

Abstract
VEGF and TGF-β1 induce angiogenesis but have opposing effects on endothelial cells. VEGF protects endothelial cells from apoptosis; TGF-β1 induces apoptosis. We have previously shown that VEGF / VEGF receptor-2 (VEGFR2) signaling mediates TGF-β1 induction of apoptosis. This finding raised an important question: Does this mechanism stimulate or inhibit angiogenesis? Here we report that VEGF-mediated apoptosis is required for TGF-β1 induction of angiogenesis. In vitro the apoptotic effect of TGF-β1 on endothelial cells is rapid and followed by a long period in which the cells are refractory to apoptosis induction by TGF-β1. Inhibition of VEGF / VEGFR2 signaling abrogates formation of cord-like structures by TGF-β1 with an effect comparable to that of z-VAD, an apoptosis inhibitor. Similarly, genetic deficiency of VEGF abolishes TGF-β1 upregulation of endothelial cell differentiation and formation of vascular structures in embryoid bodies. In vivo TGF-β1 induces endothelial cell apoptosis as rapidly as in vitro. Inhibition of VEGF blocks TGF-β1 induction of both apoptosis and angiogenesis, an effect similar to that of z-VAD. Thus, TGF-β1 induction of angiogenesis requires a rapid and transient apoptotic effect mediated by VEGF/VEGFR2. This novel, unexpected role of VEGF and VEGFR2 indicates VEGF-mediated apoptosis as a potential target to control angiogenesis.
Keywords: angiogenesis, embryoid bodies, vasculogenesis, chicken embryo chorioallantoic membrane
INTRODUCTION
Angiogenesis, the formation of capillaries from preexisting blood vessels, occurs in a variety of physiological and pathological settings, including embryonic development, wound healing and tumor growth. A number of cytokines and growth factors modulate angiogenesis. Among them, vascular endothelial growth factor (VEGF) and transforming growth factor-beta 1 (TGF-β1) play prominent roles (Ferrara, 2004; Massague et al., 2000; Presta et al., 2005).
VEGF controls a variety of endothelial cell functions involved in angiogenesis and protects endothelial cells from apoptosis (Hicklin and Ellis, 2005). VEGF transcription can be modulated by multiple stimuli (Levy et al., 1995; Liu et al., 1995; Tischer et al., 1991), among which hypoxia plays a major role (Forsythe et al., 1996; Liu et al., 1995; Shweiki et al., 1992). In addition, cytokines and growth factors upregulate VEGF expression in a variety of cell types (Goldman et al., 1993; Li et al., 1995; Pertovaara et al., 1994). Fibroblast growth factor-2 (FGF-2) and TGF-β1 induce VEGF expression in vascular endothelial cells (Ferrari et al., 2006; Seghezzi et al., 1998). VEGF activates two tyrosine kinase receptors, VEGFR-1 (flt-1) and VEGFR-2 (flk-1; KDR) (Hicklin and Ellis, 2005). VEGFR2 has been implicated in endothelial cell proliferation and survival, and VEGFR1 in chemotaxis and vascular permeability (Gille et al., 2001; Keyt et al., 1996). VEGF, VEGFR1, and VEGFR2 are indispensable for angiogenesis and their genetic deficiency causes embryonic lethality as a result of blood vessels disorganization, endothelial cell overgrowth or impaired endothelial cell development (Carmeliet et al., 1996; Ferrara et al., 1996). In tumors VEGF expression correlates with tumor vascularity and progression. Inhibition of VEGF results in decreased tumor vascularity and growth, implicating VEGF as the major tumor angiogenesis factor (Ferrara, 2004). Pharmacological treatments targeting VEGF or VEGFR-2 are currently used or in advanced clinical trials for the therapy of several malignancies (Ferrara, 2004; Ferrara and Kerbel, 2005).
TGF-β1 is an important regulator of tissue morphogenesis and a potent inhibitor of proliferation for most cell types (Massague et al., 2000). Half of mice genetically deficient in TGF-β1 die in utero and show defective vasculogenesis, a phenotype consistent with abundant TGF-β1 gene expression in endothelial precursors (Dickson et al., 1995). TGF-β1 has multiple effects on vascular endothelial cells. In vivo TGF-β1 induces angiogenesis (Madri et al., 1988; Roberts et al., 1986; Yang and Moses, 1990). However, in vitro it inhibits endothelial cell proliferation (Pollman et al., 1999b), migration and proteolytic activity, opposes the stimulatory effect of FGF-2 on these endothelial cell functions (Pepper et al., 1990; Saksela et al., 1987), and downregulates VEGFR2 expression (Mandriota et al., 1996). Notably, TGF-β1 induces endothelial cell apoptosis, opposing the prosurvival activity of VEGF (Pollman et al., 1999a; Pollman et al., 1999b). Inhibition of apoptosis abrogates TGF-β1-induced angiogenesis in vitro (Choi and Ballermann, 1995), indicating that the apoptotic effect of TGF-β1 on endothelial cells is an important component of its angiogenic activity.
During angiogenesis apoptosis is required for pruning the forming vascular network, and inhibition of apoptosis results in formation of abnormal vessels (Pollman et al., 1999a). In addition, apoptosis controls cell functions required for capillary morphogenesis in vitro and in vivo (Choi and Ballermann, 1995; Segura et al., 2002). In adult animals TGF- β1-induced endothelial cell apoptosis is required for glomerular capillary lumen formation (Fierlbeck et al., 2003).
Because of its inhibitory effects on endothelial cells it has been proposed that TGF-β1 induces angiogenesis in vivo through an indirect mechanism, by inducing expression of VEGF and/or other angiogenic factors in epithelial or other cell types (Pardali and Moustakas, 2007). However, several observations indicate that TGF-β1 has important direct effects on angiogenesis. During mouse embryogenesis ALK1 (TGF-β1 receptor I, or TβRI) expression is mainly confined to endothelial cells, and is upregulated at sites of active angiogenesis. The genetic deficiency of ALK1 causes death of mid-gestation mouse embryos from defects in angiogenesis, an effect comparable to that resulting from the genetic deficiency of TGF-β1 or of the other TβRs, ALK5 (TβRI) or TβRII (Dickson et al., 1995; Li et al., 1999; Oshima et al., 1996). In humans mutations of ALK1, the endothelial cell TGF-β1 receptor, cause hereditary hemorrhagic teleangiectasia, a condition characterized by absence of capillary bed (angiogenesis) in certain vascular districts.
VEGF and TGF-β1 are often co-expressed in tissues in which angiogenesis occurs, notably in a variety of tumors (Pardali and Moustakas, 2007). However, although numerous studies have investigated the mechanisms through which these individual growth factors control angiogenesis, their interactions at the level of endothelial cells are poorly understood. We have recently shown that TGF-β1 upregulates endothelial cell expression of VEGF, and that TGF-β1 induction of endothelial cell apoptosis is mediated by activation of VEGFR2 by VEGF (Ferrari et al., 2006). This finding raised an important question: Does this mechanism inhibit or stimulate blood vessel formation by TGF-β1? Here we report that VEGF-mediated apoptosis is required for TGF-β1 induction of angiogenesis in vitro and in vivo.
MATERIALS AND METHODS
Materials
Human purified or recombinant TGF-β1, recombinant human or mouse VEGF, goat anti-human VEGFR2 and VEGFR1 antibodies and rabbit neutralizing polyclonal antibodies to human or mouse VEGF were purchased from R&D Systems (Minneapolis, MN); z-VAD-(OMe)-FMK and DMSO from Sigma-Aldrich (St. Louis, MO). Polyclonal antibody to cleaved human caspase 3 was purchased from Cell Signaling Technologies (Beverly, MA); polyclonal antibody to total ERK-2 from Santa Cruz Biotechnology (Santa Cruz, CA); anti-mouse CD31 antibody from Pharmingen (San Jose, CA); mouse and rabbit non-immune IgG from Sigma-Aldrich, and human recombinant FGF-2 from Gibco BRL, Life Technologies, Inc., (Rockville, MD). Neutralizing monoclonal antibody to human FGF-2 (MAb 354FI) was a generous gift from Texas Biotechnology, Inc. (Houston, TX, USA).
Cells and Media
Bovine capillary endothelial cells (BCE) were isolated as described (Seghezzi et al., 1998) and grown in alpha modified minimum essential medium (αMEM; Fisher, Pittsburgh, PA, USA) supplemented with 5% donor calf serum (DCS) and L-glutamine 2 mM (Gibco BRL, Life Technologies). These cells were used between passage 6 and 15 in culture. Human umbilical vein endothelial (HUVE; Clonetics, San Diego, CA) were grown in endothelial cell basal medium-2 (EBM2; Clonetics) containing 2% fetal calf serum (FCS) and the endothelial cell growth supplements provided by the company. HUVE cells were used between passage 3 and 5 in culture. For the apoptosis assays confluent BCE or HUVE cells were starved overnight in their respective medium supplemented with 0.5% DCS or FCS, respectively, after which TGF-β1 (1 ng/ml) was added to the medium and incubation was continued for the indicated time. The time of addition of TGF-β1 was considered as time 0.
TUNEL assay for apoptosis
Endothelial cells grown on glass coverlips were stained with the FragEL DNA Fragmentation Kit for TUNEL analysis (Calbiochem; San Diego, CA), and counterstained with DAPI. TUNEL-positive cells and DAPI-stained nuclei were counted under a fluorescence microscope in ten 100× fields. The results are reported as percent apoptotic cells = (mean TUNEL-positive cells per field / mean total nuclei per field) × 100.
Embryoid bodies
Wild-type mouse embryonic stem (ES) cells and embryonic mouse fibroblast cells (EMFI) established as described (Su et al., 1999) were provided to us by Dr. A. Joyner (Memorial Sloan-Kettering Institute, New York). ES cells genetically deficient in either VEGF or VEGFR-1 or -2 were provided to us by Drs. Andras Nagy (Samuel Lunenfeld Research Institute, Toronto, Canada) (Carmeliet et al., 1996), Janet Rossant (University of Toronto, Ontario, Canada) (Fong et al., 1995) and Guo-Hua Fong (University of Connecticut) (Fong et al., 1995), respectively. ES cell culture and differentiation into embryoid bodies were performed as described (Gualandris et al., 2000a).
