

Abstract
Background
The pathology causing progressive aphasia is typically a variant of frontotemporal lobar degeneration, especially with ubiquitin-positive-inclusions (FTLD-U). Less commonly the underlying pathology is Alzheimer disease (AD).
Objective
To compare clinicopathological and MRI features of subjects with progressive aphasia and AD pathology, to subjects with aphasia and FTLD-U pathology, and subjects with typical AD.
Methods
We identified 5 subjects with aphasia and AD pathology and 5 with aphasia and FTLD-U pathology with an MRI from a total of 216 aphasia subjects. Ten subjects with typical AD clinical features and AD pathology were also identified. All subjects with AD pathology underwent pathological re-analysis with TDP-43 immunohistochemistry. Voxel-based morphometry (VBM) was used to assess patterns of grey matter atrophy in the aphasia cases with AD pathology, aphasia cases with FTLD-U, and typical AD cases with AD pathology, compared to a normal control group.
Results
All aphasic subjects had fluent speech output. However, those with AD pathology had better processing speed than those with FTLD-U pathology. Immunohistochemistry with TDP-43 antibodies was negative. VBM revealed grey matter atrophy predominantly in the temporoparietal cortices with notable sparing of the hippocampus in the aphasia with AD subjects. In comparison, the aphasic subjects with FTLD-U showed sparing of the parietal lobe. Typical AD subjects showed temporoparietal and hippocampal atrophy.
Conclusions
A temporoparietal pattern of atrophy on MRI in patients with progressive fluent aphasia and relatively preserved processing speed is suggestive of underlying AD pathology rather than FTLD-U.
Keywords: Primary progressive aphasia, Progressive non-fluent aphasia, Logopenic progressive aphasia, frontotemporal lobar degeneration with ubiquitin-only-immunoreactive changes, Voxel based morphometry
INTRODUCTION
Speech and language impairments can be the most prominent presenting symptoms of a neurodegenerative disease. The term primary progressive aphasia (PPA)1 is one of the labels used to classify patients when there is a prominent and progressive impairment of language without initial dementia. The term PPA captures patients whose language difficulties can be characterized by agrammatic and non-fluent speech, prominent anomia, fluent aphasia with comprehension deficits, or a combination or blurring of distinctions among all three features. There are also other well-publicized classification schemes2–7.
In our recent clinicopathologic and imaging study of PPA and apraxia of speech2 we demonstrated that non-fluent aphasia with apraxia of speech was associated with atrophy of the premotor and posterior inferior frontal cortices while temporal lobe atrophy correlated with progressive aphasia with ‘fluent’ speech output. The majority of our subjects with fluent aphasia had a pathological diagnosis of frontotemporal lobar degeneration with ubiquitin-only-immunoreactive changes (FTLD-U).2 None of our cases had a pathological diagnosis of Alzheimer’s disease (AD)8 which may have been due to our strict inclusion criteria. Two recent reports, however, demonstrated that a significant number of cases of progressive aphasia had AD pathology at postmortem9, 10.
We therefore set out to analyze the clinicopathological features of our cases with a progressive aphasia and AD pathology, and compare these features plus the pattern of grey matter atrophy on MRI to subjects with a progressive aphasia and FTLD-U pathology, and to subjects with typical AD where memory loss, not aphasia, is the cardinal feature.
METHODS
Case ascertainment
The Mayo Clinic medical records database was used to identify all possible cases with prominent language impairment presenting between 1984 and 2006 by using a textword and diagnostic code search for aphasic dementia, aphasia, PPA, progressive non-fluent aphasia (PNFA), semantic dementia, or apraxia of speech. A total of 5222 subjects were identified. From these 5222 subjects the medical records database was used to identify the subset that had undergone a brain autopsy at Mayo Clinic. We identified a total of 216 subjects with prominent aphasia, not necessarily meeting criteria for a diagnosis of PPA, who had an autopsy examination. The historical records of all 216 subjects were reviewed by a behavioral neurologist (K.A.J). Of the 216 subjects, 193 were excluded because they had a structural lesion that accounted for the aphasia (e.g. left middle cerebral artery territory infarct or hemorrhage). The remaining 23 subjects with an autopsy examination had a progressive aphasia from a neurodegenerative disease. Seventeen of these 23 were previously published. Of the remaining six subjects, five had AD pathology and one subject FTLD-U pathology.
All five subjects with aphasia and AD pathology had a volumetric head MRI scan. Five subjects with aphasia and FTLD-U pathology also had a volumetric head MRI scan. In addition, 10 subjects who had been given an antemortem clinical diagnosis of Alzheimer’s type dementia, with the typical presenting feature of episodic memory loss that had pathologically confirmed AD, and had a volumetric head MRI scan were randomly selected from our Alzheimer’ Disease Research Center Brain Bank. Therefore a total of 20 subjects, five with aphasia and AD pathology, five with aphasia and FTLD-U pathology, and 10 with typical AD type presentation and AD pathology, were used in this study.
Detailed demographic and clinical information, including the Mini-Mental State Examination (MMSE)11 score and the Clinical Dementia Rating (CDR) sum of boxes12 was abstracted for all 20 subjects. Speech-language pathology records of the 10 subjects with aphasia were independently reviewed by two speech-language pathologists (JRD and EAS), blinded to any autopsy information, to abstract detailed information regarding the speech and language examinations and to further delineate the speech and language characteristics. The speech-language results for the subjects with FTLD-U pathology has been previously published2. The 10 subjects with typical AD did not have speech-language pathology records but did have quantitative speech and language tests completed as part of their neuropsychological test battery. All 10 subjects with typical AD had a formal dementia evaluation by a behavioral neurologist who did not observe any deficits in comprehension and spontaneous speech in the context of the mental status exam.
All 20 subjects were also age and gender matched to a healthy control subject. All control subjects were prospectively recruited into the Mayo Clinic Alzheimer’s Disease Research Center (ADRC), or the Alzheimer’s Disease Patient Registry (ADPR), and were identified from the ADRC/ADPR database. Controls were identified as individuals who a) were independently functioning community dwellers, b) did not have active neurologic or psychiatric conditions, c) had no cognitive complaints, d) had a normal neurological and neurocognitive examination, and e) were not taking any psychoactive medications in doses that would affect cognition.
