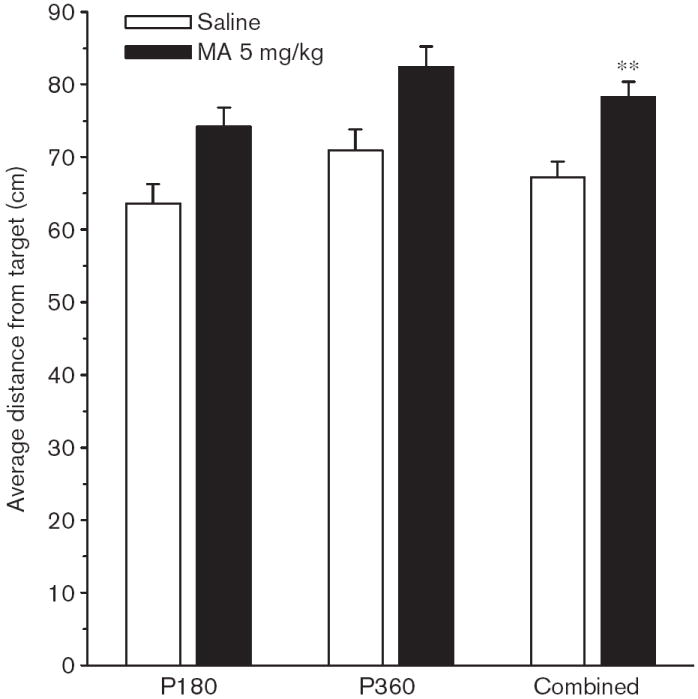
Experiment 2. (P180, P360) Morris water maze, reversal phase, probe trial, average distance: Average distance (cm) is the mean distance from the platform site recorded every 55 ms during the probe trial. Data are shown for average distances averaged across male and female rats tested at P180 and P360 and for the combination of both ages. Data are means ± SEM. Rats received 5 mg/kg methamphetamine (MA) four times a day on P11–20. **P<0.01 vs. saline.
Phase 3, reduced During the final phase of testing, when the platform was relocated to another quadrant and a smaller platform was used, there were significant main effects of Group on all measures: path length [F(1,126) = 6.33, P < 0.02]; latency [F(1,127) = 11.53, P < 0.001]; cumulative distance [F(1,126) = 7.31, P <0.01]; and first bearing [F(1,125) = 25.02, P < 0.001]. In addition, the Group × Age interaction was significant for latency [F(1,127) = 7.46, P < 0.01], but not for path length (P < 0.07), cumulative distance (P < 0.07), or first bearing (P < 0.08). The main effect of Group and the nonsignificant Group × Age interaction are illustrated in Fig. 12a–c. The primary contribution to the main effect of Group came from the longer path lengths of the MA-treated group at P360 compared with the MA-treated group at P180. There was also a significant Group × Sex interaction for path length [F(1,127) = 4.35, P < 0.05], and cumulative distance [F(1,127) = 4.52, P < 0.05], but not for latency (P > 0.10), or first bearing (P < 0.09). The interactions with Sex were attributable to significant impairments in path length and cumulative distance among female compared to male rats at both ages.
Fig. 12.

Experiment 2. (P180, P360) Morris water maze, reduced phase (phase 3), platform trials, path length: (a) Path length (cm) averaged across 4 trials/day and across sexes for the P180 groups. (b) Same as in (a) except as applied to the P360 groups. (c) Same as in (a) except that data are the combined averages of the P180 and P360 groups, averaged across days to show the main effect of Group. Data are means ± SEM. Rats received 5 mg/kg methamphetamine (MA) four times a day on P11–20. *P<0.05 vs. saline.
On the probe trial, there were significant Group main effects on average distance [F(1,119) = 10.09, P < 0.002], and site crossings [F(1,119) = 5.32, P < 0.05], but not on first bearing, or percent time or distance in the target quadrant. The effect on average distance is shown in Fig. 13. As can be seen, the MA-treated group was farther off course in swimming to the platform site than the Sal-treated group, when averaged across test ages. There were no interactions between Group and Sex.