Western blotting
Western blotting was performed as described (Ferrari et al., 2006).
siRNA transfection
Subconfluent HUVE cells were incubated with 200 pmol of human VEGFR2 or VEGFR1 siRNA oligonucleotides (Dharmacon RNA technologies, Lafayette, CO) and 4 µl of Oligofectamine (Invitrogen, Carlsbad, CA) in serum-free medium for 4 h at 37° C, after which medium supplemented with 10% serum was added (Ferrari et al., 2006). The cells were used 48 h after transfection.
Reverse transcription-polymerase chain reaction (RT-PCR)
The following primers for human VEGFR2 and VEGFR1 were synthesized by IDT DNA technologies (Coralville, IA) based on the published sequences: flt1 (s: 5’CGACCTTGGTTGTGGCTGACT; a:5’CGGTTCTGGTTGGTGGCTTTG); flk1 (s: 5’ AACAAAGTCGGGAGAGGA; a:5’ TGACAAGAAGTAGCCAGAAGA); β-actin (s: 5’ATCTGGGACCAACCTTCTAGAATGAG; a: 5’CGTCATACTCCTGCTTGCTGATCCAC) Complementary DNA (cDNA) was synthesized from 1 µg of total RNA using SuperScript II RT (Invitrogen, Carlsbad, CA) and oligo-dT 3’ primer. Two µl of cDNA was amplified by PCR as described.
Capillary cord formation in vitro
Confluent BCE or HUVE cells grown in gelatin-coated tissue culture plates were incubated in medium containing 0.5% CDS or FCS, respectively, with or without addition of TGF-β1 (1 ng/ml) and/or the indicated reagents. The cultures were photographed with an inverted phase contrast microscope (Zeiss Axiovert 25) after the indicated time of incubation. Quantification was performed by using an ocular grid and counting the number of capillary-like structures that crossed the equatorial line of the microscope field in five randomly chosen 10 × fields per sample. Each sample was analyzed in triplicate.
In vivo apoptosis and angiogenesis assays on the chicken chorioallantoic membrane (CAM)
Angiogenesis assays were performed as described (Brooks et al., 1999) using filter disks soaked with hydroxycortisone acetate (Sigma; 2.5 mg/ml in 95% ethanol). To characterize endothelial cell apoptosis CAMs were separated from the paper disks after 6 h incubation at 38° C and snap frozen in OTC. Four-micrometer sections were cut and stained with antibodies to CD31 and with the FragEL DNA Fragmentation Kit for TUNEL analysis (Calbiochem; San Diego, CA), and counterstained with DAPI. The sections were observed under a confocal microscope, and TUNEL-positive cells were counted in ten 100× fields. Five CAMs per sample were used. To characterize angiogenesis CAMs were incubated at 38° C for 72 h, and the number of branching blood vessels within the area of the filter disks was counted by stereomicroscopy as described (Brooks et al., 1999). Eight CAMs per condition were used. The CAMs were photographed with a digital camera connected to the microscope.
Statistical Analysis
t-tests on the equality of means were performed using Stata 8.
RESULTS
Endothelial cell-derived VEGF controls TGF-β1 induction of capillary morphogenesis in vitro
TGF-β1 induces vascular endothelial cell expression of VEGF, which mediates the apoptotic activity of TGF-β1 through activation of VEGFR2 (Ferrari et al., 2006). Because TGF-β1 induces angiogenesis in vitro and in vivo (Choi and Ballermann, 1995; Segura et al., 2002; Yang and Moses, 1990), we hypothesized that endothelial cell VEGF also controls the angiogenic activity of TGF-β1. As a first approach to test this hypothesis, we used a capillary morphogenesis assay by which TGF-β1 was shown to induce formation of cord-like structures in vitro (Choi and Ballermann, 1995). Confluent BCE cells grown on gelatin-coated dishes were incubated in the presence or absence of TGF-β1, and TGF-β1 was added to the medium every 24 h. Under these experimental conditions TGF-β1 induced apoptosis within 6 h to 12 h of incubation. Subsequently apoptosis decreased to control levels and further addition of TGF-β1 to the culture medium did not induce cell death (Fig. 1). In the absence of TGF-β1 the cells grew as a compact monolayer (Fig. 2 A, panel a). As described (Choi and Ballermann, 1995), addition of TGF-β1 to the culture medium induced monolayer remodeling into pre-capillary cord-like structures within 72 h to 120 h. Previous studies have shown that these capillary cords induced by TGF-β1 contain a lumen (Choi and Ballermann, 1995) (Fig. 2 A, panel b; Supplemental Figure 1).
Figure 1. Induction of endothelial cell apoptosis by TGF-β1.
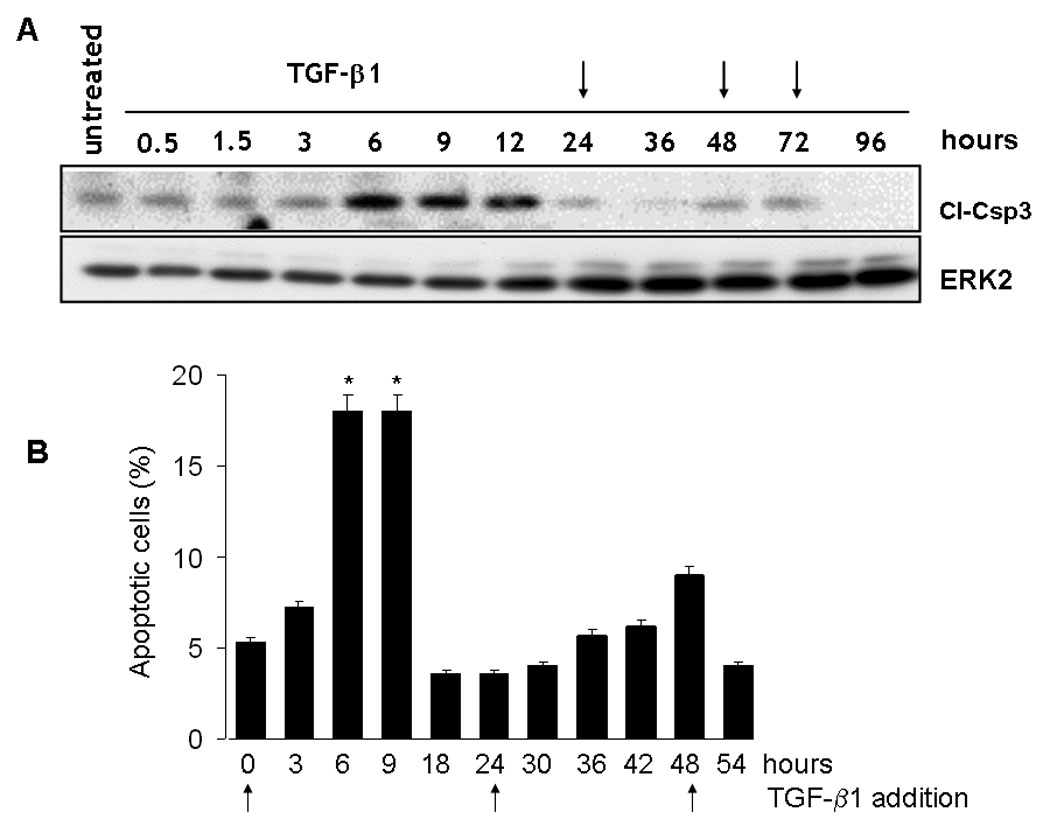
A. Western blotting analysis of caspase 3 cleavage in BCE cells incubated in medium supplemented with 0.5% DCS in the absence or presence of TGF-β1 (1 ng/ml) for the indicated time. TGF-β1 was added to the culture medium every 24 h (arrows). ERK-2: loading control. B. TUNEL assay. BCE cells were incubated for the indicated time in medium supplemented with 0.5% DCS and TGF-β1 (1 ng/ml). TGF-β1 was added to the culture medium every 24 h (arrows) and the cells were analyzed for apoptosis after 6 –12 h, a time interval during which TGF-b1 induces apoptosis in untreated cells. TUNEL staining and measurement of apotosis were carried out as described under Materials and Methods. Mean ± s.d of a representative experiments are shown. * p < 0.05 (time × vs. time 0).
Figure 2. Capillary cord formation in vitro.
