

Abstract
We numerically study the length-dependence in the bending stiffness of α-helices, coiled-coils, and a linear chain model. As the length approaches what we named the critical buckling length lc, the chain appears to become increasingly more flexible. This is due to weak non-bonded attractions that eventually lead to buckling instability and alter the chain's conformational ensemble. For α-helices and coiled-coils, lc is well less than half of their respective persistence lengths, so lc is the defining length scale for their conformations. These results elucidate the importance of weak nonspecific attractions that are inherent in many biofilaments in physiological conditions.
Persistence length lp has been considered as a fundamental length scale that determines the conformation of a uniform chain of length L. Depending on the ratio between L and lp, the chain conformation can be classified into three regimes; rod-like (L ⪡ lp), semiflexible (L ∼ lp), and flexible (L ⪢ lp). lp is determined by the bending stiffness κ of the chain and the temperature T : lp = κ/kBT (kB: Boltzmann constant) [1]. It is typically assumed that κ under a given solvent condition is constant, so is lp. For many biofilaments, lp has been experimentally measured, including DNA (53 nm) [2], intermediate filaments (1 μm) [3], F-actin (17 μm) and microtubule (5.2 mm) [4]. Atomistic simulations have also been used to calculate lp, e.g., α-helix (AH; 80−100 nm) [5], coiled-coil (CC; 150−300 nm) [6], and amyloid-like β-sheet filaments (1.2−4.8 μm) [7]. However, lp depends on L in certain cases, e.g., due to shearing or sliding among protofilaments in microtubules or in intermediate filaments [3, 8].
Here we show that length dependence of lp may be a general feature of biofilaments due to weak nonspecific attractions that are not screened in physiological conditions. In particular, attraction introduces a temperature-independent length scale, the critical buckling length lc, beyond which the chain fails to remain straight. When lc < lp, lc rather than lp governs the filament conformation, which we find to be valid for AHs and CCs.
In the rod regime, normal mode analysis (NMA) can be used to obtain κ. It has been applied to CCs [6], β-sheet filaments [7], and DNA [9]. Yet, the length dependence of κ has not been previously addressed. NMA of an elastic rod yields vibrational bending frequencies ωn (n = 1,2,3· · ·) that satisfy [10]
| (1) |
where ρl: linear mass density and the wave number kn = cn/L, where the constant cn depends on the boundary condition. When both ends of the rod move freely, cn is the solution of cos cn cosh cn = 1 [7]. For a perfectly linear elastic rod, κ is independent of n and L. However, for atomistic biofilaments, κ depends on both, and decreases for large L.
To study the length dependence of κ, we performed NMA on straight AHs of various lengths and sequences. We also tested straight leucine-zipper CCs (LZ) with ideal heptad repeats. A straight conformation is required for Eq. (1) to be valid [10]. AHs were constructed by setting the backbone dihedral angles to standard values. We built the backbone of straight LZ using the code by Offer [11] and side chains were placed based on the Protein Data Bank (PDB) structure 2ZTA to ensure a good knob-into-hole packing [12]. The filaments were then energy minimized using CHARMM [13]. A few different minimization schemes did not lead to any noticeable difference in the result.
We used the NMA facility in CHARMM to calculate ωn. There are twist and stretch modes as well, but we consider only bending modes, which are the most relevant to the chain conformation. κ(L) determined from Eq. (1) has a bell-shaped profile (Fig. 1). The initial rise of κ for short L is due to the low aspect ratio of the system that renders the continuum rod picture less accurate [9]. For shorter L there is also stronger edge effect whereby fewer atoms interact, contributing to making κ smaller. After the initial rise, κ is relatively constant or peaks, and then drops for longer L. Beyond the length where κ vanishes, ωn becomes negative, suggesting that the straight rod conformation is unstable for that mode. This occurs first for n = 1, subsequently at higher L for n = 2 and so on.
FIG. 1.