Speech-Language
Data from speech-language examination of the five subjects with aphasia and AD pathology were tabulated and analyzed for this study. Language assessment typically included several subtests from the Minnesota Test for Differential Diagnosis of Aphasia13, Part V of the Token Test14, a letter word fluency task15, and a narrative picture description (Cookie Theft) from the Boston Diagnostic Aphasia Examination16. Quantitative data from these tests were used to estimate severity of aphasia - in auditory comprehension, naming, repetition, reading comprehension and writing using a 0–4 scale (0 = normal; 4 = severe impairment) in which mid-point values (e.g., 2–3) were permitted. Independent estimates of severity by two judges (JRD and EAS) were within 0.5 points for 100% of the ratings. Judgments about apraxia of speech and dysarthria were derived from conversation, verbal responses during formal language assessment, and structured tasks for assessing apraxia of speech and dysarthria17.
Neuropsychology
All data from neuropsychological testing conducted at presentation in all 20 subjects were tabulated and analyzed. Testing included executive function (Trails Making Test B18); language functioning including naming (Boston Naming test19), lexical fluency (Controlled Oral Word Association Test20), category/semantic fluency (animals, fruit, vegetables), sentence comprehension (Multilingual Aphasia Examination Token20); reading (Wide Range Achievement Test-321 or Woodcock-Johnson-Revised22); learning and memory (Wechsler Memory Scale-Revised23); and visuoperceptual functioning (Wechsler Adult Intelligence Scale Revised- Perceptual Organization Index24). The percentile levels were either derived from norms published with the test, or from Mayo Older American Normative Studies (MOANS) norms.25, 26
Pathological examination
All 20 subjects underwent standard neuropathological examination by one of our experienced neuropathologists (JEP or DWD) as previously described27. In addition the 15 subjects with AD pathology had brain tissue histologically analyzed with the recently described antibody TDP-4328 to determine whether or not there were any pathological features of FTLD-U, or FTLD with motor neuron disease, that may be masked by the AD pathology. Semiquantitative assessment of frontal, temporal, parietal, and occipital cortex, basal nucleus, hippocampal and substantia nigral neuronal loss was conducted in all 20 subjects using hematoxylin and eosin stain on a 4-point grading scale as follows: 0= no neuronal loss; 1= mild neuronal loss usual associated with microvacuolation (for the cortical sections); 2= moderate neuronal loss associated with thinning of the cortical ribbon (for the cortical sections); and 3= end-stage neuronal loss associated with severe thinning of the cortical ribbon producing so called status spongiosis (for the cortical sections). Additional pathological features including Braak Staging29 and NIA Reagan staging8 of all 20 cases were reviewed.
MRI
T1-weighted volumetric MRI scans were acquired at 1.5T (22×16.5cm FOV, 25° flip angle, 124 contiguous 1.6mm thick coronal slices). If a patient had more than one MRI we used the closest scan of adequate quality to the time of first neurological evaluation. Voxel- based morphometry (VBM) was used to compare the patterns of grey matter atrophy in the five subjects with aphasia and AD pathology, the five with aphasia and FTLD-U pathology and the 10 with typical AD to the control group. An optimized method of VBM was applied using both customized templates and prior probability maps (priors) 30, 31, implemented using SPM2 (http://www.fil.ion.ucl.ac.uk/spm). To create the template all scans, including those from the controls, the aphasia subjects with AD pathology, the aphasia subjects with FTLD-U pathology, and the typical AD subjects, were registered to the Montreal Neurological Institute (MNI) template using a 12dof affine transformation and segmented into grey matter (GM), white matter (WM) and CSF using MNI priors. GM images were normalized to the MNI GM prior using a nonlinear discrete cosine transformation (DCT). The normalization parameters were applied to the original whole head and the images were segmented using the MNI priors. Average images were created of whole head, GM, WM and CSF, and smoothed using 8mm full-width at half-maximum (FWHM) smoothing kernel. The average whole head image becomes the customized template, and the average GM, WM and CSF images are then used as the customized prior probability maps for subsequent segmentations. All images were then registered to the customized whole brain template using a 12dof affine transformation and segmented using the customized priors. The GM images were normalized to the custom GM prior using a nonlinear DCT. The normalization parameters were then applied to the original whole head and the images were segmented once again using the customized priors. All images were modulated and smoothed with a 8mm FWHM smoothing kernel.
Two-sided T-tests were used to assess the patterns of grey matter atrophy in each of the three disease groups compared to the control group. Grey matter differences were assessed after correction for multiple comparisons using the false discovery rate (p<0.01). Direct statistical comparisons were also performed between each of the three disease groups. These between-group comparisons of grey matter differences were assessed at a threshold of p<0.005 uncorrected for multiple comparisons due to the hypothesis driven nature of these statistical tests. Only those clusters exceeding a voxel size of 100 were reported.
Statistical methods
Statistical analyses were performed utilizing the JMP computer software (JMP Software, version 6.0.0; SAS Institute Inc, Cary, NC). Although multiple tests were performed, α was set at 0.05 due to the small sample sizes and limited statistical power. Gender ratios were compared across the groups with Chi-square test. Kruskal-Wallis tests were used to compare age at onset, age at death, age at scan, time from onset to scan, MMSE and CDR sum of boxes scores at the time of scan and neuropsychometric scores across the subject groups. Because the control group was by definition cognitively normal, we excluded controls from tests of differences in cognitive scores.