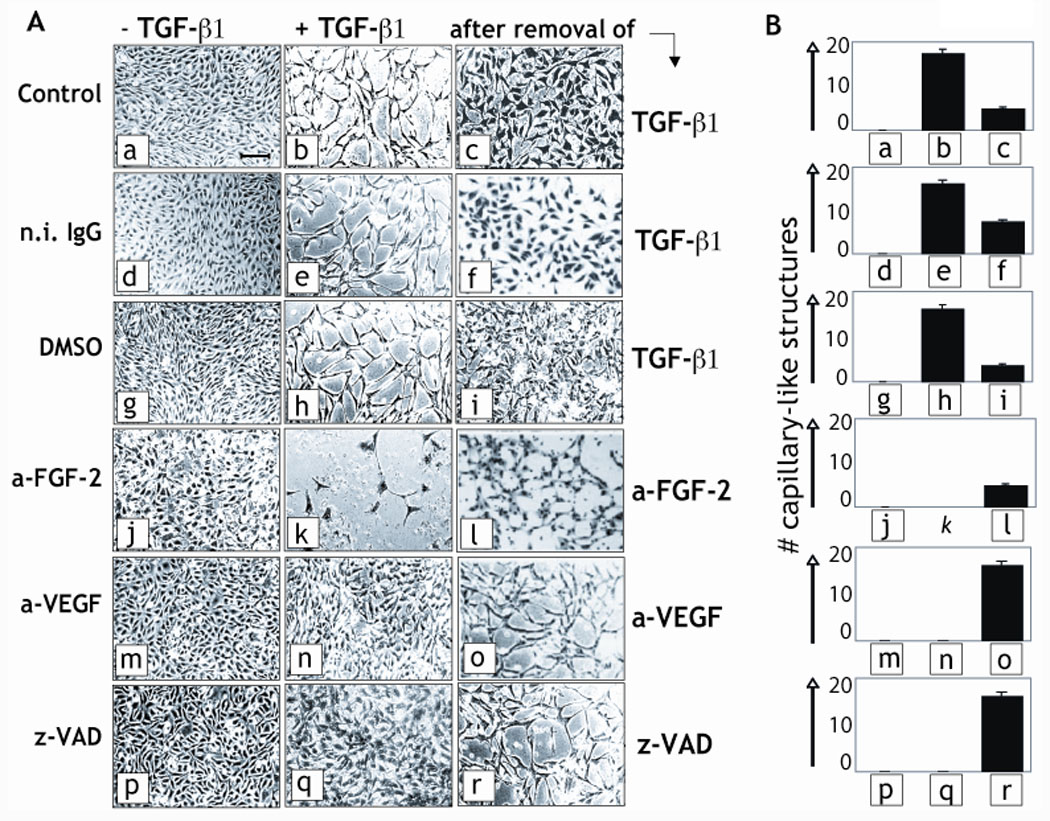
A. Confluent BCE cells grown in gelatin-coated dishes were incubated in the absence (left column) or presence of TGF-β1 (1 ng/ml; middle column) in medium supplemented with 0.5% donor calf serum and either 50 µg/ml of mouse n.i. IgG or antibodies to VEGF (a-VEGF) or FGF-2 (a-FGF-2), or with 40 µM z-VAD-(OMe)-FMK (z-VAD) or 0.2 % (v/v) DMSO (vehicle), or with no addition (Control). The cultures were stained with Giemsa and photographed after 72 h incubation. The medium was then replaced with medium with no TGF-β1 (right column, top three panels) or with medium containing TGF-β1 and n.i IgG instead of FGF-2 or VEGF MAbs, or DMSO instead of z-VAD (right column, bottom three panels, respectively). After 24 h incubation the cultures were stained with hematoxylin-eosin and photographed. Bar represents 200 µm. This experiment was repeated three times with comparable results. Magnified images of panels a and b are shown in Supplemental Fig. 1 to evidence the formation of capillary-like structures by TGF-β1. B. The results of the experiments shown in panel A were quantitated as described under Materials and Methods. The histograms represent mean ± SE. p < 0.05 (sample vs. control).
We then characterized the effect of neutralizing anti-VEGF and anti-FGF-2 antibodies on TGF-β1 induction of capillary cord formation. VEGF antibody blocks the apoptotic activity of TGF-β1 on endothelial cells, whereas antibody to FGF-2 induces apoptosis in the presence or absence of TGF-β1 (Ferrari et al., 2006; Mandriota and Pepper, 1997). Both antibodies blocked TGF-β1 induction of cord formation but with different effects. In the presence of VEGF antibody TGF-β1-treated cells (panel n) retained an intact monolayer. In contrast, addition of FGF-2 antibody to TGF-β1-treated cells caused massive cell death, with sparse, disrupted cord-like structures (panel k). This effect can be explained by previous findings that anti-FGF-2 antibody induces endothelial cell apoptosis by a VEGF-insensitive mechanism, and that FGF-2 −/− endothelial cells have higher apoptosis levels than wt cells (Ferrari et al., 2006; Mandriota and Pepper, 1997).
To analyze the role of apoptosis in TGF-β1 induction of angiogenesis we used z-VAD, a general caspase inhibitor that blocks apoptosis. z-VAD blocked induction of cord formation by TGF-β1 (Fig. 2, panel q) with an effect comparable to that of VEGF antibody. This result is consistent with previous reports showing that zVad and other apoptosis inhibitors block VEGF- and FGF-2-induced angiogenesis in other in vitro angiogenesis assays, as well as in vivo (Segura et al., 2002).
The inhibitory effect of z-VAD and of antibodies to FGF-2 or VEGF on capillary cord formation in vitro was rapidly reverted after removal of these reagents from the culture medium (Fig. 2, panels l, o, r). Similarly, removal of TGF-β1 from the culture medium caused rapid reversion of the cord-like structures into a monolayer (panels c, f, i), showing that the observed morphological changes did not reflect toxic effects of the reagents. Therefore, these results showed that endothelial cell apoptosis is an early event during induction of capillary cord formation by TGF-β1, and both upregulation and downregulation of apoptosis block the in vitro angiogenic activity of TGF-β1.
To characterize the role of VEGF receptors in TGF-β1 induction of angiogenesis we downregulated VEGFR1 and VEGFR2 by transient transfection of endothelial cells with specific siRNAs. These siRNAs effectively inhibit the expression of their respective proteins without cross-reacting, and abolish MAP kinase activation by VEGF (Ferrari et al., 2006). Transfection with VEGFR2 or VEGFR1 siRNA resulted in strong downregulation of the expression of the respective receptor for up to 96 h, without affecting the level of the other (Fig. 3 A and 3 B). Downregulation of VEGFR2 expression blocks TGF-β1 induction of endothelial cell apoptosis, whereas inhibition of VEGFR1 expression has no such effect (Ferrari et al., 2006). VEGFR2 siRNA-transfected cells did not undergo apoptosis although they were treated with TGF-β1 every 24 h for up to 72 h (Fig. 3 C), showing that downregulation of VEGFR2 expression did not provide an apoptotic stimulus per se. However, transfection with VEGFR2 siRNA abrogated both the apoptotic effect of TGF-β1 (Fig. 3 C) and TGF-β1 induction of cord formation in vitro (Fig. 3 D and E). In contrast, downregulation of VEGFR1 expression, which does not affect TGF-β1 induction of apoptosis (Ferrari et al., 2006), had no such effect (Fig. 3 D and E).
Fig. 3. Downregulation of VEGFR2 expression blocks TGF-β1 induction of capillary cord formation in vitro.
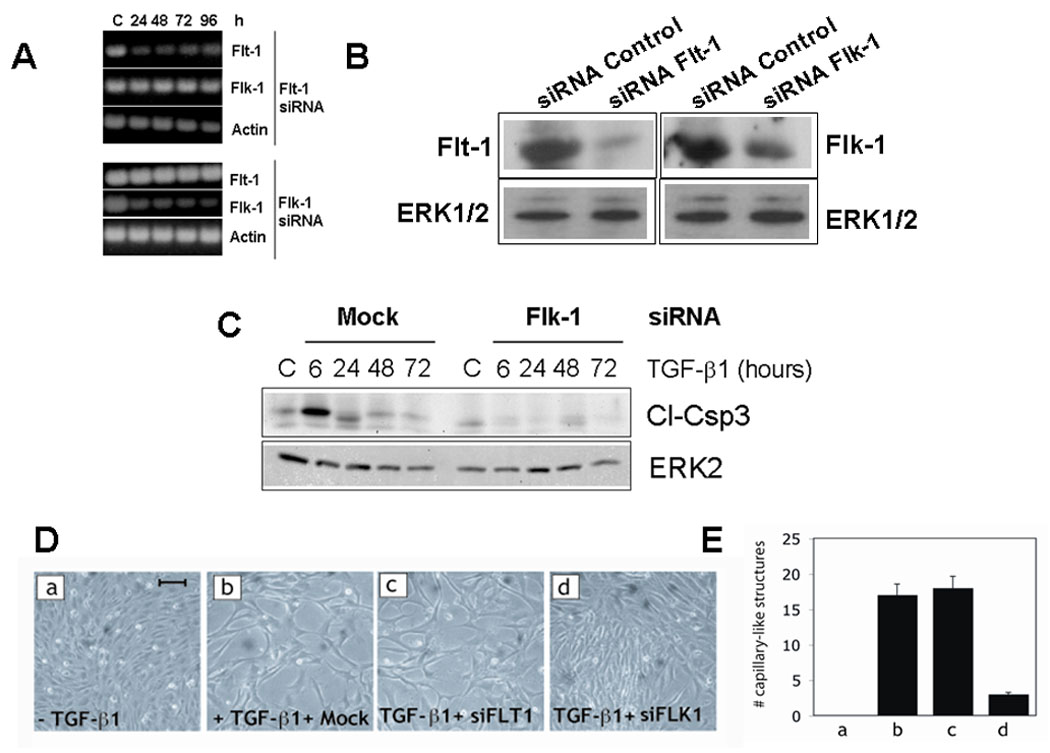
A. RT-PCR and B. Western blotting analysis of VEGFR1 (flt-1) and VEGFR2 (flk-1) mRNA expression in HUVEC transfected with VEGFR1 or VEGFR2 siRNAs. The cells were analyzed at the indicated time after siRNA transfection. ERK2 and β-actin are shown as controls. C. Western blotting analysis of caspase-3 cleavage (Cl-Csp3) in HUVEC transfected with VEGFR2 (Flk-1) siRNA or mock-transfected and incubated in medium supplemented with 0.5% FCS in the absence (C) or presence of TGF-β1 (1 ng/ml) for the indicated time. TGF-β1 was added to the culture medium every 24 h. ERK-2: loading control. D. Confluent HUVEC grown in gelatin-coated dishes and either non transfected (a) or transfected with transfection reagent alone (panel b) or with siRNAs to VEGFR1 (siFLT1; panel c) or VEGFR2 (siFLK1; panel d). The cells were photographed after 96 h incubation in medium supplemented with 0.5% FCS in the absence (a) or presence (b–d) of TGF-β1 (1 ng/ml). Bar represents 100 µm. This experiment was repeated three times with comparable results. E. The results of the experiments shown in panel A were quantitated as described under Methods. The histograms represent mean ± SE. p < 0.05 (sample vs. control).
VEGF signaling mediates the effect of TGF-β1 on formation of vascular structures in embryoid bodies
To study the role of VEGF signaling in TGF-β1 control of blood vessel formation we also used embryonic stem (ES) cells derived from wild-type (wt) mice and mice genetically deficient (KO) in either VEGF or VEGFR2 or VEGFR1. Under defined culture conditions ES cells form aggregates (embryoid bodies) and differentiate into all cell lineages (Wang et al., 1992). In embryoid bodies endothelial cells form a vascular network that can be identified by immunostaining with antibodies to endothelial cell markers such as PECAM (CD31) or ICAM-2. Blood vessel formation in embryoid bodies mimics the development of the embryonic yolk sac vasculature; it occurs through both vasculogenesis and angiogenesis, and is modulated by a variety of growth factors and cytokines including VEGF and TGF-β1 (Bautch et al., 2000; Bautch et al., 1996; Desbaillets et al., 2000; Feraud et al., 2001; Gualandris et al., 2000b; Mallet et al., 2006; Ng et al., 2004).
