

Abstract
Conditional inactivation of the ‘neuropathy target esterase’ (NTE) gene in mouse nerve cells was previously shown to result in CNS pathology comparable to the spongiform encephalopathy characteristic of prion diseases. To determine whether cellular prion protein (PrPc) is essential for development of this pathology we examined hippocampi of mice lacking NTE alone, PrPc alone or both NTE and PrPc. Light microscopic survey showed clear-cut spongiform changes in a majority of NTE-/- and NTE/PrP-/- double knockout mice but in only one PrP-/- mouse. EM analysis of spongiform lesions from NTE-/- and NTE/PrP-/- mice, and from the one affected PrP-/- mouse, revealed patches of branching tubular inclusions, comparable to the ‘tubulovesicular inclusions’ described previously in prion diseases. We conclude that spongiform pathology in conditional NTE knockout mice is not mediated by PrPc, and that tubulovesicular inclusions can be seen in spongiform encephalopathy of other etiologies and are not pathognomonic of prion disease.
Keywords: Neurodegeneration, Creutzfeldt-Jakob disease, prion, scrapie, BSE, Alzheimer's disease, sws mouse
The Neuropathy Target Esterase (NTE) gene encodes a neuronal membrane protein that was identified for its ability to be modified by organophosphate compounds (Johnson, 1990). NTE represents the mammalian homolog of the Drosophila swisscheese (sws) gene (Glynn, 2000), mutations of which produce degeneration and glial-axonal deficits. Loss of function of the NTE gene in mice produces embryonic lethality at a very early embryonic age (Winrow et al. 2003; Moser et al. 2004). A conditional mouse mutation that inactivates the NTE gene only in nerve cells resulted in prominent pathological changes in neurons and their processes in the hippocampus and thalamus (Akassoglou et al. 2004). The characteristic findings consist of vacuolation of neuronal cytoplasm, focal disappearance of cytoplasmic organelles and the appearance, in some cases, of large patches of a tubulovesicular material, unlike any normal cellular constituent, and of scattered membranes within the formerly cytoplasmic areas. Nuclei do not show these changes, and no surrounding inflammatory response was present. These changes resulted in neuropil that contained holes, producing a ‘swiss cheese-like’ appearance originally found in the sws Drosophila homologue (Kretzschmar et al. 1997).
The overall appearance of these abnormalities in the mouse corresponds closely to the spongiform encephalopathy characteristic of Creutzfeldt-Jakob disease (CJD) and other prion diseases (Lampert et al. 1969, 1972). In particular the tubulovesicular inclusions in the conditional NTE mice (Akassoglou et al. 2004) resemble the tubulovesicular structures reported in prion-infected animals by David-Ferreira et al. (1968) and by Liberski et al. (1988, 2008) in a variety of naturally occurring and experimental prion disease cases. Based on its pathology, the NTE phenotype could signify an abnormality in cellular prion protein (PrPc). Moreover, it is possible that the tubulovesicular inclusions could in fact represent conglomerates of altered PrPc.
In order to test these possibilities, we examined mice generated by cross breeding a prion-null (PrP-/-) mouse line with our nestin-Cre NTE-conditional line (Nes-cre:NTE). The resulting offspring were either PrP-/- only, NTE-/- only, heterozygous for each gene, or deficient in both PrP and neuronal NTE On examining the conditional NTE/PrP-/- (double knockout) mice in comparison with NTE-/- mice, we found comparable pathology; i.e. the conditional NTE pathology did not require a contribution of PrPc, which is absent in the double knockout mice. This implies that other mechanisms contribute to the ‘swisscheese’ pathology that are independent of prion protein aggregation.