RESULTS
Demographic and clinic features
All clinical, speech and language, and neuropsychological data reported were completed at the time of initial presentation. Demographics of all three groups are summarized in Table 1. There was no difference in demographics across the three groups including time from initial evaluation to time of death. Presenting features of the five subjects with aphasia and AD pathology are shown in Table 2. All but one subject first presented for evaluation to a behavioral neurologist more than two years after onset. In all five subjects the most prominent complaint was language impairment. By the time of presentation, however, all five had complained of and exhibited more widespread cognitive impairment. The majority had mild evidence of executive dysfunction. Two subjects were found to have features of the Gerstmann syndrome as finger agnosia, left-right confusion, and acalculia were documented in two subjects; one had complained of difficulty with calculation very early into her disease course, although the language impairment was clearly the most prominent component of her illness. Although the diagnosis of PPA was considered, all five subjects were given a descriptive diagnosis of ‘aphasic dementia’, that is, aphasia with mild dementia, to highlight the prominence of the language component in the context of more widespread cognitive impairment. All five subjects with FTLD-U pathology presented with prominent aphasia while all 10 subjects with typical AD and AD pathology presented with loss of episodic memory. None of the FTLD-U subjects were thought to have more widespread cognitive impairment by the evaluating physician and hence all five were given a clinical diagnosis of PPA. In addition, in none of those with typical AD was aphasia a prominent feature at presentation
TABLE 1.
Subject demographics of the groups
| Aphasia-AD N=5 | Aphasia-FTLDU N=5 | Typical-AD N=10 | Controls N=20 | |
|---|---|---|---|---|
| No. females (%) | 2 (40%) | 2 (40%) | 6 (60%) | 9 (45%) |
| Mean (SD) age at onset, yrs | 69 (12) | 61 (9) | 69 (9) | NA |
| Mean (SD) age at death, yrs | 77 (13) | 70 (11) | 77 (9) | NA |
| Mean (SD) age at scan, yrs. | 72 (11) | 66 (12) | 73 (9) | 69 (8) |
| Mean (SD) time from onset-scan, yrs | 3 (1) | 5 (4) | 4 (2) | NA |
| Mean (SD) time from scan-death, yrs | 6 (2) | 4 (3) | 4 (1) | NA |
| * Mean (SD) MMSE score | 22 (5) | 20 (8) | 21 (6) | 29 (1) |
| * Mean (SD) CDR sum of boxes | 4 (3) | 5 (2) | 5 (5) | 0 (0) |
Aphasia-AD = subjects with progressive aphasia and AD pathology; Aphasia-FTLDU = subjects with progressive aphasia and FTLD-U pathology; Typical-AD = subjects with a clinical and pathological diagnosis of AD; MMSE = Mini-Mental Status Examination; CDR = Clinical Dementia Rating scale
Cognitive measures recorded at the time of scan
TABLE 2.
Clinical features of the 5 cases of AD presenting as progressive aphasia
| Subject | Sex | Age at onset | Total illness duration | Time from onset to initial neurological evaluation | Symptoms at onset of disease | Additional symptoms that developed by the time of presentation | Behavioral Neurologists bedside examination findings at presentation |
|---|---|---|---|---|---|---|---|
| 1 | F | 76 | 12 | 1-year | Difficulty getting her words out | Mild difficulty with calculations, attention and possible episodic memory | Moderate aphasia and acalculia |
| 2 | M | 77 | 11 | 2-years | Difficulty with naming especially names of people | Loss of insight | Prominent aphasia, difficulty with abstract reasoning |
| 3 | F | 76 | 7 | 4-years | Word finding difficulties | Personality change and possibly mild episodic memory loss | Prominent aphasia with mild finger agnosia, left- right confusion, and subtle evidence of limb apraxia |
| 4 | F | 57 | 7 | 2 ½ -years | Difficulty with finding and sequencing words. | Possibly mild episodic memory loss | Prominent aphasia and mild difficulty with attention, learning, and recall |
| 5 | M | 54 | 9 | 3-years | Difficulties expressing his thoughts and ideas, and putting words together | Personality change, apathy and decreased attention | Prominent aphasia and mild difficulty with attention. |
Language and speech
Language and speech findings of the five subjects with aphasia and AD pathology are summarized in Table 3. In addition we provide raw scores as supplemental data (E Table 1). All subjects had language characteristics consistent with a diagnosis of aphasia and all but subject 2 (whose verbal comprehension was normal) had deficits in all tested language modalities. All had fluent narrative and conversational speech (i.e., no evidence of telegraphic/agrammatic speech or writing), with varying combinations and degrees of circumlocution, semantic or phonemic paraphasias, and a lack of specificity or paucity of specific content words. Some had pauses, hesitancy, or delayed initiation of verbal responses (Subjects 1, 4, 5), or infrequent paragrammatic errors (Subjects 2, 3). Although the aphasia was always predominant, all five subjects had subtle-to-obvious behaviors, or a profile of difficulty, that raised concerns about problems beyond the language domain (see Table 2 for description). No subject had dysarthria or apraxia of speech.
TABLE 3.
Speech-language characteristics based on formal speech-language examination at presentation (see text for description).
| Subject | Verbal comprehension** | Naming | Fluency | Repetition | Reading comprehension | Writing | Nonaphasic behavior during language testing | Dysarthria | Apraxia of speech |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 1 | 1 | Fluent (pauses for word retrieval; mild lack of content words) | 2 | 2 | 1, 2 | Concreteness; overpersonalization; disengaged from listener | None | None |
| 2 | Normal | 3 | Fluent (circumlocution; lack of content words; occasional paragrammatic errors) | 1 | 0, 1 | 0, 1 | More difficulty with abstract than stimulus-response tasks | None | None |
| 3 | 2 | 3, 4 | Fluent (frequent semantic & occasional phonemic paraphasias; occasional paragrammatic errors) | NR | 1, 2 | 2 | Tangentiality; difficulty grasping or forgetting simple task requirements | None | None |
| 4* | 1, 2 | 2, 3 | Fluent (hesitant for word retrieval efforts; frequent phonemic & occasional semantic errors) | 3, 4 | 1 | 2 | Verbal retention span and awareness of errors poor relative to aphasia severity | None | None |
| 5* | 2,3 | 3,4 | Fluent (delayed initiation & hesitancy; semantic errors) | 3 | 3 | 2 | Verbal retention span poor relative to aphasia severity | None | None |
NR = not reported
Ratings: 0 = normal; 1 = mild; 2 = moderate; 3 = marked; 4 = severe
= These subjects’ conversational speech (Subject 5 more likely than Subject 4) might meet some investigators’ definition of logopenia4.
Ratings based on 1–3 sentence level measures requiring grammatical and syntactic processing.