CD31 and ICAM-2 have different expression patterns during embryoid body differentiation. In differentiating embryoid bodies PECAM+ cells can be found that are not organized into vessels, suggesting that PECAM+ cells are endothelial cell precursors; conversely, ICAM-2+ cells are confined to patent vasculature (Bautch et al., Blood. 2000;95:1979–1987). Therefore, we used ICAM-2 as a marker of forming vascular structures in the embryoid bodies. Consistent with previous reports (Gualandris et al., 2000b; Vittet et al., 1996), wt embryoid bodies formed vascular structures that were increased by exogenous VEGF; conversely, in the presence of exogenous TGF-β1 the endothelial cells formed thick cords (Fig. 4). In VEGF-deficient embryoid bodies endothelial cells were dramatically reduced in number and formed no vessel-like structures. Addition of exogenous VEGF to these cells partially restored the wt phenotype. In contrast, treatment with TGF-β1 had no such effect. Embryoid bodies from ES cells deficient in VEGFR1 showed a vascular phenotype comparable to that of wt embryoid bodies, and responded both to exogenous VEGF and TGF-β1 in a manner similar to wt embryoid bodies. Conversely, VEGFR2 KO embryoid bodies showed few endothelial cells that were sparse or formed occasional clusters but no vascular structures, and this phenotype was not corrected by addition of either VEGF or TGF-β1 (Fig. 4).
Figure 4. VEGF signaling mediates the effect of TGF-β1 on formation of vascular structures in embryoid bodies.
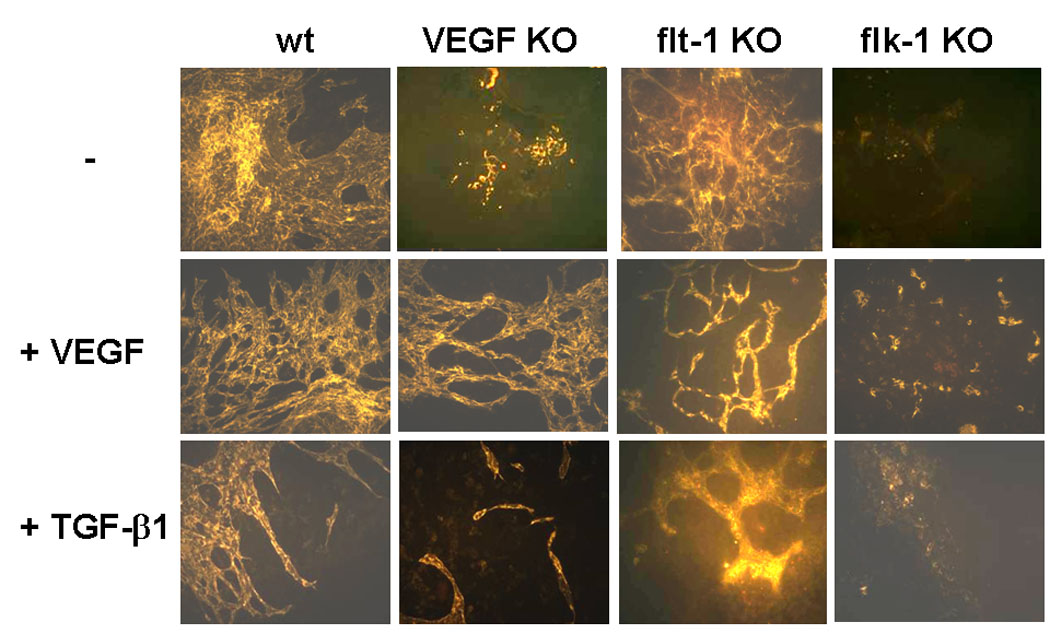
Embryonic stem cells from wt, VEGF- (VEGF KO), VEGFR1- (flt1 KO) or VEGFR2-deficient (flk-1 KO) mice were induced to differentiate in the absence (−) or presence of VEGF (30 ng/ml) or TGF-β1 (1 ng/ml). After 9-days incubation the differentiated cultures (embryoid bodies) were stained with rhodamine-labeled antibody to ICAM-2, an endothelial cell marker, and photographed with a fluorescence microscope. VEGF enhanced the formation of vascular networks in wt and VEGF-deficient embryoid bodies but not in VEGFR2-deficient embryoid bodies. Addition of TGF-β1 remodeled the capillary network in wt embryoid bodies, but had no such effect on VEGF- or VEGFR2-deficient endothelial cells. Bar represents 200 µm.
To quantitate these findings we analyzed ICAM-2 expression by RT-PCR (Fig. 5). At day 9 of differentiation, when endothelial cell differentiation is complete (Balconi et al., 2000; Vittet et al., 1996), ICAM-2 mRNA expression in VEGF-, VEGFR2- and VEGFR1-deficient embryoid bodies was dramatically reduced relative to wt embryoid bodies. Both VEGF and TGF-β1 increased ICAM-2 mRNA levels in wt embryoid bodies. VEGF also increased ICAM-2 levels in VEGF KO embryoid bodies. In contrast, TGF-β1 had no such effect, showing that the action of TGF-β1 on endothelial cell differentiation is dependent on VEGF signaling. Addition of VEGF or TGF-β1 had no significant effect on ICAM-2 mRNA levels in VEGFR1 KO and VEGFR2 KO embryoid bodies. Therefore, consistent with our results obtained by siRNA-mediated downregulation of VEGF receptors in mature endothelial cells (Fig. 4), these results showed that TGF-β1 induction of endothelial cell differentiation (vasculogenesis) requires VEGF signaling.
Figure 5. The genetic deficiency of VEGF abrogates TGF-β1 induction of endothelial cell differentiation in embryoid bodies.
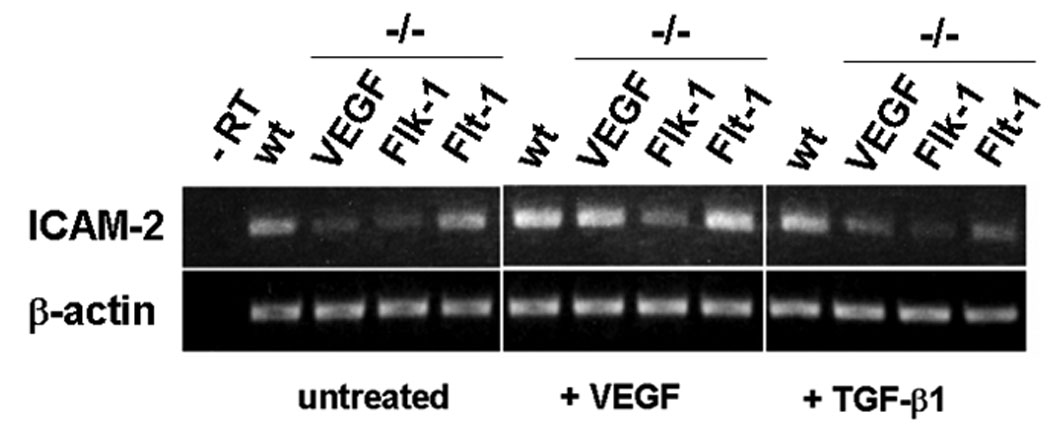
RT-PCR analysis. Embryonic stem cells from wt, VEGF- (VEGF KO), VEGFR1- (flt1 KO) or VEGFR2-deficient (flk-1 KO) mice were induced to differentiate in the absence (−) or presence of VEGF (30 ng/ml) or TGF-β1 (1 ng/ml). After 9-days of incubation the differentiated cultures (embryoid bodies) were analyzed for ICAM-2 expression. β-actin: loading control. (-RT) RT-PCR negative control.
VEGF mediates the apoptotic activity of TGF-β1 in vivo
To assess the biological significance of our findings in vivo we tested the effect of neutralizing antibody to VEGF on TGF-β1 induction of endothelial cell apoptosis in the chicken embryo choriollantoic membrane (CAM). For this purpose we treated chicken embryo CAMs with TGF-β1 in the absence or presence of neutralizing VEGF antibody. We used a TGF-β1 concentration that induces angiogenesis in the CAM (Yang and Moses, 1990). After 6 h incubation we analyzed CAM sections by TUNEL and immunostaining with antibody to CD31, an endothelial cell marker. Analysis of the sections by confocal microscopy (Fig. 6) showed that TGF-β1 strongly upregulated the number of TUNEL-positive (apoptotic) cells in the CAM with an effect comparable to that of TNF-α, a potent inducer of endothelial cell apoptosis (Ferrari et al., 2006). Virtually all apoptotic cells colocalized with CD31-positive (endothelial) cells, consistent with previous reports that TGF-β1 selectively induces apoptosis in endothelial cells but not in smooth muscle cells or pericytes (Pollman et al., 1999b). VEGF antibody strongly downregulated the number of TUNEL-positive cells induced by TGF-β1, whereas control n.i. IgG had no such effect (Fig. 6). Thus, these results showed that TGF-β1 induction of endothelial cell apoptosis in vivo requires VEGF signaling.
Figure 6. Inhibition of VEGF signaling blocks TGF-β1 induction of endothelial cell apoptosis in vivo.