Materials and methods
Generation of conditional NTE/PrP-/- mice
The PrP-/- mice were obtained from the European Mouse Mutant Archive (EMMA, Munich, Germany), as well as from David Harris (Washington University). This line was previously shown to lack prion protein completely in brain tissue by western blot analysis (Bueler et al, 1992, Fig. 3e). The NTE conditional mice (NTE f/f) were generated as previously described by engineering LoxP sites around the initiating ATG codon of the NTE gene (Akassoglou et al. 2004). To obtain mice lacking NTE in the nervous system, we crossed the NTEfl/fl with mice harboring a nestin promoter driving the cre recombinase (Tronche et al. 1999). To obtain mice deficient for prion protein and NTE, the PrP-/- mice were crossed with Nes-cre:NTE f/f mice, yielding Nes-cre:NTE f/+ PrP +/- mice and NTE f/+ PrP +/- mice. These mice were further crossed until Nes-cre:NTE f/f PrP -/- mice were obtained. A PCR assay was utilized to genotype tail DNA by using a NTE primer set (sense: 5′-GCTTAAGGGCACCTGCCAGC-3′ and antisense: 5′-GGTCTTGTAGCCTGCAGTCC-3′; yielding a + band: 300bp and a f band: 400bp), a Cre primer set (sense: 5′-GCGGTCTGGCAGTAAAAACTATC-3′ and antisense: GTGAAACAGCATTGCTGTCACTT-3′; yielding one band: 100bp), and a PrP primer set (sense: 5′-TCAGCCTAAATACTGGGCAC-3′ and antisense: 5′-GCCTAGACCACGAGAAATGC-3′; yielding a + band: 880 and a − band: 1600bp).
Individual mouse pups were identified by ear punching or toe clipping, and the genotype of each pup, based on the PCR analysis, was recorded according to the ear punch or toe clip code.
Histology and electron microscopy
Mice were anesthetized with pentobarbital and fixed by transcardiac perfusion with 3% glutaraldehyde/2% formaldehyde freshly made up from paraformaldehyde. Brains were dissected out the day following perfusion, and the cerebral hemispheres were cut into coronal slices ∼1mm thick and rinsed overnight. The slices were then quartered and postfixed in 1-2% osmium tetroxide in 0.1M cacodylate buffer (pH 7.3), after which they were dehydrated in a graded series of methanol solutions, soaked overnight in propylene oxide and embedded in Araldite. The embedded specimens were sectioned at ∼1 micron en face and the sections stained with alkaline toluidine blue for survey. Sections that contained the typical hippocampal cellular and neuropil layers were read twice for the presence of vacuolation of the neuropil comparable to that reported previously (Akassoglou et al. 2005). We compared the proportion of specimens showing vacuolation in Nes-creNTE/PrP-/- hippocampi with that in NTE-/- or PrP-/- hippocampi by Fisher's exact test (2-tailed) using Graphpad Prism software. We made the same comparison between NTE-/- hippocampi and PrP-/- hippocampi.
The mice ranged in age from 1mo to 27mo but the great majority were between 3 and 12mo old. Mean ages were: NTE knockout ∼7mo; NTE/PrP double knockout ∼5mo; PrP knockout ∼6mo. There were no apparent age-related differences in pathology within the groups.
Representative thick sections, ∼1 micron in thickness, were photographed but the hippocampi were not serially sectioned. Block faces were then trimmed, and sections ∼0.1 micron thick were cut with a diamond knife and mounted on formvar/carbon coated copper grids, stained with potassium permanganate and ethanolic uranyl acetate and examined in a Philips or JEOL transmission microscope at 60 or 80kV. The diameter of the individual elements comprising the tubulovesicular inclusions was measured in digitized images using image J.
Results
Brain-specific inactivation of the NTE gene in the Nes-cre:NTEfl/fl mice resulted in hippocampal vacuolation along with intracellular tubulovesicular aggregates, sharply angulated membranes and focal breakdown of cytoplasmic organelles, resulting in a picture comparable to spongiform encephalopathy (Akassoglou et al. 2004). These findings raised the possibility that the pathology resulting from mutation of NTE could be related to prion-induced spongiform encephalopathy. The present study was designed to assess whether prion pathology could account for the phenotype observed in the conditional NTE knockout mice. In order to address this question, we examined hippocampi of Nes-creNTE/PrP-/- mice to determine whether focal vacuolation was caused by a prion-dependent mechanism. Previous biochemical analysis of lysates from the conditional Nes-cre:NTEfl/fl showed that expression of NTE protein was decreased over 90% in the cerebrum (Akassoglou et al. 2004).