Neuropsychology
The neuropsychological findings are summarized in Table 4. Test scores were not statistically different across the three groups with the exception of the test for Visual Reproduction Memory23 which was worse in the typical AD group compared to the FTLD-U group (p<0.05). It was observed however that subjects with aphasia and AD pathology had very similar scores across the different cognitive domains tested when compared to the subjects with aphasia and FTLD-U pathology. The one exception was that the subjects with aphasia and FTLD-U pathology showed a trend to perform worst than those with aphasia and AD pathology on the Controlled Oral Word Association Test20 (p=0.07). The pattern of language impairment for the subjects with aphasia and AD pathology was similar to the pattern of language impairment for the typical AD subjects, yet the typical AD subjects had more severe memory impairment with a trend for a lower 30-minute Delay and recognition memory scores on the Auditory Verbal Learning Test23 (p=0.09 for both). Overall the two groups of subjects with aphasia performed worse on the Boston Naming Test19 compared to the subjects with typical AD (p=0.08), however the subjects with typical AD performed worst on all tests of memory compared to the two groups of subjects with aphasia. There was also a trend for subjects with typical AD to have a worst perceptual organization index compared to subjects with aphasia and AD (p=0.07) and subjects with FTLD-U (p=0.05).
TABLE 4.
Neuropsychometric performances at presentation
| Aphasia-AD | Aphasia-FTLDU | Typical-AD | P value | |
|---|---|---|---|---|
| Time from onset to testing (years) | 2.5 [1–4] | 2 [1–4] | 2 [1–7] | ns |
| Age at testing in years | 77 [57–80] | 62 [55–76] | 75 [52–82] | ns |
| Years of education | 16 [15–18] | 14 [12–16] | 14 [8–18] | ns |
| Executive Functioning (Mean =10, SD =3) | ||||
| Trails Making Test B | 2.5* [2–3] | 2* [2–4] | 2*[2–14] | ns |
| Language Functioning (Mean = 10, SD =3) | ||||
| Boston Naming Test | 4* [2–6] | 2* [1–10] | 5* [2–11] | ns |
| Controlled Oral Word Association Test | 9 [4–11] | 2* [2–8] | 8 [2–12] | 0.09 |
| Category/Semantic Fluency | 4*[2–6] | 2*[1–7] | 3*[2–7] | ns |
| Multilingual Aphasia Examination Token 5*[3–7] | 5*[1–12] | 5*[3–13] | ns | |
| Reading Mean =100, SD =15 | ||||
| Wide Range Achievement Test-3 or Woodcock-Johnson-Revised | α110 [95–112] or β90 [85–95] | α99 [95–100] or β91[91–91] | α89 [86–110] | ns |
| Learning and Memory (Mean = 10, SD =3) | ||||
| WMS-R Logical Memory I Immediate Recall | 4* [1–8] | 4* [2–9] | 2.5* [2–5] | ns |
| WMS-R Logical Memory II Delayed Recall | 5* [2–11] | 4.5* [2–8] | 4* [2–7] | ns |
| % retention | 60 [40–100] | 66.5 [29–100] | 45 [0–100] | ns |
| ζ WMS-R Visual Reproduction Immediate Memory | 7 [3–9] | 10.5[8–11] | 5* [2–10] | 0.03 |
| WMS-R Visual Reproduction II Delayed Memory | 6* [2–9] | 7 [6–8] | 4* [2–9] | ns |
| % retention | 56 [0–93] | 50 [35–68] | 19 [0–61] | ns |
| Auditory Verbal Learning Test Trial 1 | 5* [2–7] | 6*[2–7] | 5* [2–7] | ns |
| Auditory Verbal Learning Test 30-minute Delay | 6* [5–11] | 5.5* [2–7] | 5* [2–7] | ns |
| % retention | 44 [0–100] | 50[0–100] | 0 [0–100] | ns |
| Recognition | 6* [5–13] | 5.5* [3–8] | 3* [2–8] | ns |
| Visuoperceptual Functioning (Mean =100, SD =15) | ||||
| WAIS-R Perceptual Organization Index | 106 [88–114] | 95 [89–118] | 89 [72–95] | 0.08 |
Note: Data shown in cells are median scores and range. WAIS-R = Wechsler Adult Intelligence Scale-Revised; WMS-R = Wechsler Memory Scale-Revised. Standard scores less than 85 and scaled scores less than 7 are considered impaired, with lower scores reflecting greater impairment. An asterisk (*) next to scores signals impaired performance. α = Reading test completed was the Wide Range Achievement Test-3; β = Reading test completed was the Woodcock-Johnson-Revised
Significantly different between the Aphasia-FTLDU group and the typical AD group (p<0.5 using Mann-Whitney U test)
Aphasia-AD = subjects with progressive aphasia and AD pathology; Aphasia-FTLDU = subjects with progressive aphasia and FTLD-U pathology; Typical-AD = subjects with a clinical and pathological diagnosis of AD
Pathological findings
Grossly all 20 subjects had mild-moderate generalized cerebral atrophy. In two subjects with aphasia and AD pathology and all 10 subjects with typical AD and AD pathology there was moderate-severe medial temporal lobe atrophy. In all 15 subjects with AD pathology there were widespread neocortical neurofibrillary tangles of Braak and Braak stage VI29. There were also moderate-frequent neuritic and diffuse neocortical plaques in all 15 subjects with AD pathology, therefore, all 15 met NIA Reagan criteria for high probability AD8. The distribution of Alzheimer’s pathology was atypical in only one subject with aphasia and AD pathology with relative sparing of the hippocampus proper which showed only mild neuronal loss, although there was a high density of neurofibrillary tangles. In two subjects with aphasia and AD pathology there was gliosis in the globus pallidum. Two subjects with AD pathology, one with aphasia and one typical AD had brainstem and limbic Lewy bodies consistent with transitional or limbic Lewy body disease. Immunohistochemistry with TDP-43 antibodies28 was negative in all 15 subjects with AD pathology. The five subjects with FTLD-U were Braak Stage ≤ II29 and NIA -Reagan low probability of AD8. Semiquantitative analysis of regional neuronal loss across all three groups is shown in Table 5.