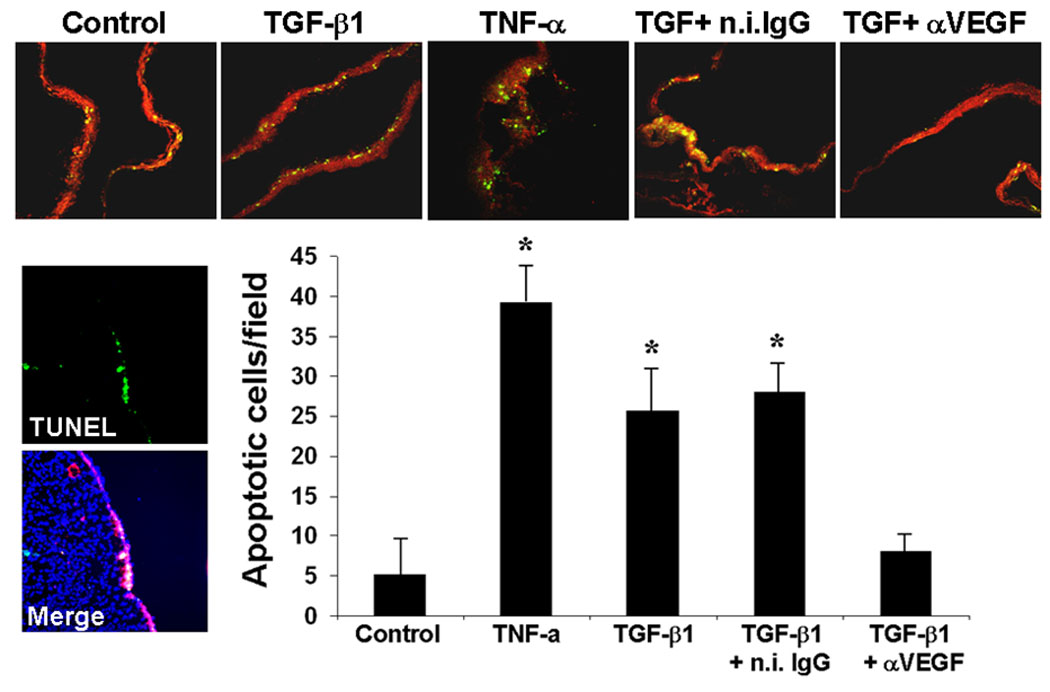
Confocal micrographs (100 ×) of CAMs incubated for 6 h with either PBS (Control) or TGF-β1 (100 ng) in the presence or absence of isotype-matched non-immune IgG or anti-mouse VEGF antibody (5 µg). A CAM incubated with TNF-α (100 ng + 1 mg of cycloheximide) is shown as a positive control for apoptosis. CD31-positive endothelial cells are stained in red, TUNEL-positive cells are stained in yellow-green. Lower left panels. TUNEL: section stained only for TUNEL; Merge: high-power (200 ×) image of the same field with merged DAPI and CD31 staining and with TUNEL staining shows colocalization of TUNEL-positive cells with CD31-positive cells. Filter disks soaked with hydroxycortisone acetate (Sigma; 2.5 mg/ml in 95% ethanol) plus or minus the indicated reagents were applied on 10-day old CAMs as described (Brooks et al., 1999). After 6 h incubation at 38° C the CAMs were separated from the paper disks, snap frozen in OTC and 4 µm sections were cut. Five CAMs per sample were used. B. TUNEL-positive cells colocalized with CD31-positive cells were counted in low-power (100 ×) fields of sections from all the five CAMs of each sample. The histogram shows mean number of TUNEL-positive nuclei / field ± s.d. * p < 0.01 (sample vs. control).
VEGF mediates TGF-β1 induction of angiogenesis in vivo
We then tested the effect of neutralizing anti-VEGF antibody on TGF-β1 induction of blood vessel formation in the CAM. As described (Yang and Moses, 1990), TGF-β1 induced formation of new capillaries from pre-existing CAM vessels with an effect comparable to that of FGF-2. The vessels formed in the presence of TGF-β1 were relatively large and had few branches, whereas those induced by FGF-2 were thin and had many branches (Fig. 7 A and B) (Yang and Moses, 1990). To confirm that our VEGF antibody neutralized chicken VEGF, we tested if it blocked angiogenesis induction in the CAM by FGF-2. We have previously shown that FGF-2 upregulates endothelial cell expression of VEGF, and that antibody to VEGF blocks FGF-2 induction of angiogenesis in vivo (Ferrari et al., 2006; Seghezzi et al., 1998). VEGF antibody blocked the angiogenic effect of FGF-2 in the CAM, showing that it neutralized chicken VEGF (Fig. 7 A and B). We therefore tested the effect of VEGF antibody on TGF-β1 induction of angiogenesis. TGF-β1 increased the number of CAM vessels approximately 2.5-fold, an effect comparable to that obtained with FGF-2 and consistent with previous findings (Yang and Moses, 1990). Antibody to VEGF completely abrogated this effect, showing that TGF-β1 induces angiogenesis in vivo through the autocrine and/or paracrine action of VEGF.
Figure 7. A and B. Inhibition of VEGF signaling blocks TGF-β1 induction of angiogenesis in vivo.
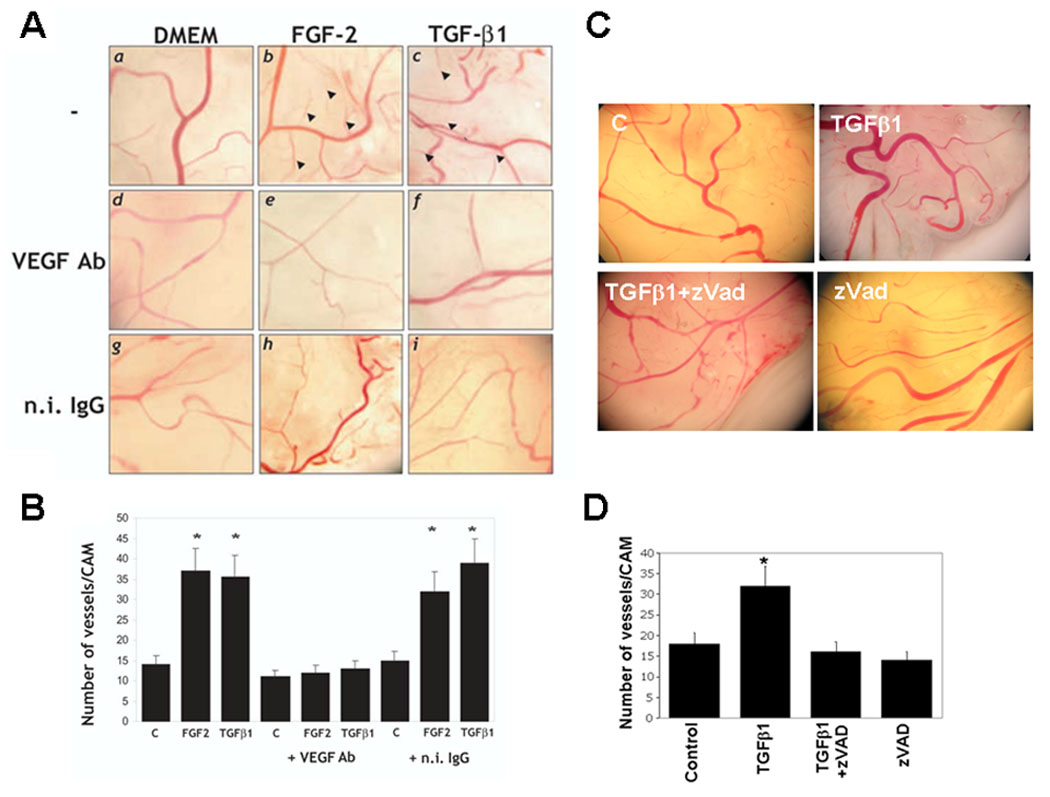
A. Chorioallantoic membranes of 10-day old chicken embryos incubated with either hrFGF-2 (50 ng) or hr TGF-β1 (100 ng) or with control medium (DMEM) in the absence (−) or presence of 5 µg of anti-mouse VEGF antibody (VEGF Ab) or n.i. IgG. Representative images from one experiment are shown. Arrowheads indicate newly formed branching vessels. B. Number of branching new vessels in CAMs treated with the indicated reagents as described above. The histograms represent mean ± SE of the number of vessels/CAM determined in three independent experiments (8 CAMs/sample/experiment). *: p < 0.05 (sample vs. control). C. and D. Inhibition of apoptosis blocks TGF-β1 induction of angiogenesis in vivo. C. Chorioallantoic membranes of 10-day old embryos untreated (Control), treated with z-VAD-(OMe)-FMK (z-VAD) or incubated with TGF-β1 alone or with TGF-β1 and z-VAD. D. Number of branching new vessels in CAMs treated with the indicated reagents as described above. The histograms represent mean ± SE of the number of vessels/CAM determined in three independent experiments (8 CAMs/sample/experiment). *: p < 0.05 (sample vs. control).
Because VEGF mediates the apoptotic activity of TGF-β1 on the CAM endothelial cells, these results indicated that the rapid apoptotic effect mediated by VEGF is required for TGF-β1 induction of angiogenesis in vivo. Therefore, we treated CAMs with TGF-β1 in the presence or absence of z-VAD, a general caspase inhibitor that blocks in vitro angiogenesis (Fig. 2, panel q). As shown in Figure 7 C and D, z-VAD completely abrogated the angiogenic activity of TGF-β1 with an effect comparable to that of VEGF antibody. In our assays the growth factors were administered to the CAM in the presence of hydroxycortisone acetate (2.5 mg/ml) as described (Brooks et al., 1999), to block the inflammatory response. Therefore, under our experimental conditions, endothelial cell apoptosis did not result from the inflammatory response, and VEGF derived not from inflammatory cells but from the epithelial and/or endothelial cells of the CAM. Thus, these results showed that rapid and transient upregulation of VEGF- VEGFR2-mediated endothelial cell apoptosis is required for TGF-β1 induction of angiogenesis in vivo.
DISCUSSION
We have previously shown that the rapid induction of endothelial cell apoptosis by TGF-β1 is mediated by the autocrine or paracrine activation of VEGFR2 by endothelial cell VEGF (Ferrari et al., 2006). This finding raised a fundamental question: Does VEGF-mediated apoptosis promote or inhibit TGF-β1 induction of angiogenesis? The data reported here show that VEGF-mediated apoptosis is an early event required for induction of blood vessel formation by TGF-β1. Therefore, endothelial cell apoptosis is not only necessary for pruning the forming vascular network during the late stages of angiogenesis; it also occurs in the initial steps of, and is required for angiogenesis to proceed.