Fig. 13.
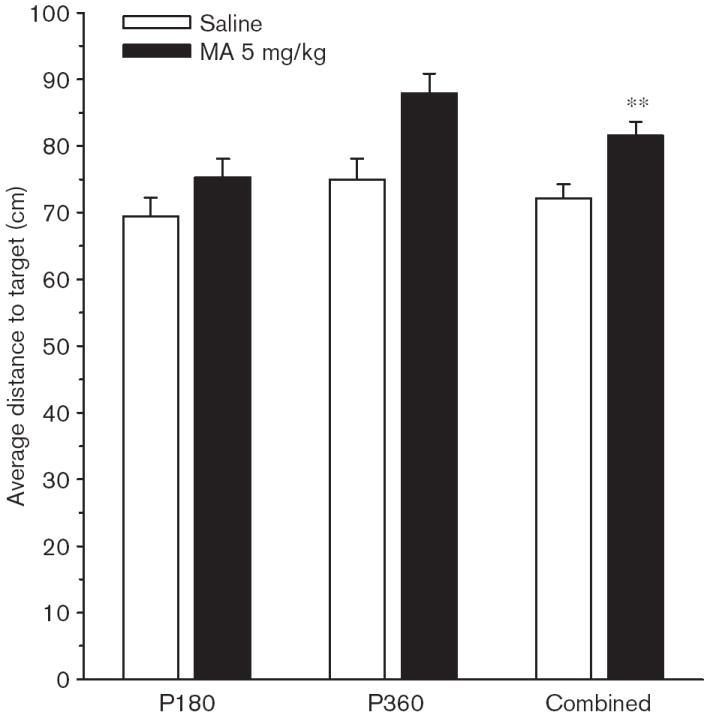
Experiment 2. (P180, P360) Morris water maze, reduced phase (phase 3), probe trial, average distance: Average distance (cm) is the mean distance from the platform site recorded every 55 ms. Data are shown as average distances averaged across male and female rats tested at P180 and P360 and across the combination of both ages. Data are means ± SEM. Rats received 5 mg/kg methamphetamine (MA) four times a day on P11–20. **P<0.01 vs. saline.
Discussion
The present experiment showed the following: (i) MWM spatial learning and reference memory deficits we had shown previously after P11–20 MA treatment and assessed at P60–90 are also observed at other ages (both earlier and later); (ii) P11–20 MA-induced MWM learning and memory deficits emerge early after the cessation of treatment and are present when testing begins as early as P30 or P40; (iii) there is little difference in the effect of MA whether testing begins on P30 or P40; (iv) P11–20 MA-induced MWM learning and memory deficits are long lasting and evident in groups tested beginning at P180 or P360; and (v) there is little difference in the effect of MA whether testing begins on P180 or P360. In the latter case, although the MA effects are obtained at both of these ages, the effects seem to be more pronounced at P360 than at P180, although the interaction was not significant. Whether this is an indication that the deficit worsens very slightly (but not significantly) with age or is the product of a less than perfect sampling distribution of treated animals at the two ages is unclear. Another possibility is that this difference is related to the size of the animals in relation to the platform. At P180, male rats had difficulty climbing onto the platform. This was especially noticeable during the reduced trial phase when the platform was 75% smaller. P180 animals climbed onto the platform but often fell off, forcing them to relocate the platform (although the trial had officially ended). At P360, the animals were even larger and could not stand on the smaller platform and merely held onto it to end the trial. It might be that falling off the platform among the P180 animals interfered with their learning. Examination of Fig 12a shows that saline-treated animals’ learning was progressing well on days 1 and 2 but deteriorated on day 3. From day 3 onward the saline-treated group’s performance resembled that of the MA-treated group. A predicted line for the saline-treated group based on their days 1 and 2 and extrapolated to days 3–5 or based on the actual performance of the P360 saline-treated group would be well below that of the MA-treated group, suggesting that some factor disrupted control performance on the reduced platform trials.