TABLE 5.
Semiquantitation of neuronal loss
| Subject | Frontal | Temporal | Parietal | Occipital | Basal Nucleus | Hippocampus | Substantia Nigra |
|---|---|---|---|---|---|---|---|
| Aphasia-AD | 1.5(1–2) | 2 (2–2) | 1.5(1–3) | 1(0–2) | 2(1–2) | 1(0.5–2) | 1.5(1–2) |
| Aphasia-FTLDU | 1.5 (2–3) | 2 (2–3) | 1 (1–2) | 1(0–1) | 1(1–2) | 2(2–3) | 1 (1–2) |
| Typical AD | 1 (1–2) | 2 (2–3) | 2 (2–3) | 1 (1–2) | 2(1–3) | 2(1–3) | 1 (1–2) |
0= no neuronal loss; 1= mild neuronal loss usual associated with microvacuolation (for the cortical sections); 2= moderate neuronal loss associated with thinning of the cortical ribbon (for the cortical sections); and 3= end-stage neuronal loss associated with severe thinning of the cortical ribbon producing so called status spongiosis (for the cortical sections).
Data reported as median and range
MRI
The group of subjects with aphasia and AD pathology showed a predominantly left-sided pattern of temporoparietal grey matter loss compared to controls (Figure 1A). The grey matter loss in the temporal lobes included the left posterior inferior, middle and superior temporal gyri with remarkable sparing of the medial and anterior temporal pole. A small amount of grey matter loss was also identified in the frontal lobes. The right hemisphere showed very little involvement with areas of loss only identified in the posterior temporal lobe and parietal lobe.
Figure 1.
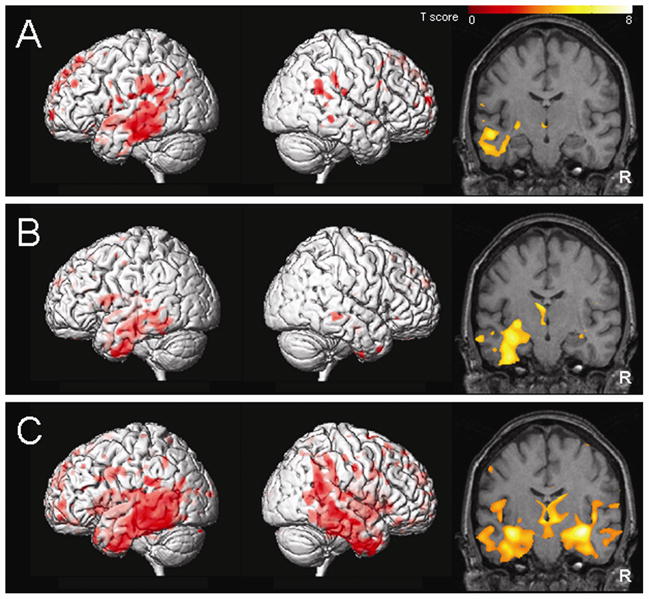
The patterns of grey matter loss identified by voxel-based morphometry in the aphasic subjects with AD pathology (A), the aphasic subjects with FTLD-U pathology (B), and the subjects with clinical and pathological AD (C), compared to controls (corrected for multiple comparisons, p<0.01). The results are shown both on a 3D surface render to illustrate the patterns of cortical grey matter loss, and on a representative coronal slice (y=−15) to illustrate the involvement of the hippocampus. R = right.
The group of subjects with aphasia and FTLD-U pathology also showed a left-sided pattern of atrophy predominantly involving the temporal lobe, including the amygdala, hippocampus, inferior and middle temporal gyri and fusiform gyrus, compared to controls (Figure 1B). The regions of loss extended back into the posterior temporal lobe but did not involve the parietal lobes. Grey matter loss was also identified in the frontal lobes and anterior insula.
In contrast to both aphasic groups the pattern of grey matter loss was bilateral in the subjects with clinical and pathological AD, compared to controls (Figure 1C). Grey matter loss particularly affected the medial temporal lobes and the temporoparietal association neocortex, although the posterior cingulate, frontal lobes, and posterior insula were also involved.
Direct statistical comparisons were also performed across the disease groups. The group of subjects with typical AD showed greater involvement of the right anterior hippocampus and amygdala than the aphasia subjects with AD pathology, and greater involvement of the posterior cingulate and parietal lobe than the subjects with FTLD-U. The aphasia subjects with AD pathology showed greater grey matter loss in regions of the left lateral frontal and parietal lobes compared to FTLD-U, and in the left lateral frontal and temporal lobes compared to AD. The FTLD-U group showed greater grey matter loss in the medial frontal lobes and anterior insula than both the aphasia with AD pathology group and the typical AD group, and greater anterior temporal lobe atrophy than the aphasia with AD pathology group. The coordinates of these regions are shown in Table 6.
TABLE 6.
Regions of grey matter loss identified in the three subject groups when compared directly to each of the other two subject groups
| Subject Group | Comparison group | Region | Coordinates | Z score | ||
|---|---|---|---|---|---|---|
| x | y | z | ||||
| Aphasia-AD | Aphasia-FTLDU | L lateral frontal lobe R parietal lobe |
−45 32 |
44 −77 |
27 8 |
3.79 3.18 |
| AD-AD | L lateral frontal lobe L posterior temporal lobe |
−44 −51 |
43 −22 |
28 −12 |
3.79 3.15 |
|
| Aphasia-FTLDU | Aphasia-AD | Medial frontal lobe L anterior insula L amygdala |
−2 −27 −21 |
15 29 7 |
47 3 −34 |
3.96 3.47 3.06 |
| AD-AD | L anterior insula Medial frontal lobe |
−26 −3 |
26 42 |
5 24 |
3.46 3.25 |
|
| Typical AD | Aphasia-AD | R hippocampus/amygdala | 30 | −9 | −19 | 4.23 |
| Aphasia-FTLDU | R posterior cingulate R parietal lobe |
9 44 |
−63 −51 |
5 8 |
3.65 3.32 |
|
This table demonstrates the regions of the brain that showed greater grey matter loss in the subject group than in the comparison group. Voxel coordinates are in millimeters after transformation into standard Montreal Neurologic Institute stereotactic space.