Light microscopy
The results of our light microscopic survey of the conditional NTE single knockout mice (Nes-creNTE) in comparison with NTE/PrP double knockout mice (Nes-creNTE/PrP-/-) and PrP-/- mice are summarized in Table 1. Of 21 NTE/PrP double knockouts examined, 11 (52%) showed vacuolation in focal regions of the of the hippocampus (Fig. 1A). Overall, the findings in the double knockouts were comparable to those reported previously in NTE single knockouts (Akassoglou et al. 2004, Fig. 2C) and in sws fly brains (Kretzschmar et al. 1997, Fig. 1C). Of 15 PrP-/- mice, in contrast, only one (7%) showed hippocampal vacuolation. Of the 14 NTE knockout mice examined, 10 (71%) showed hippocampal vacuolation, consistent with our earlier report (Akassoglou et al. 2004).
Table 1.
Vacuolation was seen in 10 out of 14 Nes-cre:NTEfl/fl single knockout hippocampi and in 11 out of 21 Nes-cre:NTEfl/fl/PrP-/- double knockout hippocampi but in only 1 out of 15 PrP-/- knockout hippocampi. Statistical comparison of the respective groups showed that the NTE and NTE/PrP double knockout populations are not significantly different from one another, but both do differ significantly from the PrP knockout group (**).
| HIPPOCAMPAL VACUOLATION | ||||||
|---|---|---|---|---|---|---|
| NTE-/- | NTE/PrP-/- | PrP-/- | ||||
| # | (%) | # | (%) | # | (%) | |
| Vacuolated | 10 | (71%) | 11 | (52%) | 1 | (7%) |
| Total | 14 | 21 | 15 | |||
| ____p= .311 (ns)_____ | ||||||
| _______________p= .0013**__________________ | ||||||
| _____p= .0097**___________ | ||||||
Fig. 1.
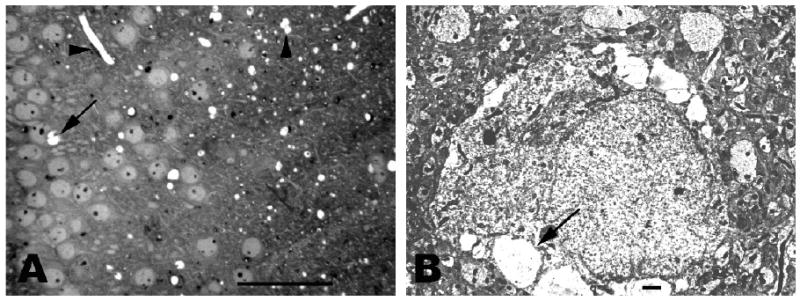
Fig. 1A. Photomicrograph from 2mo Nes-cre:NTEfl/fl/PrP-/- double knockout hippocampus showing marked vacuolation of neuropil (right). Vertical arrowhead at upper right indicates three neuropil vacuoles that appear to have fused. Horizontal arrowhead at upper left indicates dense endothelial cell nucleus in the wall of an elongated capillary. Vacuolation of cytoplasm in a hippocampal pyramid is shown at middle left (arrow). Bar = 100 microns.
Fig. 1B. Electron micrograph of neuron from 2 mo Nes-cre:NTEfl/fl/PrP-/- double knockout hippocampus. Nucleus and cytoplasm appear normal, as does the surrounding neuropil. Multiple large vacuoles, one indicated by arrow, are present along the periphery of the perikaryal cytoplasm. Bar = 1 micron.
Fig. 2.