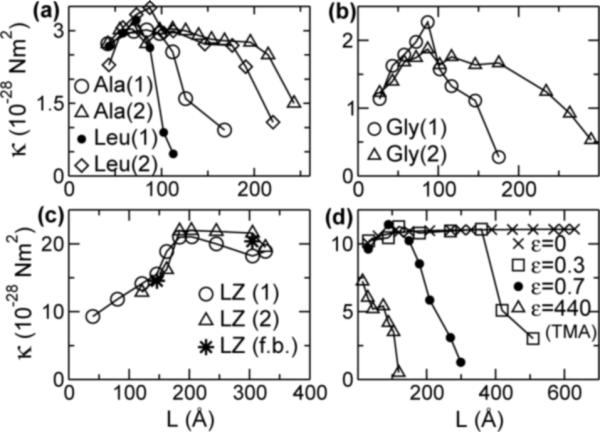
κ vs. L. (a) Poly-Ala, poly-Leu, and (b) poly-Gly AHs, (c) LZ, and (d) 1D chain (ε in units of kcal/mol). All are from NMA (n = 1, 2, in parentheses; and n = 1 in (d)), except (c) f.b.: forced-bending simulation, (d) TMA for ε = 440. Solvent models used are: (a,b,d) RDIE, and (c) ACE2 [14]. Similar plots are obtained for AHs in VAC, with κ overall larger (cf., Fig. 4). κ further dropped for LZ at 487 Å, but is not plotted since the chain bent slightly during the initial energy minimization.
The bell-shaped profile of κ was observed over a range of amino acid sequences in different solvation environments. For AH, we tested poly-alanine (poly-Ala), poly-glycine (poly-Gly), poly-leucine (poly-Leu), and poly-lysine (poly-Lys), either in vacuum (VAC) or in a distance-dependent dielectric (RDIE) medium that mimics the solvation effect, all of which showed bell-shaped profiles. In the plateau region, κ is roughly similar among AHs tested, except for poly-Gly, which gives a smaller value (Fig. 1a,b). Insensitivity of κ on the amino acid sequence of AH agrees with a previous report [5]. Some of these AHs may not fold or are unstable in solution (e.g., poly-Gly). However, in folded forms, their elastic properties (∼curvature of the energy well) are mainly governed by the directional and short-ranged backbone hydrogen bond network that forms the elastic core of the filament [7]. Side chain interactions determine thermal stability of the structure (∼depth of the energy well). This is reflected by the κ of poly-Gly AH being comparable to those of others despite having no side chains (cf., Fig. 4). There is a stronger dependence on the solvation model; κ is nearly twice larger in VAC than in RDIE. In VAC, slower decaying electrostatic interactions in backbone hydrogen bonds render AHs stiffer. For LZ, since they are more stable in solvated environments, we used the ACE2 continuum solvent model in CHARMM [14] that captures the solvation effect better.
FIG 4.
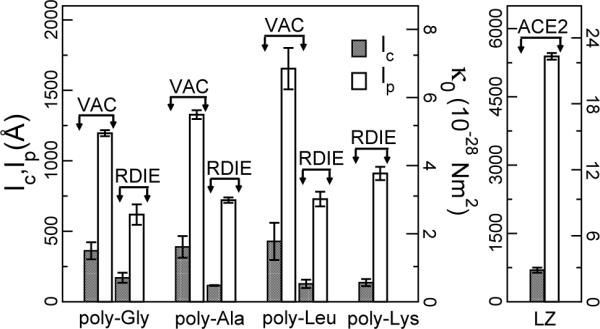
lc, lp, and κ0 of AH and LZ determined from the X − Y plots for n = 1 (Fig. 3). For LZ, labels of the y-axes follow those of AH on respective sides.
The drop of κ for longer chains is due to weak non-bonded attractions among atoms that increase as the chain bends and eventually lead to buckling instability. To check, we constructed a bead on a chain model (1D chain) where beads have the mass of a carbon atom, and are connected by harmonic springs of rest length b = 1 Å and spring constant Kb = 8000 kcal/mol·Å2 (unless otherwise noted). Bending stiffness is prescribed by a harmonic potential for the bond angle θ, U(θ) = Kθ(θ – π)2, with Kθ = 800 kcal/mol·rad2 (unless otherwise noted). Non-bonded attraction is incorporated via the Lennard-Jones potential for the distance r between any two non-neighbor beads
| (2) |
NMA shows that when ε = 0, κ does not drop, which also agrees with the analytic expression (Fig. 1d). κ drops for ε = 0.3 kcal/mol and further for ε = 0.7 kcal/mol, confirming that the drop is indeed due to the attractive force. In the plot of log ω1 vs. log L, deviation of the slope from −2 occurs at smaller L for larger ε (Fig. 2a).
FIG. 2.

(Color online). Whole-chain analysis (NMA and TMA) vs. local fluctuation analysis. (a) Deviation from linear elasticity in 1D chain, occurring from shorter L for larger ε. (b) Symbols: κ obtained from local fluctuation analysis, which are independent of L. Square: AH (in VAC; open: 58.6 Å, solid: 381 Å), Triangle: 1D chain (ε = 440; open: 74 Å, solid: 119 Å). Dashed and dotted horizontal lines: κ obtained from TMA, which decreases with L. (c) Sample snapshots of long chains during MD, which do not collapse.