Aphasia-AD = subjects with progressive aphasia and AD pathology; Aphasia-FTLDU = subjects with progressive aphasia and FTLD-U pathology; Typical-AD = subjects with a clinical and pathological diagnosis of AD; L = left; R = right
DISCUSSION
In this study we demonstrate that when progressive aphasia is secondary to AD pathology the pattern of grey matter loss early in the disease course appear to be different from that of typical AD and also different from that of progressive aphasia with FTLD-U pathology. We also show that there is no secondary FTLD-U pathology.
The medial temporal lobes were relatively spared on MRI at the time of presentation in patients who present with progressive aphasia and AD pathology32. This should not be surprising since episodic memory loss was not the most dominant feature of their illness at presentation. In contrast, our subjects with typical presentation of episodic memory loss and AD pathology showed severe involvement of the medial temporal lobe. In addition, we also found that the posterior cingulate region was heavily involved in our typical AD subjects as has been previously reported33, 34, yet was relatively spared in our five aphasic subjects with AD pathology. Frontal lobe atrophy was observed in both groups although the direct statistical comparison suggested that the frontal lobe loss was slightly more severe in the subjects with aphasia and AD pathology. Frontal lobe dysfunction was documented on presentation and confirmed with neuropsychometric testing in the aphasic group.
The pathology that most frequently underlies progressive fluent aphasia has been shown to be FTLD-U2, 10, 35 or dementia lacking distinctive histology36. Our progressive aphasia subjects with AD pathology also had fluent speech output. Therefore, when a patient presents with a fluent aphasia (i.e. without apraxia of speech or agrammatism, or loss of syntax), the differential diagnosis should be first FTLD-U pathology and secondly AD. Frontotemporal lobar degeneration with ubiquitin-only immunoreactive changes has replaced dementia lacking distinctive histology with the advent of ubiquitin and TDP-43 immunohistochemistry37, 38. We have shown that the pattern of atrophy early in the disease course is different when FTLD-U and AD pathology underlie progressive aphasia. In our subjects with FTLD-U pathology there was left predominantly anterior temporal lobe atrophy with sparing of the parietal lobe. In contrast, in our progressive aphasic subjects with AD pathology the temporoparietal association neocortex was heavily involved with less involvement of the anterior temporal lobes. The temporoparietal atrophy in the aphasic subjects with AD pathology correlated with the fact that some of our subjects had features of Gerstmann syndrome which is typically associated with parietal lobe injury. Some medial frontal lobe atrophy was also observed in the aphasic subjects with FTLD-U. However, the frontal loss in more lateral regions was less severe than in the other two patient groups. This is interesting since the Controlled Word Association Test is a test of processing speed and the FTLD-U group was the only group of the three that performed poorly on the Controlled Word Association Test20. This suggests that the Controlled Word Association Test may be helpful in differentiating FTLD-U from AD as the cause of progressive fluent aphasia.
The anterior insula was also found to be involved in the subjects with fluent aphasia and FTLD-U pathology. The anterior insula has previously been implicated in the non-fluent variant of FTLD4, 39 and particularly in apraxia of speech40. However, other studies have found no evidence for an association between insula atrophy and apraxia of speech2 and have also found atrophy of the insula in fluent aphasia cases41. It is possible that apparent insula atrophy may simply reflect widening of the perisylvian fissure due to atrophy in the frontal lobe, since anterior insula atrophy has also been observed in non-aphasic cases of FTLD42. The anatomical complexity of this region makes it difficult for VBM to accurately localize change.
All five subjects with progressive aphasia and AD had evidence of anomia, comprehension deficits, and fluent speech in ordinary conversation and on formal speech and language evaluation. None of our subjects had nonfluent speech, apraxia of speech, or agrammatism, as two studies reported10, 43 The clinical presentation in the five subjects with aphasia and AD pathology were felt to be somewhat atypical for PPA by the evaluating physicians, although PPA was still in the differential, since in all five subjects aphasia was not an isolated feature at the time of presentation. Neuropsychological examination also demonstrated more widespread cognitive impairment. Similar findings of more widespread cognitive impairment in patients that present with progressive fluent aphasia and is found to have AD pathology have been previously reported43.
Language impairment including confrontation naming 42 and sentence comprehension 44 has been demonstrated in typical AD subjects. Our subjects with typical AD had formal testing of language and indeed performance in confrontation naming, semantic fluency, and sentence comprehension were below average similar to our progressive aphasia with AD subjects. Yet, all of our typical AD subjects had a formal dementia evaluation by a behavioral neurologist who did not appreciate any obvious deficits in conversational speech with respect to prosody, melody, articulation, grammatical form and qualitatively in the rate of word production while our progressive aphasia with AD pathology subjects had obvious speech and language deficits on interview with a behavioral neurologist. This suggests one of two possibilities. First, our neuropsychological tests of language are not able to differentiate between primary language deficits and language deficits that may be occurring as a result of memory loss. Second, neuropsychological testing is more sensitive to language impairment in AD subjects than bedside cognitive or mental status testing. Hence all subjects with AD regardless of presenting features have language impairment as suggested42, 44, but on routine evaluation the language impairment is being overshadowed by the memory loss. Therefore, it may not be the language deficits that make our aphasia with AD group standout, but rather it is the absence of prominent episodic memory loss and visual perceptual deficits.
One of the other interesting features of this study that requires more analysis was the similarity in the temporoparietal pattern of atrophy found in our progressive aphasia and typical AD pathology group, and the temporoparietal atrophy reported in logopenic PPA4. These similarities support the suggestion that AD pathology may underlie logopenic PPA4. This suggestion is also further strengthened by the fact that a possible one or two of our five subjects with AD pathology may meet criteria for logopenic PPA.4 Although in our aphasia group with AD pathology we did not find any hippocampal volume loss as was reported with logopenic PPA4, this difference could be explained by the fact that the time from disease onset to scan was longer in their study which suggests that their subjects were further along in their disease course.