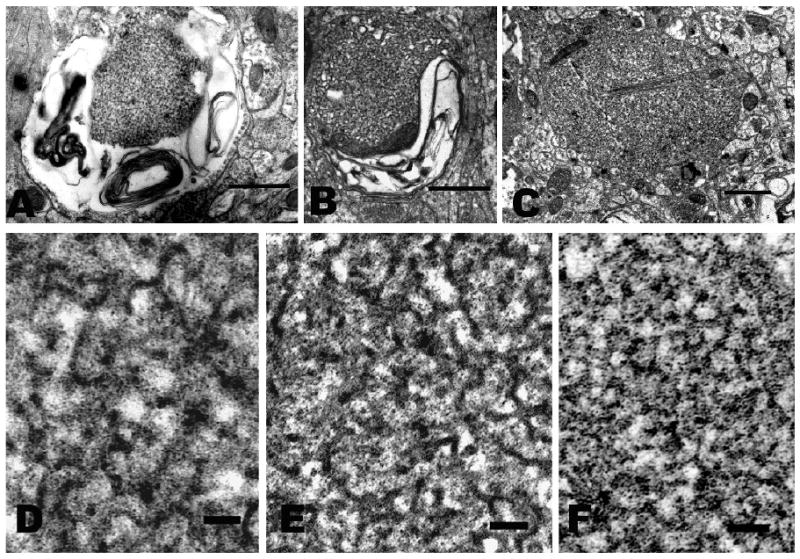
Electron micrographs of tubulovesicular inclusions in cell processes within hippocampal neuropil.
A-C. Survey views. In all, the neighboring cellular processes are well fixed and closely apposed to the degenerating one with no sign of inflammatory cells. Bar = 1 micron.
B-F. High-magnification images of ‘tubulovesicular’ inclusions showing narrow, branching tubules. Bar = 0.1 micron.
A, D. NTE/PrP double knockout (6 mo Nes-cre:NTEfl/fl/PrP-/-). The swollen process in A contains a tubulovesicular inclusion surrounded by scattered membranous debris.
B, E. NTE single knockout (27 mo Nes-cre:NTEfl/fl). The process in B contains membranous debris surrounding a tubulovesicular inclusion.
C, F. PrP knockout (4 mo .PrP-/-). The cell process in C contains residual mitochondria (upper left), filament bundles (center) and a tubulovesicular inclusion.
Statistical analysis showed that the NTE-/- single knockout and NTE/PrP-/- double knockout populations were not significantly different (p= .311). Comparison of double knockouts with the PrP single knockouts, however, showed the respective populations to be significantly different (p= .0097). Similarly the NTE knockout population differed significantly from the the PrP knockouts (p= .0013). Thus the absence of PrPc did not prevent spongiform encephalopathy from developing in more than half of the NTE/PrP-/- double knockouts as well as in a single case of the PrP knockout.
We did not see changes in the pathology with age, as might be expected with neurodegeneration. Although we do not know the reason for this, the apparent lack of progression and the lack of obvious neurological findings in these mutants could reflect ongoing neurogenesis in the hippocampus (cf. Gould, 2007) that keeps pace with cell loss.
The fact that significant vacuolation was not seen in nearly half of the double knockouts and 30% of the NTE knockouts could reflect the penetrance of the gene in that the defects do not appear uniformly and do not show up in all sections. Rather they occur in a spotty distribution, comparable in that respect to the hippocampal lesions of scrapie (DeArmond et al, 1987), which were also described as spotty. Serial sectioning of the NTE-/- and double knockout hippocampi might have revealed a larger proportion that were vacuolated. However, our goal was to determine whether spongiform changes occur in a significant number of hippocampi and not necessarily in all of the samples.