Because of the multiple inhibitory effects of TGF- β1 on endothelial cells, it has been proposed that TGF- β1 induces angiogenesis in vivo indirectly through its action on immune response cells. However, a variety of findings indicate that TGF- β1 has important, direct effects on angiogenesis in vivo. Half of the mice genetically deficient in TGF-β1 die in utero and show defective vasculogenesis and angiogenesis, a phenotype consistent with abundant TGF-β1 expression in endothelial precursors. Importantly, during mouse embryogenesis ALK1 (TGF-β receptor I) expression is mainly confined to endothelial cells, and is upregulated at sites of active angiogenesis. Mice genetically deficient in ALK1 die from defects in angiogenesis, as do mice deficient in either ALK5 (TGF-β receptor I) or TGF-β receptor II (Dickson et al., 1995; Li et al., 1999; Oshima et al., 1996). In humans mutations of ALK1, the endothelial cell TGF-β1 receptor, cause hereditary hemorrhagic teleangiectasia, a condition characterized by absence of capillary bed (angiogenesis) in certain vascular districts. In addition, in adult animals TGF- β1 induces endothelial cell apoptosis, a process required for glomerular capillary formation (Fierlbeck et al., 2003).
Our finding that TGF-β1-induced endothelial cell apoptosis occurs rapidly and is followed by a long period in which the cells are refractory to the apoptotic effect of TGF-β1 can be explained by several, non-mutually exclusive mechanisms, including proteasomal degradation of phosphorylated Smad2 (Lo and Massague, 1999; Zhang et al., 2001), downregulation of TGF-β receptor (TGF-βR) and/or VEGFR2 expression (Anders et al., 1997; Mandriota et al., 1996; Minami et al., 2001). Our observation is also consistent with the finding that induction of endothelial cell death is followed by increased proliferation and resistance to apoptosis (Sakao et al., 2005). A variety of tumors contain high levels of both TGF-β1 and VEGF, which do not result in massive endothelial cell apoptosis. Our finding of the transient nature of the apoptotic effect of TGF-β1-induced apoptosis, and the known effect of TGF-β1 on TGF-β receptor and VEGFR2 expression can explain the apparent lack of endothelial cell apoptosis in these tumors.
Under our experimental conditions siRNA-mediated downregulation of VEGFR2 expression did not result in endothelial cell apoptosis in vitro for at least 72 h; similarly, inhibition of VEGF did not cause rapid endothelial cell apoptosis in vivo. These findings are seemingly in contrast with the current concept that VEGF/VEGFR2 signaling is required for endothelial cell survival in vitro and in vivo (Gerber et al., 1998a; Gerber et al., 1998b; Lee et al., 2007; Sweeney et al., 2002). However, several reports have shown that inhibition of VEGF – VEGFR2 signaling by antibodies or chemical inhibitors of the receptor does not cause rapid apoptosis of cultured endothelial cells in the absence of a pro-apoptotic stimulus (Geng et al., 2001; Lu et al., 2005; Sakao et al., 2007). The genetic deficiency of VEGF in cultured endothelial cells results in apoptosis only after 72 h incubation in serum-free medium, a pro-apoptotic culture condition (Gerber et al., 1998a; Gerber et al., 1998b; Lee et al., 2007). Our observation that downregulation of VEGFR2 expression does not provide an apoptotic stimulus per se is also consistent with the recent finding that Notch-1 downregulates VEGFR2 expression without inducing endothelial cell apoptosis (Shawber et al., 2007). In addition, to the best of our knowledge, our previous (Ferrari et al., 2006) and present work is the first that tested the effect of siRNA-mediated downregulation of VEGFR2 expression on endothelial cell apoptosis. The specificity of the effect of the VEGFR2 siRNAs we used is shown by their lack of downregulation of VEGFR1 expression. Their functional efficacy was shown by our previous results showing that transfection with VEGFR2 siRNAs abolish MAPK activation in response to VEGF (Ferrari et al., 2006). Our finding that inhibition of VEGF - VEGFR2 signaling does not induce rapid endothelial cell apoptosis is also supported by our analysis of endothelial cell apoptosis in the CAM. In these experiments TGF-β1 induced endothelial cell apoptosis as rapidly (6 h) as in vitro. Inhibition of VEGF by neutralizing antibody did not increase endothelial cell apoptosis in the presence of TGF-β1; on the contrary, it blocked the apoptotic effect of TGF-β1. Based on our data we cannot exclude that inhibition of VEGF or VEGFR2 expression for a longer time would result in apoptosis in the absence of TGF-β1. However, our results show that the rapid apoptotic effect of TGF-β1 on endothelial cells requires VEGF signaling and is necessary for the progression of the angiogenic process. Thus, VEGF-VEGFR2 signaling not only provides endothelial cells with pro-survival and proliferative/migratory stimuli required for vessel formation but also mediates the initial apoptotic effect necessary for TGF-β1 induction of angiogenesis.
Inhibition of VEGF signaling – which blocks TGF-β1 induction of apoptosis - blocked endothelial cell cord formation in vitro with no apparent morphological changes in the cell monolayer. Conversely, inhibition of FGF-2 - which induces apoptosis - resulted in few scattered and disrupted cord-like structures. Thus, both inhibition of, and excess apoptosis block angiogenesis, indicating that an optimal level of apoptosis is required for vessel formation.
A previous report has shown that TGF-β1/ALK1 (TGF-β receptor I) signaling in endothelial cells stimulates angiogenesis through Smad activation (Goumans et al., 2002). Our data are not in contrast with this finding as our results do not imply that apoptosis is Smad-independent. Indeed, Smad signaling could act upstream of VEGF (e.g. by promoting VEGF expression). Several papers have shown that TGF-β1 induction of VEGF in a variety of cell types other than endothelial cells is Smad-dependent. One article (Bostrom et al., 2004) reported that overexpression of, or stimulation of ALK-1 results in Smad activation and upregulation of VEGF expression in endothelial cells. However, the possibility that in these cells TGF-β1 induction of VEGF expression is mediated by a Smad-independent mechanism (e.g. p38MAPK) was not explored. We are currently investigating this hypothesis.
Our experiments on endothelial cell differentiation in embryoid bodies provide genetic evidence for a role of VEGF signaling in TGF-β1 induction of vasculogenesis. Consistent with previous observations (Feraud et al., 2001; Gualandris et al., 2000b), both VEGF and TGF-β1 increased endothelial cell differentiation and organization in embryoid bodies. However, we found that TGF-β1 has no such effect on embryoid bodies genetically deficient in VEGF. A previous report has shown that TGF-β1 increases endothelial cell differentiation in a VEGF-independent manner and inhibits endothelial tube formation in embryoid bodies (Mallet et al., 2006). Two reasons can explain this discrepancy with our results. The independence of TGF-β1 action from VEGF was shown by using a neutralizing antibody added a day 0 of differentiation; in contrast, we used cells genetically deficient in VEGF or VEGF receptors. In addition, the authors used PECAM as a marker of endothelial cell differentiation, whereas we used ICAM-2. These endothelial cell markers have different expression patterns during embryoid body differentiation. In differentiating embryoid bodies PECAM+ cells can be found that are not organized into vessels, suggesting that PECAM+ cells are endothelial cell precursors; conversely, ICAM-2+ cells are confined to patent vasculature (Bautch et al., 2000).
The genetic deficiency of VEGFR2 caused a dramatic decrease in endothelial cell differentiation, which was not increased by either VEGF or TGF-β1. Thus, the very low number of endothelial cells in VEGFR2-deficient embryoid bodies did not allow any conclusion as to the role of VEGFR2 in TGF-β1 induction of vascular structures in embryoid bodies. However, our data obtained with ES cells genetically deficient in VEGF or VEGFR1 are consistent with our results obtained with mature endothelial cells, showing the requirement of VEGF-VEGFR2 signaling for TGF-β1 induction of angiogenesis in vitro and in vivo. This mechanism can therefore play a significant role in the control of endothelial stem cell differentiation during angiogenesis in vivo.
Our findings that TGF-β1 induces apoptosis and angiogenesis in vitro and in vivo through the autocrine or paracrine activation of VEGFR2 by VEGF indicate a potential pharmacological approach to anti-angiogenesis therapy. High levels of VEGF, VEGFR2 and TGF-β1 are present in many tumors. Crosstalk between the signaling pathways activated by these growth factors controls endothelial cell apoptosis (Ferrari et al., 2006) and angiogenesis. Understanding the mechanism(s) that modulate this crosstalk can permit the development of novel pharmacological tools to convert VEGF, a potent angiogenesis inducer and survival factor, into an inducer of uncontrolled endothelial cell apoptosis, and therefore into a potent anti-angiogenesis factor.
Supplementary Material
Supplemental Figure 1. In vitro angiogenesis. Confluent BCE cells grown in gelatin-coated dishes were incubated in the absence (upper panel) or presence of TGF-β1 (1 ng/ml; lower panel) in medium supplemented with 0.5% calf serum. The cultures were photographed after 72 h incubation. Shown are enlarged images of panel a and b of Fig. 2.
ACKNOWLEDGEMENTS
We are grateful to Drs. A. Joyner, Andras Nagy, Janet Rossant and Guo-Hua Fong for their generous gifts of wt and mutant ES cells. This work was supported by grants NIH R01 HL070203 and R01 HL070203-03S1 to P.M., and by funds from the Department of Cardiothoracic Surgery of NYU School of Medicine.