The finding that neonatal MA treatment results in spatial learning and reference memory deficits in the MWM suggests that MA can affect hippocampal development. Many experiments using lesions, NMDA antagonists, and gene targeting lead to deficits in MWM platform and probe trial performance (see Brandeis et al., 1989; D’Hooge and de Deyn, 2001; Burgess et al., 2002; Morris et al., 2003 for reviews). Consistent with this, we have shown that animals exposed to MA from P11–20 exhibit decreased spine density in the hippocampus (Williams et al., 2004a). Although treatments that disrupt hippocampal function reliably disrupt MWM performance, treatments that disrupt MWM performance are not necessarily restricted to the hippocampus, because injury to other brain regions can also result in impaired MWM performance (Brandeis et al., 1989).
We have shown here and previously that P11–20 MA treatment does not impair swimming speed (Vorhees et al., 2000; Williams et al., 2003b, 2004b). Moreover, path length and cumulative distance measures are insensitive to differences in swimming speed and these indices clearly showed impairments of spatial learning and memory. Although it is the case that MA-treated offspring exhibit task-specific as well as spatial impairments during the acquisition phase, task-specific effects do not contribute to impairments during reversal (Williams et al., 2002). We have also shown that MA-treated offspring do not show impairments on early trials on day 1 of testing, indicating that they do not have preexisting performance differences (Vorhees et al., 2000). Increased thigmotaxis during the acquisition phase can be attributed to lack of acquiring task-specific skill learning (Williams et al., 2002), as seen in the present experiment, but increased thigmotaxis during the reduced platform phase represents a very different behavior. From observing the swim paths of animals during this phase of testing, it is evident that MA-treated offspring enter the periphery only after they fail to find the platform where it had been located previously. The reason MA-treated offspring spend more time in the periphery of the maze on these trials is because they adapt more slowly to the new location of the platform than controls.
Drug studies in animals present complex problems for determining appropriate doses. Ideally, one would have physiologically-based pharmacokinetic data but such data are difficult to obtain. Pharmacokinetic data have been obtained in adult (Cho et al., 2001), neonatal (Cappon and Vorhees, 2001), and fetal rats (Acuff-Smith et al., 1995), but human fetal data have not been available. Another problem is species differences in metabolism (De La Torre and Farre, 2004). Human pharmacokinetic data come from low-dose experiments whereas abuse-related intake can only be extrapolated, with the assumption of linear pharmacokinetics, from levels in individuals arrested by police. In a recent report based on 105 arrestees suspected of drug intoxication, for example, the authors assumed that the users took the drug intravenously in four evenly spaced doses per day for 4 consecutive days (Melega et al., 2007). This model yielded an estimated median dose of 52 mg/dose, leading to an estimated 260 mg/dose for a high-dose user. On a weight-adjusted basis, these doses would (for a woman weighing 60 kg) be 0.87 mg/kg for a median user and 4.33 mg/kg for a high-dose user. These are similar to the doses used in the present work. Also, as rats metabolize MA more rapidly than humans (elimination half-life in humans is ~ 12 h whereas in rats it is ~ 1 h)(Cho et al., 2001) total exposure is higher in humans than in rats unless rats are dosed more often.
The situation is further complicated because we are modeling prenatal exposure and little is known about the doses pregnant MA users take. To model the second half of human intrauterine brain development, rats have to be exposed postnatally (Clancy et al., 2007). This eliminates the placenta and might change the dynamics of the exposure. MA readily crosses the placenta and reaches equilibrium with maternal serum, but exposure is relatively prolonged because the rate of clearance from the fetal compartment is slower (Burchfield et al., 1991).