The histopathological findings in the AD cases presenting with aphasia were more widespread than the imaging findings. This suggests that the pathological process spread beyond language-related regions and did not remain atypical with hippocampal sparing throughout the entire disease course. In addition, TDP-43 immunohistochemistry was negative demonstrating the aphasia in our subjects with AD pathology was not due to coexisting FTLD-U pathology28. It was important to perform TDP-43 analysis to rule out underlying FTLD-U pathology since ubiquitin immunohistochemistry is not specific and also highlights AD-type lesions.
Supplementary Material
Acknowledgments
This study was supported by the NIH Roadmap Multidisciplinary Clinical Research Career Development Award Grant (K12/NICHD)-HD49078, by grants P50 AG16574, U01 AG06786 and R01 AG11378 from the National Institute on Aging, Bethesda MD and the generous support of the Robert H. and Clarice Smith and Abigail Van Buren Alzheimer’s Disease Research Program of the Mayo Foundation, U.S.A.
DSK has been a consultant to GE HealthCare, GlaxoSmithKline and Myriad Pharmaceuticals, has served on a Data Safety Monitoring Board for Sanofi-Aventis and Neurochem Pharmaceuticals, and is an investigator in a clinical trial sponsored by Elan Pharmaceuticals. RCP has been a consultant to GE Healthcare and on the data safety monitoring board in a clinical trial sponsored by Elan Pharmaceuticals. BFB and NGR receive grant support as investigators in a clinical trial sponsored by Myriad Pharmaceuticals.
Footnotes
Disclosures: DSK has been a consultant to GE HealthCare, GlaxoSmithKline and Myriad Pharmaceuticals, has served on a Data Safety Monitoring Board for Sanofi-Aventis and Neurochem Pharmaceuticals, and is an investigator in a clinical trial sponsored by Elan Pharmaceuticals. RCP has been a consultant to GE Healthcare and has served on a data safety monitoring board in a clinical trial sponsored by Elan Pharmaceuticals. BFB and NGR receive grant support as investigators in a clinical trial sponsored by Myriad Pharmaceuticals
References
- 1.Mesulam MM. Slowly progressive aphasia without generalized dementia. Ann Neurol. 1982;11:592–598. doi: 10.1002/ana.410110607. [DOI] [PubMed] [Google Scholar]
- 2.Josephs KA, Duffy JR, Strand EA, et al. Clinicopathological and imaging correlates of progressive aphasia and apraxia of speech. Brain. 2006;129:1385–1398. doi: 10.1093/brain/awl078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Neary D, Snowden JS, Gustafson L, et al. Frontotemporal lobar degeneration: a consensus on clinical diagnostic criteria. Neurology. 1998;51:1546–1554. doi: 10.1212/wnl.51.6.1546. [DOI] [PubMed] [Google Scholar]
- 4.Gorno-Tempini ML, Dronkers NF, Rankin KP, et al. Cognition and anatomy in three variants of primary progressive aphasia. Ann Neurol. 2004;55:335–346. doi: 10.1002/ana.10825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kertesz A, Davidson W, McCabe P, et al. Primary progressive aphasia: diagnosis, varieties, evolution. J Int Neuropsychol Soc. 2003;9:710–719. doi: 10.1017/S1355617703950041. [DOI] [PubMed] [Google Scholar]
- 6.Snowden JS, Neary D, Mann DM, et al. Progressive language disorder due to lobar atrophy. Ann Neurol. 1992;31:174–183. doi: 10.1002/ana.410310208. [DOI] [PubMed] [Google Scholar]
- 7.Grossman M, Ash S. Primary progressive aphasia: a review. Neurocase. 2004;10:3–18. doi: 10.1080/13554790490960440. [DOI] [PubMed] [Google Scholar]
- 8.Working Group. Consensus recommendation for the postmortem diagnosis of Alzheimer’s disease. The National Institute on Aging, and Reagan Institute Working Group on Diagnostic Criteria for Neuropathologic Assessment of Alzheimer’s Disease. Neurobiol Aging. 1997;18 (Suppl):S1–S2. [PubMed] [Google Scholar]
- 9.Kertesz A, McMonagle P, Blair M, et al. The evolution and pathology of frontotemporal dementia. Brain. 2005;128:1996–2005. doi: 10.1093/brain/awh598. [DOI] [PubMed] [Google Scholar]
- 10.Knibb JA, Xuereb JH, Patterson K, Hodges JR. Clinical and pathological characterization of progressive aphasia. Ann Neurol. 2006;59:156–165. doi: 10.1002/ana.20700. [DOI] [PubMed] [Google Scholar]
- 11.Folstein MF, Folstein SE, McHugh PR. “Mini-mental state”. A practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res. 1975;12:189–198. doi: 10.1016/0022-3956(75)90026-6. [DOI] [PubMed] [Google Scholar]
- 12.Hughes C, Berg L, Danziger W, et al. A new clinical scale for the staging of dementia. Br J Psychiatry. 1982;140:566–572. doi: 10.1192/bjp.140.6.566. [DOI] [PubMed] [Google Scholar]
- 13.Schuell H. The minnesota test for differential diagnosis of aphasia. Minnesota: University of Minnesota Press; 1972. [DOI] [PubMed] [Google Scholar]
- 14.DeRenzi E, Vignolo L. The token test: A sensitive test to detect receptive disturbances in aphasics. Brain. 1962;85:665–678. doi: 10.1093/brain/85.4.665. [DOI] [PubMed] [Google Scholar]
- 15.Wertz R, Keith R, Custer D. Normal and aphasic behaviour on a measure of auditory input and a measure of verbal output. Annual Convention of the American Speech and Hearing Association; Chicago, IL. 1971. [Google Scholar]
- 16.Goodglass H, Kaplan E, Barresi B. The Boston Diagnostic Aphasia Examination. 3. Philadelphia: Lippincott Williams & Wilkins; 2001. [Google Scholar]