Electron microscopy
The Nes-cre:NTEfl/fl lesions previously examined by electron microscopy (Akassoglou et al. 2004) showed several characteristic ultrastructural findings: Within focal regions of the hippocampal neuropil we found swollen neuronal processes. The normal cytoplasmic elements were missing and were replaced by vacuoles containing membrane fragments and abnormal aggregates of inclusions that displayed a ‘branching reticular appearance’ and formed ‘tubular networks’. Neuronal perikarya displayed focal regions in which cytoplasm was replaced by empty vacuoles extending from the nuclear envelope to the plasma membrane often adjacent to normal-looking cytoplasm. In no case, was there evidence of surrounding edema or cellular inflammatory response; i.e. the degenerating cells or processes were surrounded by entirely normal-looking tissue. The pathology was limited to neurons; examination of oligodendrocytes and astrocytes showed no apparent defects.
In the conditional NTE/PrP-/- double knockout mice, we found pathology comparable to that observed previously in the Nes-cre:NTEfl/fl single knockout mice. Fig. 1B shows a hippocampal neuron surrounded by normal-looking, well-preserved neuropil. The cell nucleus is normal in appearance and most of the cytoplasm shows typical, normal-looking organelles including mitochondria, granular endoplasmic reticulum and Golgi apparatus. However, near the periphery of the cell, conspicuous large vacuoles are present, mostly empty but containing some membrane fragments and particulate debris. The surrounding neuropil closely apposes the cell body with no signs of extracellular edema or inflammatory cells.
Similar abnormalities are also present in isolated neuronal processes within the neuropil. Some of these processes display swelling and conspicuous membrane fragments (Fig. 2A) in which a laminar structure can be seen (‘myelin figure’), comparable to those in the NTE single knockout (Fig. 2B) and to the membranous inclusions described originally by Lampert et al (1969, 1972) in prion diseases. In addition small numbers of the affected processes contain inclusions comprised of a meshwork of branching tubules (Figs. 2A, D), reminiscent of the ‘tubulovesicular’ structures previously reported in prion diseases (David-Ferreira 1968; Liberski et al. 1988) in appearance and comparable to those in the NTE knockout (Figs. 2B, E). Indeed, although the appearance of these structures undoubtedly varies with fixation method, the image in Fig. 2D is nearly identical to that in Fig. 4 of David-Ferreira et al. (1968). In our specimens the outer diameter of the tubules measured ∼23nm. These images are comparable also to those in the NTE single knockout hippocampus (Figs. 2B, E). We have not seen inclusions of this kind in the cell bodies of hippocampal pyramids.
Since the tubulovesicular structures are much smaller in diameter than the section thickness, their entire diameter is usually included within the section, resulting in superimposition of the top and bottom regions and the side walls. This could account for their dense appearance and their indistinct margins when they are cut longitudinally. They also appear dense when cut across and appear as circular profiles, however (Fig. 2D-F). This suggests that they are in fact comprised of an electron dense material throughout or that they contain ‘a dense material,’ as suggested by David-Ferreira et al. (1968, Fig. 4) in their description of scrapie-infected mouse CNS.
The tubulovesicular structures in prion diseases have been reported to be ∼ 20-50nm in diameter (David-Ferreira 1968; Liberski et ala. 1988) and ∼27-35nm in diameter (Liberski et al. 2008). Potential differences in size could be significant or could possibly reflect differences in calibration or in preparative method, causing swelling or shrinkage. In the neuropil too, no evidence of inflammation or edema surrounding the affected processes was observed.
Although we did not expect to see spongiform pathology in mice lacking PrP alone, a previous study of PrP-null mice also showed “… coarse vacuolation of … the hippocampus…” in a minority of the animals (Bueler et al. 1992), consistent with our observation of spongiform pathology in one PrP-null mouse.
Side-by-side comparison of a degenerating process from that mouse with those from NTE-/- and double knockout mice showed comparable pathology. The tubulovesicular inclusion in Figs. 2C, F (PrP-/-) closely resembles that in Figs. 2A, D (double knockout)) as well as that seen in the Nes-cre: NTEfl/fl single knockout mice reported previously (Akassoglou et al. 2004), illustrated also in Figs 2B, E. These structures have not been reported in hippocampi from normal controls. Thus tubulovesicular inclusions appear to be a feature of spongiform encephalopathy even in the absence of PrPc.