Another approach to extrapolating doses is by interspecies scaling (Mordenti and Cappell, 1989). Body size influences physiological processes including metabolic rate, oxygen consumption, CO2 clearance, and there is a systematic relationship between body mass multiplied by an empirically determined scaling coefficient raised to the power 0.67 to 0.75 and these physiological processes (West et al., 2002). The validity of scaling formulas has been widely demonstrated for processes ranging from cellular to organismal levels and among different species from tree shrews to elephants. Pharmaceutical applications of this general principle have been used to extrapolate from doses obtained in laboratory animal studies to those used in initial human trials. Based on doses in the present study, the formula dosehuman = doserat × [(weighthuman/weightrat)7] suggests that the dose used in rats in the present experiment is at the lower end of the human abuse spectrum.
Some concerns have, nevertheless, been raised about the use of allometric dose scaling (De La Torre and Farre, 2004; Baumann et al., 2007). For example, De La Torre and Farre (2004) note that species differences in the metabolism of ± 3,4-methylenedioxymethamphetamine caused by differences in cytochrome P450 result in an active principal metabolite, methylenedioxyamphetamine, in rats but to a relatively inactive principal metabolite, dihydroxymethamphetamine, in humans. In addition, De La Torre and Farre (2004) note that, following doses > 10 mg/kg in adult rats, brain concentrations of the parent drug appear to rise nonlinearly. Our previous work, however, suggests that this nonlinearity might not occur in neonatal rats treated with MA (Cappon and Vorhees, 2001). Baumann et al. (2007) recently raised concerns with allometric scaling for extensively metabolized agents such as amphetamines and argued instead for a pharmacologically based approach. They noted, for example, that the neurochemical and physiological actions of ± 3,4-methylenedioxymethamphetamine occur at doses of 0.3–2.5 mg/kg in rats and at a dose of ~ 1.5 mg/kg in humans. It is, however, worth questioning the relevance of pharmacological endpoints for understanding intake in drug-abusing populations. For example, low doses of MA do not produce reductions in brain monoamines but it has been shown that MA users exhibit large reductions in caudate-putamen and globus pallidus DA postmortem (Wilson et al., 1996; Moszczynska et al., 2004), supporting the use of high doses of MA in laboratory studies.
For studies in neonatal animals, such as reported here, the issue of dose relevance remains controversial. Certainly, it would be helpful to analyze cord blood, meconium, or hair from women using MA near the time of delivery. Until such data are available, choosing doses for laboratory studies remains necessarily imprecise; however, it should be noted that we have published data previously on the effects of P11–20 MA using 4 doses/day of 5 mg/kg/day, as used here, and also using several lower doses (i.e. 5.0, 2.5, 1.25, or 0.625 mg/kg). The long-term effects of MA on MWM learning even at these lower doses were similar to those reported here (Williams et al., 2004b).
Acknowledgments
The research reported herein was supported by grants DA006733 (CVV) and DA014269 (MTW) from the National Institutes of Health, US Department of Health and Human Services.