- 17.Duffy JR. Motor speech disorders; Substrates, differential diagnosis, and management. San Louis: Mosby; 2005. [Google Scholar]
- 18.Reitan R. Validity of the Trail-Making Test as an indication of organic brain damage. Percept Mot Skills. 1958;8:271–276. [Google Scholar]
- 19.Kaplan E, Goodglass H, Weintraub S. The Boston Naming Test. Boston: Veterans Administration Medical Center; 1978. [Google Scholar]
- 20.Benton A, Hamsher K. Multilingual Aphasia Examination (Manual) Iowa City: University of Iowa; 1978. [Google Scholar]
- 21.Wilkinson G. Wrat-3: The wide range achivement test administration manual. Wilmington: Wide Range; 1993. [Google Scholar]
- 22.Woodcock R, Johnson M. Woodcook-Johnson psyco-Educational-Battery-Revised. Itasca: Riverside; 1989. [Google Scholar]
- 23.Wechsler D. Wechsler Memory Scale-Revised (Manual) New York: Psychological Corporation; 1987. [Google Scholar]
- 24.Wechsler D. Wechsler Adult Intelligence Scale Revised (Manual) San Antonia: The Psychological Corporation; 1981. [Google Scholar]
- 25.Ivnik RJ, Malec J, Smith GE, et al. Neuropsychological tests’ norms above age 55. COWAT, BNT, MAE token, WRAT-R reading, AMNART, STROOP, TMT and JLO. The Clinical Neuropsychologist. 1996;10:262–278. [Google Scholar]
- 26.Ivnik RJ, Malec J, Smith GE, et al. Mayo’s Older American NOrmative Studies. WAIS-R, WMS-R, and AVLT norms for ages 56–97. The Clinical Neuropsychologist. 1992;6 (Supplement):1–104. [Google Scholar]
- 27.Knopman DS, Boeve BF, Parisi JE, et al. Antemortem diagnosis of frontotemporal lobar degeneration. Ann Neurol. 2005;57:480–488. doi: 10.1002/ana.20425. [DOI] [PubMed] [Google Scholar]
- 28.Neumann M, Sampathu DM, Kwong LK, et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science. 2006;314:130–133. doi: 10.1126/science.1134108. [DOI] [PubMed] [Google Scholar]
- 29.Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol (Berl) 1991;82:239–259. doi: 10.1007/BF00308809. [DOI] [PubMed] [Google Scholar]
- 30.Ashburner J, Friston KJ. Voxel-based morphometry--the methods. Neuroimage. 2000;11:805–821. doi: 10.1006/nimg.2000.0582. [DOI] [PubMed] [Google Scholar]
- 31.Senjem ML, Gunter JL, Shiung MM, et al. Comparison of different methodological implementations of voxel-based morphometry in neurodegenerative disease. Neuroimage. 2005;26:600–608. doi: 10.1016/j.neuroimage.2005.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mesulam M. Primary progressive aphasia pathology. Ann Neurol. 2006 doi: 10.1002/ana.20940. [DOI] [PubMed] [Google Scholar]
- 33.Baron JC, Chetelat G, Desgranges B, et al. In vivo mapping of gray matter loss with voxel-based morphometry in mild Alzheimer’s disease. Neuroimage. 2001;14:298–309. doi: 10.1006/nimg.2001.0848. [DOI] [PubMed] [Google Scholar]
- 34.Boxer AL, Rankin KP, Miller BL, et al. Cinguloparietal atrophy distinguishes Alzheimer disease from semantic dementia. Archives of Neurology. 2003;60:949–956. doi: 10.1001/archneur.60.7.949. [DOI] [PubMed] [Google Scholar]
- 35.Davies RR, Hodges JR, Kril JJ, et al. The pathological basis of semantic dementia. Brain. 2005;128:1984–1995. doi: 10.1093/brain/awh582. [DOI] [PubMed] [Google Scholar]
- 36.Hodges JR, Davies RR, Xuereb JH, et al. Clinicopathological correlates in frontotemporal dementia. Ann Neurol. 2004;56:399–406. doi: 10.1002/ana.20203. [DOI] [PubMed] [Google Scholar]
- 37.Lipton AM, White CL, 3rd, Bigio EH. Frontotemporal lobar degeneration with motor neuron disease-type inclusions predominates in 76 cases of frontotemporal degeneration. Acta Neuropathol (Berl) 2004;108:379–385. doi: 10.1007/s00401-004-0900-9. [DOI] [PubMed] [Google Scholar]
- 38.Josephs KA, Holton JL, Rossor MN, et al. Frontotemporal lobar degeneration and ubiquitin immunohistochemistry. Neuropathol Appl Neurobiol. 2004;30:369–373. doi: 10.1111/j.1365-2990.2003.00545.x. [DOI] [PubMed] [Google Scholar]
- 39.Nestor PJ, Graham NL, Fryer TD, et al. Progressive non-fluent aphasia is associated with hypometabolism centred on the left anterior insula. Brain. 2003;126:2406–2418. doi: 10.1093/brain/awg240. [DOI] [PubMed] [Google Scholar]
- 40.Dronkers NF. A new brain region for coordinating speech articulation. Nature. 1996;384:159–161. doi: 10.1038/384159a0. [DOI] [PubMed] [Google Scholar]
- 41.Mummery CJ, Patterson K, Price CJ, et al. A voxel-based morphometry study of semantic dementia: relationship between temporal lobe atrophy and semantic memory. Ann Neurol. 2000;47:36–45. [PubMed] [Google Scholar]
- 42.Grossman M, McMillan C, Moore P, et al. What’s in a name: voxel-based morphometric analyses of MRI and naming difficulty in Alzheimer’s disease, frontotemporal dementia and corticobasal degeneration. Brain. 2004;127:628–649. doi: 10.1093/brain/awh075. [DOI] [PubMed] [Google Scholar]
- 43.Galton CJ, Patterson K, Xuereb JH, Hodges JR. Atypical and typical presentations of Alzheimer’s disease: a clinical, neuropsychological, neuroimaging and pathological study of 13 cases. Brain. 2000;123(Pt 3):484–498. doi: 10.1093/brain/123.3.484. [DOI] [PubMed] [Google Scholar]
- 44.Grossman M, White-Devine T. Sentence comprehension in Alzheimer’s disease. Brain Lang. 1998;62:186–201. doi: 10.1006/brln.1997.1898. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.