Discussion
Our principal finding is that the spongiform encephalopathy previously observed in Nes-cre:NTEfl/fl mice occurs in the Nes-cre:NTEfl/fl/PrP-deficient mice as well. Thus, the possibility raised previously that the pathology we found could signify abnormal prion protein was not validated by this genetic analysis since comparable pathology developed when PrPc was absent, i.e. when the NTE mutation was introduced into PrP-/- mice. Therefore, the vacuolation and other pathologic changes caused by the selective loss of NTE in mice do not appear to require the presence of PrPc.
Our data also demonstrate that the tubulovesicular inclusions we see in the degenerating neuronal processes can not represent aggregates of prion protein. The identity of the tubulovesicular structures in prion disease models was investigated previously, and in that study as well, the authors concluded that these distinctive stuctures did not represent prion protein (PrPsc) based on immunogold studies (Liberski et al. 1997). What these aggreagates do represent remains unclear (Liberski et al. 2008).
The report by David-Ferreira et al (1968) distinguishes between two types of inclusions found in CNS processes of scrapie-infected mice: 1. granules and rods “with electron lucent centers and with walls approximately 95A in thickness”, and 2. “a close-meshed network of varicose tubules 200-500A in diameter which were filled with a moderately dense substance”.
Our images obtained from NTE single knockout mice and NTE/PrP double knockout mice as well as one PrP knockout mouse showed no examples of the first type of inclusion, i.e. the granules and rods, but did show inclusions of the second type, i.e. the varicose tubules. We conclude, therefore, that these ‘tubulovesicular inclusions’ are not peculiar to prion disease.
Our interpretation of the pathology we see is that the cells involved are undergoing autolysis, i.e. proteolysis of normal cytoplasmic components resulting in the generation of lower molecular weight breakdown products. Our assumption is that some of these breakdown products coalesce to form tubulovesicular aggregates, and the phospholipid components form myelin figures. Moreover, the breakdown results in an increase in the number of osmotically active intracellular particles leading to influx of water and swelling of the cellular processes
We assume that these pathological changes must take place rapidly, since once the plasma membrane itself breaks down, there is no longer an osmotically active barrier separating intracellular from extracllular compartments, and the cell contents would then be released into the surrounding extracellular space and dissipate there, leaving behind only an insoluble residue. Thus the swollen cell processes that characterize the spongiform encephalopathy we see probably represent a snapshot of a dynamic process that proceeds relatively rapidly in the individual cells and their processes. The relatively low frequency of appearance of the tubulovesicular inclusions could indicate that these structures too represent a short-lived stage in cytolysis, after which they break down further and become soluble.
How the loss of NTE protein causes or accelerates this kind of degeneration is not known. The NTE gene encodes an esterase enzyme activity, as does its Drosophila homologue, sws. Analysis of the sws mutation in Drosophila indicates that sws is involved in glial-neuronal signaling, or possibly in recognizing a ligand at the neuronal surface (Glynn 1999). Sws/NTE may act primarily during development; so the prominence of lesions in the hippocampus may reflect the neurogenesis believed to occur there (Gould 2007), although pathological changes were also seen in other regions of the brain (Akassoglou et al. 2004). In addition, recent genetic studies have correlated motor neuron defects with mutations in the human NTE gene (Rainer et al 2008). Inappropriate signaling, or lack of signaling, may trigger the degenerative changes that are observed in both vertebrates and flies. Alternatively, loss of NTE may lead to abnormalities in the endoplasmic reticulum (ER), where there is normally an abundance of intracellular NTE protein (Glynn 1998; Akassoglou et al. 2004). Changes in NTE levels may result in problems in protein folding, transport or degradation originating in the ER.