References
- Acevedo SF, de EI, Raber J. Sex- and histamine-dependent long-term cognitive effects of methamphetamine exposure. Neuropsychopharmacology. 2007;32:665–672. doi: 10.1038/sj.npp.1301091. [DOI] [PubMed] [Google Scholar]
- Acuff-Smith KD, Schilling MA, Fisher JE, Vorhees CV. Stage-specific effects of prenatal D-methamphetamine exposure on behavioral and eye development in rats. Neurotoxicol Teratol. 1995;18:199–215. doi: 10.1016/0892-0362(95)02015-2. [DOI] [PubMed] [Google Scholar]
- Barr AM, Panenka WJ, MacEwan GW, Thornton AE, Lang DJ, Honer WG, Lecomte T. The need for speed: an update on methamphetamine addiction. J Psychiatry Neurosci. 2006;31:301–313. [PMC free article] [PubMed] [Google Scholar]
- Baumann MH, Wang X, Rothman RB. 3,4-Methylenedioxymethamphetamine (MDMA) neurotoxicity in rats: a reappraisal of past and present findings. Psychopharmacology (Berl) 2007;189:407–424. doi: 10.1007/s00213-006-0322-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayer SA, Altman J, Russo RJ, Zhang X. Timetables of neurogenesis in the human brain based on experimentally determined patterns in the rat. Neurotoxicology. 1993;14:83–144. [PubMed] [Google Scholar]
- Brandeis R, Brandys Y, Yehuda S. The use of the Morris water maze in the study of memory and learning. Int J Neurosci. 1989;48:29–69. doi: 10.3109/00207458909002151. [DOI] [PubMed] [Google Scholar]
- Burchfield DJ, Lucas VW, Abrams RM, Miller RL, deVane CL. Disposition and pharmacodynamics of methamphetamine in pregnant sheep. J Am Med Assoc. 1991;265:1968–1973. [PubMed] [Google Scholar]
- Burgess N, Maguire EA, O’Keefe J. The human hippocampus and spatial and episodic memory. Neuron. 2002;35:625–641. doi: 10.1016/s0896-6273(02)00830-9. [DOI] [PubMed] [Google Scholar]
- Cain DP, Saucier D. The neuroscience of spatial navigation: focus on behavior yields advances. Rev Neurosci. 1996;7:215–231. doi: 10.1515/revneuro.1996.7.3.215. [DOI] [PubMed] [Google Scholar]
- Cain DP, Saucier D, Hall J, Hargreaves EL, Boon F. Detailed behavioral analysis of water maze acquisition under APV or CNQX: contribution of sensorimotor disturbances to drug-induced acquisition deficits. Behav Neurosci. 1996;110:86–102. doi: 10.1037//0735-7044.110.1.86. [DOI] [PubMed] [Google Scholar]
- Cappon GD, Vorhees CV. Plasma and brain methamphetamine concentrations in neonatal rats. Neurotoxicol Teratol. 2001;23:81–88. doi: 10.1016/s0892-0362(00)00118-5. [DOI] [PubMed] [Google Scholar]
- Chang L, Smith LM, LoPresti C, Yonekura ML, Kuo J, Walot I, Ernst T. Smaller subcortical volumes and cognitive deficits in children with prenatal methamphetamine exposure. Psychiatr Res: Neuroimaging. 2004;132:95–106. doi: 10.1016/j.pscychresns.2004.06.004. [DOI] [PubMed] [Google Scholar]
- Cho AK, Melega WP, Kuczenski R, Segal DS. Relevance of pharmacokinetic parameters in animal models of methamphetamine abuse. Synapse. 2001;39:161–166. doi: 10.1002/1098-2396(200102)39:2<161::AID-SYN7>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- Chomchai C, Na MN, Watanarungsan P, Yossuck P, Chomchai S. Methamphetamine abuse during pregnancy and its health impact on neonates born at Siriraj Hospital, Bangkok, Thailand. Southeast Asian J Trop Med Public Health. 2004;35:228–231. [PubMed] [Google Scholar]
- Clancy B, Darlington RB, Finlay BL. Translating developmental time across mammalian species. Neuroscience. 2001;105:7–17. doi: 10.1016/s0306-4522(01)00171-3. [DOI] [PubMed] [Google Scholar]
- Clancy B, Kersh B, Hyde J, Darlington RB, Anand KJS, Findlay BL. Web-based method for translating neurodevelopment from laboratory species to humans. Neuroinformatics. 2007;5:79–94. doi: 10.1385/ni:5:1:79. [DOI] [PubMed] [Google Scholar]