Although NMA efficiently characterizes biofilament elasticity [6, 7, 9], it uses a harmonic approximation around the conformational energy minimum. To check if the drop in κ also occurs without the harmonic approximation, we performed thermal motion analysis (TMA) [7] whereby lp (hence κ) is estimated by applying the worm-like chain model to the end-to-end distance R [15]
| (3) |
where 〈R2〉 is the time average. We performed Langevin dynamics simulations on the 1D chain at 300 K, with ε = 440 kcal/mol, Kb = 4000 kcal/mol·Å2 and Kθ = 400 kcal/mol·rad2. We tested the range L = 29−119 Å for several hundred nanoseconds. Drop in κ again occurred (Fig. 1d). However, κ measured in TMA with ε = 0 did not show a drop (data not shown).
Remarkably, when we calculated κ from the variance of θ along the chain and by applying the equipartition theorem, no drop was observed (Fig. 2b). Similarly, we performed molecular dynamics (MD) simulations of poly-Ala in VAC for 20 ns, and measured κ using a local fluctuation analysis developed in [5], which did not show a drop. On the other hand, TMA of the same MD trajectories did show a drop in κ for longer AHs (Fig. 2b). We observed a similar behavior in MD simulations of AHs constructed using the sequence of the single α-helix (SAH) domain of myosin X [23], which remained folded in the ACE2 continuum solvent environment (data not shown). Note that even when the apparent κ obtained from global analyses (NMA or TMA) is small, the chain does not collapse (Fig. 2c). Therefore, longer chains do not actually become locally flexible. Instead, weak attractions alter their conformational ensemble, and make them appear to be more flexible.
To additionally check our calculation, we performed an explicit-water MD simulation of a 44-Å poly-Ala AH for 3.1 ns. While the simulation time was too short to apply TMA, local fluctuation analysis as above [5] yielded κ = 3.5 × 10−28 Nm2, slightly above the plateau value in RDIE (cf., Fig. 4). For LZ, we performed the ‘forced bending’ simulation where a transverse force was incrementally applied at the free end of a cantilevered LZ [7]. It remained linearly elastic up to a 32.9% strain, and gave κ similar to those from NMA (Fig. 1c).
Above results suggest that non-bonded attraction cannot be ignored for sufficiently long filaments. Even when forces balance in a straight configuration, attraction between two points on the chain increases as it bends. As non-bonded attraction is usually weak, restoring force prevails for short chains. However, beyond a certain length, the filament develops buckling instability, which can be described as [10]
| (4) |
where κ0 is the length-independent, local bending stiffness. The critical buckling force fc represents non-bonded attraction. Setting fc = 0 in Eq. (4) restores Eq. (1). Since kn = cn/L, for a given fc,
| (5) |
such that for L > lc, < 0, i.e., there is no stable wave solution. Since cn grows with n, higher modes have larger lc, as in Fig. 1. Using Eq. (4), we replot the data with X ≡ L−2 vs. (Fig. 3). From the linear fit, κ0, fc, and lc can be retrieved (Fig. 4). κ0 thus retrieved is indeed close to the plateau value of κ(L) in Fig. 1.
FIG. 3.
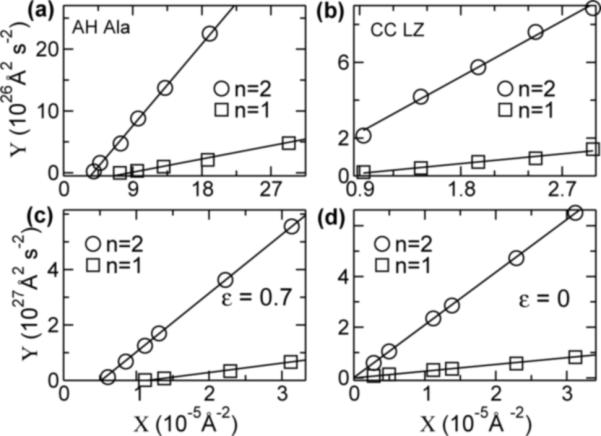
X − Y plot (X ≡ L−2, ). Solid line: linear fit. The initial rise phase in Fig. 1 was not used in the fit. Only portions near the origin are shown. In (d), lines converge to the origin, hence fc ≈ 0.