The mechanisms underlying prion disease pathology remain unclear (Aguzzi et al. 2007), but the resemblance between the pathology found in both NTE-deficient mice and conditional NTE/PrP double knockout mice and that in prion diseases suggests the possibility that the degeneration seen in prion diseases might be mediated through an effect on NTE or, alternatively, that prion diseases and NTE deficiency may ultimately act through a common pathway. One might expect that a variety of different neurodegenerative diseases would also act through a common pathway. Spongiform changes are known to occur in a number of dementias (Table 16-5 in Ironside et al, 2008). Yet, surprisingly, the spongiform pathology seen in prion diseases and in mice deficient in NTE is not present in others, including Alzheimer's. Thus the neurodegeneration resulting from lack of NTE is a distinctive outcome comparable to that in prion diseases but different from that observed in a number of other neurological diseases.
Summary and conclusions
The microscopic and ultrastructural pathology seen in the hippocampus of NTE/PrP-/- double knockout mice matches that of the Nes-cre: NTEfl/fl/PrP+ single knockout mice examined previously.
The swellings seen are consistent with proteolysis of cytoplasmic components producing high concentrations of protein fragments that increase the osmolality of the neuronal cytoplasm and draw water into the ghost profiles. The tubulovesicular inclusions may represent aggregates of breakdown products generated by cytoplasmic autolysis.
Since tubulovesicular inclusions occur in the double knockout as well as in the PrP knockout they cannot represent patches of altered prion protein.
The pathology we see in our double knockout mice, which lack PrPc, cannot be considered pathognomonic of prion disease pathology.
The close correspondence between the NTE/PrP double knockout pathology and that seen previously in prion diseases suggests that both may operate through a common pathway and raises the possibility that prion disease pathology is mediated by NTE.
Acknowledgments
The authors are indebted to Dr.Martin Sadowsky for helpful comments about the manuscript and to Chris Petzold for expert technical assistance. This study was supported by grants from the NIH (NS 051282 to MC and NS 037475 to JR) and National MS Society (RG 3618 to JR).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aguzzi A, Heikenwalder M, Polymenidou M. Insights into prion strains and neurotoxicity. Nature Revs Mol Cell Biol. 2007:550–561. doi: 10.1038/nrm2204. [DOI] [PubMed] [Google Scholar]
- Akassoglou K, Malester B, Xu J, Tessarollo L, Rosenbluth J, Chao MV. Brain-specific deletion of neuropathy target esterase/swisscheese results in neurodegeneration. Proc Natl Acad Sci U S A. 2004;101:5075–5080. doi: 10.1073/pnas.0401030101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Büeler H, Fischer M, Lang Y, Bluethmann H, Lipp HP, DeArmond SJ, Prusiner SB Aguet M, Weissmann C. Normal development and behaviour of mice lacking the neuronal cell-surface PrP protein. Nature. 1992;356:577–82. doi: 10.1038/356577a0. [DOI] [PubMed] [Google Scholar]
- David-Ferreira JF, David-Ferreira KL, Gibbs CJ, Jr, Morris JA. Scrapie in mice: ultrastructural observations in the cerebral cortex. Proc Soc Exp Biol Med. 1968;127:313–320. doi: 10.3181/00379727-127-32680. [DOI] [PubMed] [Google Scholar]
- DeArmond SJ, Mobley WC, DeMott DL, Barry RA, Beckstead JH, Prusiner SB. Changes in the localization of brain prion proteins during scrapie infection. Neurology. 1987;37:1271–80. doi: 10.1212/wnl.37.8.1271. [DOI] [PubMed] [Google Scholar]