- Clancy B, Finlay BL, Darlington RB, Anand KJ. Extrapolating brain development from experimental species to humans. Neurotoxicology. 2007 doi: 10.1016/j.neuro.2007.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crawford CA, Williams MT, Newman ER, McDougall SA, Vorhees CV. Methamphetamine exposure during the preweanling period causes prolonged changes in dorsal striatial protein kinase A activity, dopamine D2-like binding sites, and dopamine content. Synapse. 2003;48:131–137. doi: 10.1002/syn.10197. [DOI] [PubMed] [Google Scholar]
- D’Hooge R, de Deyn PP. Applications of the Morris water maze in the study of learning and memory. Brain Res Rev. 2001;36:60–90. doi: 10.1016/s0165-0173(01)00067-4. [DOI] [PubMed] [Google Scholar]
- Day NL, Cottreau CM, Richardson GA. The epidemiology of alcohol, marijuana, and cocaine use among women of childbearing age and pregnant women. Clin Obstet Gynecol. 1993;36:232–245. doi: 10.1097/00003081-199306000-00005. [DOI] [PubMed] [Google Scholar]
- De La Torre R, Farre M. Neurotoxicity of MDMA (ecstasy): the limitations of scaling from animals to humans. Trends Pharmacol Sci. 2004;25:505–508. doi: 10.1016/j.tips.2004.08.001. [DOI] [PubMed] [Google Scholar]
- Dixon SD. Effects of transplacental exposure to cocaine and methamphetamine on the neonate. Western J Med. 1989;150:436–442. [PMC free article] [PubMed] [Google Scholar]
- Dixon SD, Bejar R. Echoencephalographic findings in neonates associated with maternal cocaine and methamphetamine use: incidence and clinical correlates. J Pediatr. 1989;115:770–778. doi: 10.1016/s0022-3476(89)80661-4. [DOI] [PubMed] [Google Scholar]
- Herlenius E, Lagercrantz H. Development of neurotransmitter systems during critical periods. Exp Neurol. 2004;190:S8–S21. doi: 10.1016/j.expneurol.2004.03.027. [DOI] [PubMed] [Google Scholar]
- Johnston LD, O’Malley PM, Bachman JG, Schulenberg JE. Monitoring the future national survey results on drug abuse, 1975–2005. Volume I: secondary school students. Bethesda, MD: National Institute on Drug Abuse, US Department of Health and Human Services; 2006a. [Google Scholar]
- Johnston LD, O’Malley PM, Bachman JG, Schulenberg JE. Monitoring the future national survey results on drug abuse, 1975–2005. Volume II: college students and adults age 19–45. Bethesda, MD: National Institute on Drug Abuse, US Department of Health and Human Services; 2006b. [Google Scholar]
- Lester BM, LaGasse LL, Seifer R. Cocaine exposure and children: the meaning of subtle effects. Science. 1998;282:633–634. doi: 10.1126/science.282.5389.633. [DOI] [PubMed] [Google Scholar]
- Little BB, Snell LM, Gilstrap LC. Methamphetamine abuse during pregnancy: outcome and fetal effects. Obstet Gynecol. 1988;72:541–544. [PubMed] [Google Scholar]
- Melega WP, Cho AK, Harvey D, Lacan G. Methamphetamine blood concentrations in human abusers: application to pharmacokinetic modeling. Synapse. 2007;61:216–220. doi: 10.1002/syn.20365. [DOI] [PubMed] [Google Scholar]
- Meredith CW, Jaffe C, ng-Lee K, Saxon A J. Implications of chronic methamphetamine use: a literature review. Harv Rev Psychiatry. 2005;13:141–154. doi: 10.1080/10673220591003605. [DOI] [PubMed] [Google Scholar]
- Mordenti J, Cappell W. The use of interspecies scaling in toxicokinetics. In: Yacobi A, Kelly J, Batra V, editors. Toxicokinetics in new drug development. New York: Pergamon Press; 1989. pp. 42–96. [Google Scholar]