REFERENCES
- Anders RA, Arline SL, Dore JJ, Leof EB. Distinct endocytic responses of heteromeric and homomeric transforming growth factor beta receptors. Mol Biol Cell. 1997;8(11):2133–2143. doi: 10.1091/mbc.8.11.2133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balconi G, Spagnuolo R, Dejana E. Development of endothelial cell lines from embryonic stem cells: A tool for studying genetically manipulated endothelial cells in vitro. Arterioscler Thromb Vasc Biol. 2000;20(6):1443–1451. doi: 10.1161/01.atv.20.6.1443. [DOI] [PubMed] [Google Scholar]
- Bautch VL, Redick SD, Scalia A, Harmaty M, Carmeliet P, Rapoport R. Characterization of the vasculogenic block in the absence of vascular endothelial growth factor-A. Blood. 2000;95(6):1979–1987. [PubMed] [Google Scholar]
- Bautch VL, Stanford WL, Rapoport R, Russell S, Byrum RS, Futch TA. Blood island formation in attached cultures of murine embryonic stem cells. Dev Dyn. 1996;205(1):1–12. doi: 10.1002/(SICI)1097-0177(199601)205:1<1::AID-AJA1>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- Bostrom K, Zebboudj AF, Yao Y, Lin TS, Torres A. Matrix GLA protein stimulates VEGF expression through increased transforming growth factor-beta1 activity in endothelial cells. J Biol Chem. 2004;279(51):52904–52913. doi: 10.1074/jbc.M406868200. [DOI] [PubMed] [Google Scholar]
- Brooks PC, Montgomery AM, Cheresh DA. Use of the 10-day-old chick embryo model for studying angiogenesis. Methods Mol Biol. 1999;129:257–269. doi: 10.1385/1-59259-249-X:257. [DOI] [PubMed] [Google Scholar]
- Carmeliet P, Ferreira V, Breier G, Pollefeyt S, Kieckens L, Gertsenstein M, Fahrig M, Vandenhoeck A, Harpal K, Eberhardt C, Declercq C, Pawling J, Moons L, Collen D, Risau W, Nagy A. Abnormal blood vessel development and lethality in embryos lacking a single VEGF allele. Nature. 1996;380(6573):435–439. doi: 10.1038/380435a0. [DOI] [PubMed] [Google Scholar]
- Choi ME, Ballermann BJ. Inhibition of capillary morphogenesis and associated apoptosis by dominant negative mutant transforming growth factor-beta receptors. J Biol Chem. 1995;270(36):21144–21150. doi: 10.1074/jbc.270.36.21144. [DOI] [PubMed] [Google Scholar]
- Desbaillets I, Ziegler U, Groscurth P, Gassmann M. Embryoid bodies: an in vitro model of mouse embryogenesis. Exp Physiol. 2000;85(6):645–651. [PubMed] [Google Scholar]
- Dickson MC, Martin JS, Cousins FM, Kulkarni AB, Karlsson S, Akhurst RJ. Defective haematopoiesis and vasculogenesis in transforming growth factor-beta 1 knock out mice. Development. 1995;121(6):1845–1854. doi: 10.1242/dev.121.6.1845. [DOI] [PubMed] [Google Scholar]
- Feraud O, Cao Y, Vittet D. Embryonic stem cell-derived embryoid bodies development in collagen gels recapitulates sprouting angiogenesis. Lab Invest. 2001;81(12):1669–1681. doi: 10.1038/labinvest.3780380. [DOI] [PubMed] [Google Scholar]
- Ferrara N. Vascular endothelial growth factor: basic science and clinical progress. Endocr Rev. 2004;25(4):581–611. doi: 10.1210/er.2003-0027. [DOI] [PubMed] [Google Scholar]
- Ferrara N, Carver-Moore K, Chen H, Dowd M, Lu L, O'Shea KS, Powell-Braxton L, Hillan KJ, Moore MW. Heterozygous embryonic lethality induced by targeted inactivation of the VEGF gene. Nature. 1996;380(6573):439–442. doi: 10.1038/380439a0. [DOI] [PubMed] [Google Scholar]
- Ferrara N, Kerbel RS. Angiogenesis as a therapeutic target. Nature. 2005;438(7070):967–974. doi: 10.1038/nature04483. [DOI] [PubMed] [Google Scholar]
- Ferrari G, Pintucci G, Seghezzi G, Hyman K, Galloway AC, Mignatti P. VEGF, a prosurvival factor, acts in concert with TGF-beta1 to induce endothelial cell apoptosis. Proc Natl Acad Sci U S A. 2006;103(46):17260–17265. doi: 10.1073/pnas.0605556103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fierlbeck W, Liu A, Coyle R, Ballermann BJ. Endothelial cell apoptosis during glomerular capillary lumen formation in vivo. J Am Soc Nephrol. 2003;14(5):1349–1354. doi: 10.1097/01.asn.0000061779.70530.06. [DOI] [PubMed] [Google Scholar]
- Fong GH, Rossant J, Gertsenstein M, Breitman ML. Role of the Flt-1 receptor tyrosine kinase in regulating the assembly of vascular endothelium. Nature. 1995;376(6535):66–70. doi: 10.1038/376066a0. [DOI] [PubMed] [Google Scholar]
- Forsythe JA, Jiang BH, Iyer NV, Agani F, Leung SW, Koos RD, Semenza GL. Activation of vascular endothelial growth factor gene transcription by hypoxia-inducible factor 1. Mol Cell Biol. 1996;16(9):4604–4613. doi: 10.1128/mcb.16.9.4604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geng L, Donnelly E, McMahon G, Lin PC, Sierra-Rivera E, Oshinka H, Hallahan DE. Inhibition of vascular endothelial growth factor receptor signaling leads to reversal of tumor resistance to radiotherapy. Cancer Res. 2001;61(6):2413–2419. [PubMed] [Google Scholar]
- Gerber H-P, Dixit V, Ferrara N. Vascular Endothelial Growth Factor Induces Expression of the Antiapoptotic Proteins Bcl-2 and A1 in Vascular Endothelial Cells. J Biol Chem. 1998a;273(21):13313–13316. doi: 10.1074/jbc.273.21.13313. [DOI] [PubMed] [Google Scholar]
- Gerber H-P, McMurtrey A, Kowalski J, Yan M, Keyt BA, Dixit V, Ferrara N. Vascular Endothelial Growth Factor Regulates Endothelial Cell Survival through the Phosphatidylinositol 3'-Kinase/Akt Signal Transduction Pathway. REQUIREMENT FOR Flk-1/KDR ACTIVATION. J Biol Chem. 1998b;273(46):30336–30343. doi: 10.1074/jbc.273.46.30336. [DOI] [PubMed] [Google Scholar]
- Gille H, Kowalski J, Li B, LeCouter J, Moffat B, Zioncheck TF, Pelletier N, Ferrara N. Analysis of biological effects and signaling properties of flt-1 (vegfr- 1) and kdr (vegfr-2). a reassessment using novel receptor-specific vascular endothelial growth factor mutants. J Biol Chem. 2001;276(5):3222–3230. doi: 10.1074/jbc.M002016200. [DOI] [PubMed] [Google Scholar]
- Goldman CK, Kim J, Wong WL, King V, Brock T, Gillespie GY. Epidermal growth factor stimulates vascular endothelial growth factor production by human malignant glioma cells: a model of glioblastoma multiforme pathophysiology. Mol Biol Cell. 1993;4(1):121–133. doi: 10.1091/mbc.4.1.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goumans MJ, Valdimarsdottir G, Itoh S, Rosendahl A, Sideras P, ten Dijke P. Balancing the activation state of the endothelium via two distinct TGF-beta type I receptors. Embo J. 2002;21(7):1743–1753. doi: 10.1093/emboj/21.7.1743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gualandris A, Annes JP, Arese M, Noguera I, Jurukovski V, Rifkin DB. The latent transforming growth factor-beta-binding protein-1 promotes in vitro differentiation of embryonic stem cells into endothelium. Mol Biol Cell. 2000a;11(12):4295–4308. doi: 10.1091/mbc.11.12.4295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gualandris A, Annes JP, Arese M, Noguera I, Jurukovski V, Rifkin DB. The latent transforming growth factor-beta-binding protein-1 promotes In vitro differentiation of embryonic stem cells into endothelium [In Process Citation] Mol Biol Cell. 2000b;11(12):4295–4308. doi: 10.1091/mbc.11.12.4295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hicklin DJ, Ellis LM. Role of the vascular endothelial growth factor pathway in tumor growth and angiogenesis. J Clin Oncol. 2005;23(5):1011–1027. doi: 10.1200/JCO.2005.06.081. [DOI] [PubMed] [Google Scholar]
- Keyt BA, Nguyen HV, Berleau LT, Duarte CM, Park J, Chen H, Ferrara N. Identification of vascular endothelial growth factor determinants for binding KDR and FLT-1 receptors. Generation of receptor-selective VEGF variants by site-directed mutagenesis. J Biol Chem. 1996;271(10):5638–5646. doi: 10.1074/jbc.271.10.5638. [DOI] [PubMed] [Google Scholar]
- Lee S, Chen TT, Barber CL, Jordan MC, Murdock J, Desai S, Ferrara N, Nagy A, Roos KP, Iruela-Arispe ML. Autocrine VEGF signaling is required for vascular homeostasis. Cell. 2007;130(4):691–703. doi: 10.1016/j.cell.2007.06.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy AP, Levy NS, Wegner S, Goldberg MA. Transcriptional regulation of the rat vascular endothelial growth factor gene by hypoxia. J Biol Chem. 1995;270(22):13333–13340. doi: 10.1074/jbc.270.22.13333. [DOI] [PubMed] [Google Scholar]
- Li DY, Sorensen LK, Brooke BS, Urness LD, Davis EC, Taylor DG, Boak BB, Wendel DP. Defective angiogenesis in mice lacking endoglin. Science. 1999;284(5419):1534–1537. doi: 10.1126/science.284.5419.1534. [DOI] [PubMed] [Google Scholar]
- Li J, Perrella MA, Tsai JC, Yet SF, Hsieh CM, Yoshizumi M, Patterson C, Endege WO, Zhou F, Lee ME. Induction of vascular endothelial growth factor gene expression by interleukin-1 beta in rat aortic smooth muscle cells. J Biol Chem. 1995;270(1):308–312. doi: 10.1074/jbc.270.1.308. [DOI] [PubMed] [Google Scholar]
- Liu Y, Cox SR, Morita T, Kourembanas S. Hypoxia regulates vascular endothelial growth factor gene expression in endothelial cells Identification of a 5' enhancer. Circ Res. 1995;77(3):638–643. doi: 10.1161/01.res.77.3.638. [DOI] [PubMed] [Google Scholar]
- Lo RS, Massague J. Ubiquitin-dependent degradation of TGF-beta-activated smad2. Nat Cell Biol. 1999;1(8):472–478. doi: 10.1038/70258. [DOI] [PubMed] [Google Scholar]