While we address the length dependence of κ, for a given L, our results agree favorably with previous ones. A local fluctuation analysis of 78-residue (113 Å) long poly-Ala AH yielded κ = 4.91 × 10−28 Nm2 in VAC [5], while we get (5.44−5.65)×10−28 Nm2. As for CC, a previous study of the myosin S2 domain (PDB 1NKN, 76 residues) reports κ = 7.20×10−28 Nm2 [6]. This was measured on a bent structure, with the end to end distance (96 Å) used for L. But Eq. (1) applies only to straight rods. Upon rebuilding a straight CC using the S2 sequence (L = 105 Å), we get κ = 10.9 × 10−28 ≈ (105/96)4 × 7.20 × 10−28 (Nm2) (cf., Eq. (1)). Also, residues 106−224 of tropomyosin CC (L = 168 Å; from PDB 2B9C) has κ = 9.22 × 10−28 Nm2. In contrast, a 84-residue LZ (L = 121 Å; rise phase in Fig. 1c) has κ = 14.3 × 10−28 Nm2. Higher stiffness of LZ may be attributed to the good knob-into-hole packing of side chains that contribute to its elastic core. This was further tested on cortexillin [16], a stiff CC (PDB 1D7M, L = 146 Å): κ = 15.2 × 10−28 Nm2, comparable to a LZ with a similar length. Compared to AH, stiffness of CC thus has a stronger sequence-dependence.
From the NMA of myosin S2 and the fragment of tropomyosin considered above, we get lp as 260 nm and 223 nm, respectively, which are 1.8 to 3.5 times longer than experimental estimates [17-19]. As irregularities in the heptad periodicity may locally destabilize the molecule [20, 21], our calculation based on completely folded CCs in ACE2 provides an upper bound of lp. Previously, mechanical properties of a single AH and the CC geometry were used to estimate lp ≃ 150 nm [22]. The smaller value in this continuum rod model may be because the deformation-dependence of energy by the knob-into-hole packing was ignored.
An important outcome of the present work is that for AHs and CCs, lc < lp (Fig. 4), suggesting that lc rather than lp is the length scale governing the chain conformation. In cases when an AH or a CC need to remain straight, they should be shorter than lc (∼12 nm for AH and <∼70 nm for CC). A good example is the SAH domain of myosin X (L = 5.3 nm), that forms a part of the lever arm [23]. Also, the 50-nm long CC stalk of kinesin has a hinge in the middle [24], so that the respective CC segments are well shorter than lc.
As long as there is non-bonded attraction, filaments buckle for L ≥ lc. For thin flexible polymers (e.g., polystyrene), lp is only several chemical bonds in length. In this case, non-bonded attraction can be easily offset by changing the solvent quality, where the distinction between good and poor solvent regimes is well-established [25]. Biofilaments are stiffer, with lp far greater than the chemical bond length or the size of solvation shells around polar groups of the chain (a few Å). Hence, changes in solvent quality is not as effective in canceling attractive forces. Furthermore, non-bonded attraction, which is typically the van der Waals force, is screened little in physiological conditions, in contrast to Coulombic forces [26]. Attraction-induced critical buckling should thus be more relevant to biofilaments. However, other interactions may come into play. Strong negative charges on DNA may generate repulsive forces that offset the weak attraction, making it difficult to observe buckling instability. Longer microtubules appear to be stiffer, although the study in [8] tested L in the range of 0.1lp and does not preclude buckling for longer L. It would be interesting to see if lc is less than lp in F-actin, which has only two protofilaments and the shearing effect may not be as pronounced as in microtubules.
Since lc is temperature independent (other than through κ0 and fc that may have weak T-dependence), the L > lc regime is not in conformational equilibrium and the chain will be trapped in a subset of conformations that may depend on the way how the system is prepared. In the case of a polymer network, the importance of attractive interaction has been previously considered: no matter how small, due to abundant contacts among polymers, nonspecific attractions eventually lead to non-equilibrium or glassy behavior [25, 27]. The present work shows that, even for a single biofilament, weak attractive force can profoundly affect its conformation.
Acknowledgments
We acknowledge Gerald Offer for providing the CC-building code. This work was supported by the National Institute of Health (Grant number: R21NS058604).