- Morris RG, Moser EI, Riedel G, Martin SJ, Sandin J, Day M, O’Carroll C. Elements of a neurobiological theory of the hippocampus: the role of activity-dependent synaptic plasticity in memory. Philos Trans R Soc Lond B Biol Sci. 2003;358:773–786. doi: 10.1098/rstb.2002.1264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moszczynska A, Fitzmaurice P, Ang L, Kalasinsky K, Schmunk GA, Peretti FJ, et al. Why is parkinsonism not a feature of human methamphetamine users? Brain. 2004;127:363–370. doi: 10.1093/brain/awh046. [DOI] [PubMed] [Google Scholar]
- Oro AS, Dixon SD. Perinatal cocaine and methamphetamine exposure: maternal and neonatal correlates. J Pediatr. 1987;111:571–578. doi: 10.1016/s0022-3476(87)80125-7. [DOI] [PubMed] [Google Scholar]
- Rice D, Barone S., Jr Critical periods of vulnerability for the developing nervous system: evidence from humans and animal models. Environ Health Perspect. 2000;108:511–533. doi: 10.1289/ehp.00108s3511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson GA, Conroy ML, Day NL. Prenatal cocaine exposure: effects on the development of school-age children. Neurotoxicol Teratol. 1996a;18:627–634. doi: 10.1016/s0892-0362(96)00121-3. [DOI] [PubMed] [Google Scholar]
- Richardson GA, Hamel SC, Goldschmidt L, Day NL. The effects of prenatal cocaine use on neonatal neurobehavioral status. Neurotoxicol Teratol. 1996b;18:519–528. doi: 10.1016/0892-0362(96)00062-1. [DOI] [PubMed] [Google Scholar]
- Sapolsky RM, Meaney MJ. Maturation of the adrenocortical stress response: neuroendocrine control mechanisms and the stress hyporesponsive period. Brain Res Rev. 1986;11:65–76. doi: 10.1016/s0006-8993(86)80190-1. [DOI] [PubMed] [Google Scholar]
- Schaefer TL, Ehrman LA, Gudelsky GA, Vorhees CV, Williams MT. Comparison of monoamine and corticosterone levels 24 h following (+)methamphetamine, (+ / −)3,4-methylenedioxymethamphetamine, cocaine, (+)fenfluramine or (+ / −)methylphenidate administration in the neonatal rat. J Neurochem. 2006;98:1369–1378. doi: 10.1111/j.1471-4159.2006.04034.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith LM, Chang L, Yonekura ML, Grob C, Osborn D, Ernst T. Brain proton magnetic resonance spectroscopy in children exposed to methamphetamine in utero. Neurology. 2001;57:255–260. doi: 10.1212/wnl.57.2.255. [DOI] [PubMed] [Google Scholar]
- Smith LM, LaGasse LL, Derauf C, Grant P, Shah R, Arria A, et al. The infant development, environment, and lifestyle study: effects of prenatal methamphetamine exposure, polydrug exposure, and poverty on intrauterine growth. Pediatrics. 2006;118:1149–1156. doi: 10.1542/peds.2005-2564. [DOI] [PubMed] [Google Scholar]
- Struthers JM, Hansen RL. Visual recognition memory in drug-exposed infants. Dev Behav Pediatr. 1992;13:108–111. doi: 10.1097/00004703-199204000-00005. [DOI] [PubMed] [Google Scholar]
- Vorhees CV, Williams MT. Morris water maze: procedures for assessing spatial and related forms of learning and memory. Nat Protocols. 2006;1:848–858. doi: 10.1038/nprot.2006.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vorhees CV, Ahrens KG, Acuff-Smith KD, Schilling MA, Fisher JE. Methamphetamine exposure during early postnatal development in rats: I. acoustic startle augmentation and spatial learning deficits. Psychopharmacology. 1994a;114:392–401. doi: 10.1007/BF02249328. [DOI] [PubMed] [Google Scholar]
- Vorhees CV, Ahrens KG, Acuff-Smith KD, Schilling MA, Fisher JE. Methamphetamine exposure during early postnatal development in rats: II. hypoactivity and altered responses to pharmacological challenge. Psychopharmacology. 1994b;114:402–408. doi: 10.1007/BF02249329. [DOI] [PubMed] [Google Scholar]