- Glynn P. Neuropathy target esterase. Biochem J. 1999;344:625–631. [PMC free article] [PubMed] [Google Scholar]
- Glynn P. Neural development and neurodegeneration: two faces of neuropathy target esterase. Prog Neurobiol. 2000;61:61–74. doi: 10.1016/s0301-0082(99)00043-x. [DOI] [PubMed] [Google Scholar]
- Gould E. How widespread is adult neurogenesis in mammals? Nat Rev Neurosci. 2007;8:481–488. doi: 10.1038/nrn2147. [DOI] [PubMed] [Google Scholar]
- Ironside JW, Ghetti B, Head MW, Piccardo P, Will RG. Prion Diseases. In: Love S, Louis DN, Ellison D, editors. Greenfield's Neuropathology. Hodder-Arnold; London: 2008. pp. 1197–1274. [Google Scholar]
- Johnson MK. Organophosphates and delayed neuropathy—Is NTE alive and well? Toxicol Appl Pharm. 1990;102:385–399. doi: 10.1016/0041-008x(90)90036-t. [DOI] [PubMed] [Google Scholar]
- Kretzschmar D, Hasan G, Sharma S, Heisenberg M, Benzer S. The swiss cheese mutant causes glial hyperwrapping and brain degeneration in Drosophila. J Neurosci. 1997;17:7425–7452. doi: 10.1523/JNEUROSCI.17-19-07425.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lampert PW, Earle KM, Gibbs CJ, Jr, Gajdusek DC. Experimental kuru encephalopathy in chimpanzees and spider monkeys. Electron microscopic studies. J Neuropathol Exp Neurol. 1969;28:353–70. doi: 10.1097/00005072-196907000-00001. [DOI] [PubMed] [Google Scholar]
- Lampert PW, Gajdusek DC, Gibbs CJ., Jr Subacute spongiform virus encephalopathies. Scrapie, Kuru and Creutzfeldt-Jakob disease: a review. Am J Pathol. 1972;68:626–652. [PMC free article] [PubMed] [Google Scholar]
- Liberski PP, Jeffrey M, Goodsir C. Tubulovesicular structures are not labeled using antibodies to prion protein (PrP) with the immunogold electron microscopy techniques. Acta Neuropthol. 1997;93:260–4. doi: 10.1007/s004010050612. [DOI] [PubMed] [Google Scholar]
- Liberski PP, Sikorska B, Hauw JJ, Kopp N, Streichenberger N, Giraud P, Budka H, Boellaard JW, Brown P. Tubulovesicular structures are a consistent (and unexplained) finding in the brains of humans with prion diseases. Virus Res. 2008;132:226–8. doi: 10.1016/j.virusres.2007.11.008. [DOI] [PubMed] [Google Scholar]
- Liberski PP, Yanagihara R, Gibbs CJ, Jr, Gajdusek DC. Tubulovesicular structures in experimental Creutzfeldt-Jakob disease and scrapie. Intervirol. 1988;29:115–9. doi: 10.1159/000150036. [DOI] [PubMed] [Google Scholar]
- Moser M, Li Y, Vaupel K, Kretzschmar D, Kluge R, Glynn P, Buettner R. Placental failure and impaired vasculogenesis result in embryonic lethality for neuropathy target esterase-deficient mice. Mol Cell Biol. 2004;24:1667–79. doi: 10.1128/MCB.24.4.1667-1679.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rainer S, Bui M, Mark E, Thomas D, Tokarz D, Ming L, Delaney C, Richardson R, Albers J, Matsunami N, et al. Neuropathy Target Esterase gene mutations cause motor neuron disease. Amer J Human Genetics. 2008;82:780–785. doi: 10.1016/j.ajhg.2007.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tronche F, Kellendonk C, Kretz O, Gass P, Anlag K, Orban P, Bock R, Klein R, Schutz G. Disruption of the glucocorticoid receptor gene in the nervous system results in reduced anxiety. Nature Genet. 1999;23:99–103. doi: 10.1038/12703. [DOI] [PubMed] [Google Scholar]
- Winrow CJ, Hemming ML, Allen DM, Quistad GB, Casida JE, Barlow C. Loss of neuropathy target esterase in mice links organophosphate exposure to hyperactivity. Nat Genet. 2003;33:477–85. doi: 10.1038/ng1131. [DOI] [PubMed] [Google Scholar]