References
- 1.Howard J. Mechanics of Motor Proteins and the Cytoskeleton. Sinauer Associates, Inc; 2001. [Google Scholar]
- 2.Rivetti C, Guthold M, Bustamante C. J. Mol. Biol. 1996;264:919. doi: 10.1006/jmbi.1996.0687. [DOI] [PubMed] [Google Scholar]
- 3.Mücke N, Kreplak L, Kirmse R, Wedig T, Herrmann H, Aebi U, Langowski J. J. Mol. Biol. 2004;335:1241. doi: 10.1016/j.jmb.2003.11.038. [DOI] [PubMed] [Google Scholar]
- 4.Gittes F. J. Cell Biol. 1993;120:923. doi: 10.1083/jcb.120.4.923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Choe S, Sun SX. J. Chem. Phys. 2005;122:244912. doi: 10.1063/1.1940048. [DOI] [PubMed] [Google Scholar]
- 6.Adamovic I, Mijailovich S, Karplus M. Biophys. J. 2008;94:3779. doi: 10.1529/biophysj.107.122028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Park J, Kahng B, Kamm RD, Hwang W. Biophys. J. 2006;90:2510. doi: 10.1529/biophysj.105.074906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pampaloni F, Lattanzi G, Jonas A, Surrey T, Frey E, Florin E. Proc. Natl. Acad. Sci. USA. 2006;103:10248. doi: 10.1073/pnas.0603931103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Matsumoto A, Go N. J. Chem. Phys. 1999;110:11070. [Google Scholar]
- 10.Landau LD, Lifshitz EM. Theory of Elasticity. 3rd ed. Pergamon Press; 1986. [Google Scholar]
- 11.Offer G, Sessions R. J. Mol. Biol. 1995;249:967. doi: 10.1006/jmbi.1995.0352. [DOI] [PubMed] [Google Scholar]
- 12.Crick FHC. Acta Crystallographica. 1953;365 [Google Scholar]
- 13.Brooks B, Bruccoleri R, Olafson B, States D, Swaminathan S, Karplus M. J. Comp. Chem. 1983;4:187. [Google Scholar]
- 14.Schaefer M, Karplus M. J. Phys. Chem. 1996;100:1578. [Google Scholar]
- 15.Marko J, Siggia E. Macromolecules. 1995;28:8759. [Google Scholar]
- 16.Burkhard P, Kammerer R, Steinmetz M, Bourenkov G, Aebi U. Structure. 2000;8:223. doi: 10.1016/s0969-2126(00)00100-3. [DOI] [PubMed] [Google Scholar]
- 17.Hvidt S, Nestler FHM, Greaser ML, Ferry JD. Biochemistry. 1982;21:4064. doi: 10.1021/bi00260a024. [DOI] [PubMed] [Google Scholar]
- 18.Uyeda T, Abramson P, Spudich J, Howard J. Proc. Natl. Acad. Sci. USA. 1996;93:4459. doi: 10.1073/pnas.93.9.4459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Phillips G, Jr, Chacko S. Biopolymers. 1996;38:89. doi: 10.1002/(SICI)1097-0282(199601)38:1%3C89::AID-BIP7%3E3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- 20.Brown J, Cohen C, Parry D. Proteins. 1996;26:134. doi: 10.1002/(SICI)1097-0134(199610)26:2<134::AID-PROT3>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- 21.van Noort J, van der Heijden T, de Jager M, Wyman C, Kanaar R, Dekker C. Proc. Natl. Acad. Sci. USA. 2003;100:7581. doi: 10.1073/pnas.1330706100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wolgemuth C, Sun S. Phys. Rev. Lett. 2006;97:248101. doi: 10.1103/PhysRevLett.97.248101. [DOI] [PubMed] [Google Scholar]
- 23.Knight P, Thirumurugan K, Xu Y, Wang F, Kalverda A, Stafford W, III, Sellers J, Peckham M. J. Biol. Chem. 2005;280:34702. doi: 10.1074/jbc.M504887200. [DOI] [PubMed] [Google Scholar]
- 24.de Cuevas M, Tao T, Goldstein L. J. Cell Biol. 1992;116:957. doi: 10.1083/jcb.116.4.957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.de Gennes P-G. Scaling Concepts in Polymer Physics. Cornell University Press; Ithaca, NY: 1979. [Google Scholar]
- 26.Israelachvili JN. Intermolecular and Surface Forces. 2nd ed. Academic Press; 1992. [Google Scholar]
- 27.Fabry B, Maksym G, Butler J, Glogauer M, Navajas D, Fredberg J. Phys. Rev. Lett. 2001;87:148102. doi: 10.1103/PhysRevLett.87.148102. [DOI] [PubMed] [Google Scholar]