- Vorhees CV, Reed TM, Schilling MA, Fisher JE, Moran MS, Cappon GD, Nebert DW. CYP2D1 polymorphism in methamphetamine-treated rats: genetic differences in neonatal mortality and a comparison of spatial learning and acoustic startle. Neurotoxicol Teratol. 1998;20:265–273. doi: 10.1016/s0892-0362(97)00129-3. [DOI] [PubMed] [Google Scholar]
- Vorhees CV, Morford LL, Inman SL, Reed TM, Schilling MA, Cappon GD, et al. Genetic differences in spatial learning between Dark Agouti and Sprague–Dawley strains: possible correlation with CYP2D2 polymorphism in rats treated neonatally with methamphetamine. Pharmacogenetics. 1999;9:171–181. [PubMed] [Google Scholar]
- Vorhees CV, Inman-Wood SL, Morford LL, Broening HW, Fukumura M, Moran MS. Adult learning deficits after neonatal exposure to D-methamphetamine: selective effects on spatial navigation and memory. J Neurosci. 2000;20:4732–4739. doi: 10.1523/JNEUROSCI.20-12-04732.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West GB, Woodruff WH, Brown JH. Allometric scaling of metabolic rate from molecules and mitochondria to cells and mammals. Proc Natl Acad Sci U S A. 2002;99(Suppl 1):2473–2478. doi: 10.1073/pnas.012579799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West JR, Pierce DR. Perinatal alcohol exposure and neuronal damage. In: West JR, editor. Alcohol and brain development. New York: Oxford University Press; 1987. pp. 120–157. [Google Scholar]
- Williams MT, Inman-Wood SL, Morford LL, McCrea AE, Ruttle AM, Moran MS, et al. Preweaning treatment with methamphetamine induces increases in both corticosterone and ACTH in rats. Neurotoxicol Teratol. 2000;22:751–759. doi: 10.1016/s0892-0362(00)00091-x. [DOI] [PubMed] [Google Scholar]
- Williams MT, Vorhees CV, Boon F, Saber AJ, Cain DP. Methamphetamine exposure from postnatal days 11 to 20 causes impairments in both behavioral strategies and spatial learning in adult rats. Brain Res. 2002;958:312–321. doi: 10.1016/s0006-8993(02)03620-x. [DOI] [PubMed] [Google Scholar]
- Williams MT, Moran MS, Vorhees CV. Refining the critical period for methamphetamine-induced spatial deficits in the Morris water maze. Psychopharmacology. 2003a;168:329–338. doi: 10.1007/s00213-003-1433-y. [DOI] [PubMed] [Google Scholar]
- Williams MT, Morford LL, Wood SL, Wallace TL, Fukumura M, Broening HW, Vorhees CV. Developmental d-methamphetamine treatment selectively induces spatial navigation impairments in reference memory in the Morris water maze while sparing working memory. Synapse. 2003b;48:138–148. doi: 10.1002/syn.10159. [DOI] [PubMed] [Google Scholar]
- Williams MT, Brown RW, Vorhees CV. Neonatal methamphetamine administration induces region-specific long-term neuronal morphological changes in the rat hippocampus, nucleus accumbens and parietal cortex. Eur J Neurosci. 2004a;19:3165–3171. doi: 10.1111/j.0953-816X.2004.03405.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams MT, Moran MS, Vorhees CV. Behavioral and growth effects induced by low dose methamphetamine administration during the neonatal period in rats. Int J Dev Neurosci. 2004b;22:273–283. doi: 10.1016/j.ijdevneu.2004.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams MT, Schaefer TL, Furay AR, Ehrman LA, Vorhees CV. Ontogeny of the adrenal response to (+)-methamphetamine in neonatal rats: the effect of prior drug exposure. Stress. 2006;9:153–163. doi: 10.1080/10253890600902842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson JM, Kalasinsky KS, Levey AI, Bergeron C, Reiber G, Anthony RM, et al. Striatal dopamine nerve terminal markers in human, chronic methamphetamine users. Nature Med. 1996;2:699–703. doi: 10.1038/nm0696-699. [DOI] [PubMed] [Google Scholar]