- Lu H, Lin C, Zheng Z, Li S, Guo S, Zhang X, Fu M, Liang X, Wu M. Angiogenesis inhibitor Z24 induces endothelial cell apoptosis and suppresses tumor growth and metastasis. J Pharmacol Sci. 2005;97(4):533–540. doi: 10.1254/jphs.fp0040761. [DOI] [PubMed] [Google Scholar]
- Madri JA, Pratt BM, Tucker AM. Phenotypic modulation of endothelial cells by transforming growth factor-beta depends upon the composition and organization of the extracellular matrix. J Cell Biol. 1988;106(4):1375–1384. doi: 10.1083/jcb.106.4.1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallet C, Vittet D, Feige JJ, Bailly S. TGFbeta1 induces vasculogenesis and inhibits angiogenic sprouting in an embryonic stem cell differentiation model: respective contribution of ALK1 and ALK5. Stem Cells. 2006;24(11):2420–2427. doi: 10.1634/stemcells.2005-0494. [DOI] [PubMed] [Google Scholar]
- Mandriota SJ, Menoud PA, Pepper MS. Transforming growth factor beta 1 down-regulates vascular endothelial growth factor receptor 2/flk-1 expression in vascular endothelial cells. J Biol Chem. 1996;271(19):11500–11505. doi: 10.1074/jbc.271.19.11500. [DOI] [PubMed] [Google Scholar]
- Mandriota SJ, Pepper MS. Vascular endothelial growth factor-induced in vitro angiogenesis and plasminogen activator expression are dependent on endogenous basic fibroblast growth factor. J Cell Sci. 1997;110(Pt 18):2293–2302. doi: 10.1242/jcs.110.18.2293. [DOI] [PubMed] [Google Scholar]
- Massague J, Blain SW, Lo RS. TGFbeta signaling in growth control, cancer, and heritable disorders. Cell. 2000;103(2):295–309. doi: 10.1016/s0092-8674(00)00121-5. [DOI] [PubMed] [Google Scholar]
- Minami T, Rosenberg RD, Aird WC. Transforming growth factor-beta 1-mediated inhibition of the flk-1/KDR gene is mediated by a 5'-untranslated region palindromic GATA site. J Biol Chem. 2001;276(7):5395–5402. doi: 10.1074/jbc.M008798200. [DOI] [PubMed] [Google Scholar]
- Ng YS, Ramsauer M, Loureiro RM, D'Amore PA. Identification of genes involved in VEGF-mediated vascular morphogenesis using embryonic stem cell-derived cystic embryoid bodies. Lab Invest. 2004;84(9):1209–1218. doi: 10.1038/labinvest.3700150. [DOI] [PubMed] [Google Scholar]
- Oshima M, Oshima H, Taketo MM. TGF-beta receptor type II deficiency results in defects of yolk sac hematopoiesis and vasculogenesis. Dev Biol. 1996;179(1):297–302. doi: 10.1006/dbio.1996.0259. [DOI] [PubMed] [Google Scholar]
- Pardali K, Moustakas A. Actions of TGF-beta as tumor suppressor and pro-metastatic factor in human cancer. Biochim Biophys Acta. 2007;1775(1):21–62. doi: 10.1016/j.bbcan.2006.06.004. [DOI] [PubMed] [Google Scholar]
- Pepper MS, Belin D, Montesano R, Orci L, Vassalli JD. Transforming growth factor-beta 1 modulates basic fibroblast growth factor-induced proteolytic and angiogenic properties of endothelial cells in vitro. J Cell Biol. 1990;111(2):743–755. doi: 10.1083/jcb.111.2.743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pertovaara L, Kaipainen A, Mustonen T, Orpana A, Ferrara N, Saksela O, Alitalo K. Vascular endothelial growth factor is induced in response to transforming growth factor-beta in fibroblastic and epithelial cells. J Biol Chem. 1994;269(9):6271–6274. [PubMed] [Google Scholar]
- Pollman MJ, Naumovski L, Gibbons GH. Endothelial cell apoptosis in capillary network remodeling. J Cell Physiol. 1999a;178(3):359–370. doi: 10.1002/(SICI)1097-4652(199903)178:3<359::AID-JCP10>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- Pollman MJ, Naumovski L, Gibbons GH. Vascular cell apoptosis: cell type-specific modulation by transforming growth factor-beta1 in endothelial cells versus smooth muscle cells. Circulation. 1999b;99(15):2019–2026. doi: 10.1161/01.cir.99.15.2019. [DOI] [PubMed] [Google Scholar]
- Presta M, Dell'Era P, Mitola S, Moroni E, Ronca R, Rusnati M. Fibroblast growth factor/fibroblast growth factor receptor system in angiogenesis. Cytokine Growth Factor Rev. 2005;16(2):159–178. doi: 10.1016/j.cytogfr.2005.01.004. [DOI] [PubMed] [Google Scholar]
- Roberts AB, Sporn MB, Assoian RK, Smith JM, Roche NS, Wakefield LM, Heine UI, Liotta LA, Falanga V, Kehrl JH, et al. Transforming growth factor type beta: rapid induction of fibrosis and angiogenesis in vivo and stimulation of collagen formation in vitro. Proc Natl Acad Sci U S A. 1986;83(12):4167–4171. doi: 10.1073/pnas.83.12.4167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakao S, Taraseviciene-Stewart L, Cool CD, Tada Y, Kasahara Y, Kurosu K, Tanabe N, Takiguchi Y, Tatsumi K, Kuriyama T, Voelkel NF. VEGF-R blockade causes endothelial cell apoptosis, expansion of surviving CD34+ precursor cells and transdifferentiation to smooth muscle-like and neuronal-like cells. Faseb J. 2007;21(13):3640–3652. doi: 10.1096/fj.07-8432com. [DOI] [PubMed] [Google Scholar]
- Sakao S, Taraseviciene-Stewart L, Lee JD, Wood K, Cool CD, Voelkel NF. Initial apoptosis is followed by increased proliferation of apoptosis-resistant endothelial cells. Faseb J. 2005;19(9):1178–1180. doi: 10.1096/fj.04-3261fje. [DOI] [PubMed] [Google Scholar]
- Saksela O, Moscatelli D, Rifkin DB. The opposing effects of basic fibroblast growth factor and transforming growth factor beta on the regulation of plasminogen activator activity in capillary endothelial cells. J Cell Biol. 1987;105(2):957–963. doi: 10.1083/jcb.105.2.957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seghezzi G, Patel S, Ren CJ, Gualandris A, Pintucci G, Robbins ES, Shapiro RL, Galloway AC, Rifkin DB, Mignatti P. Fibroblast growth factor-2 (FGF-2) induces vascular endothelial growth factor (VEGF) expression in the endothelial cells of forming capillaries: an autocrine mechanism contributing to angiogenesis. J Cell Biol. 1998;141(7):1659–1673. doi: 10.1083/jcb.141.7.1659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segura I, Serrano A, De Buitrago GG, Gonzalez MA, Abad JL, Claveria C, Gomez L, Bernad A, Martinez AC, Riese HH. Inhibition of programmed cell death impairs in vitro vascular-like structure formation and reduces in vivo angiogenesis. Faseb J. 2002;16(8):833–841. doi: 10.1096/fj.01-0819com. [DOI] [PubMed] [Google Scholar]
- Shawber CJ, Funahashi Y, Francisco E, Vorontchikhina M, Kitamura Y, Stowell SA, Borisenko V, Feirt N, Podgrabinska S, Shiraishi K, Chawengsaksophak K, Rossant J, Accili D, Skobe M, Kitajewski J. Notch alters VEGF responsiveness in human and murine endothelial cells by direct regulation of VEGFR-3 expression. J Clin Invest. 2007;117(11):3369–3382. doi: 10.1172/JCI24311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shweiki D, Itin A, Soffer D, Keshet E. Vascular endothelial growth factor induced by hypoxia may mediate hypoxia-initiated angiogenesis. Nature. 1992;359(6398):843–845. doi: 10.1038/359843a0. [DOI] [PubMed] [Google Scholar]
- Su J, Muranjan M, Sap J. Receptor protein tyrosine phosphatase alpha activates Src-family kinases and controls integrin-mediated responses in fibroblasts. Curr Biol. 1999;9(10):505–511. doi: 10.1016/s0960-9822(99)80234-6. [DOI] [PubMed] [Google Scholar]
- Sweeney P, Karashima T, Kim SJ, Kedar D, Mian B, Huang S, Baker C, Fan Z, Hicklin DJ, Pettaway CA, Dinney CP. Anti-vascular endothelial growth factor receptor 2 antibody reduces tumorigenicity and metastasis in orthotopic prostate cancer xenografts via induction of endothelial cell apoptosis and reduction of endothelial cell matrix metalloproteinase type 9 production. Clin Cancer Res. 2002;8(8):2714–2724. [PubMed] [Google Scholar]
- Tischer E, Mitchell R, Hartman T, Silva M, Gospodarowicz D, Fiddes JC, Abraham JA. The human gene for vascular endothelial growth factor. Multiple protein forms are encoded through alternative exon splicing. J Biol Chem. 1991;266(18):11947–11954. [PubMed] [Google Scholar]
- Vittet D, Prandini MH, Berthier R, Schweitzer A, Martin-Sisteron H, Uzan G, Dejana E. Embryonic stem cells differentiate in vitro to endothelial cells through successive maturation steps. Blood. 1996;88(9):3424–3431. [PubMed] [Google Scholar]
- Wang R, Clark R, Bautch VL. Embryonic stem cell-derived cystic embryoid bodies form vascular channels: an in vitro model of blood vessel development. Development. 1992;114(2):303–316. doi: 10.1242/dev.114.2.303. [DOI] [PubMed] [Google Scholar]
- Yang EY, Moses HL. Transforming growth factor beta 1-induced changes in cell migration, proliferation, and angiogenesis in the chicken chorioallantoic membrane. J Cell Biol. 1990;111(2):731–741. doi: 10.1083/jcb.111.2.731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Chang C, Gehling DJ, Hemmati-Brivanlou A, Derynck R. Regulation of Smad degradation and activity by Smurf2, an E3 ubiquitin ligase. Proc Natl Acad Sci U S A. 2001;98(3):974–979. doi: 10.1073/pnas.98.3.974. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure 1. In vitro angiogenesis. Confluent BCE cells grown in gelatin-coated dishes were incubated in the absence (upper panel) or presence of TGF-β1 (1 ng/ml; lower panel) in medium supplemented with 0.5% calf serum. The cultures were photographed after 72 h incubation. Shown are enlarged images of panel a and b of Fig. 2.