

Abstract
The neuronal α4β2 nicotinic acetylcholine receptor (nAChR) is a target for general anesthetics. Currently available experimental structural information is inadequate to understand where anesthetics bind and how they modulate the receptor motions essential to function. Using our published open-channel structure model of α4β2 nAChR, we identified and evaluated six amphiphilic interaction sites for the volatile anesthetic halothane via flexible ligand docking and subsequent 20-ns molecular dynamics simulations. Halothane binding energies ranged from −6.8 to −2.4 kcal/mol. The primary binding sites were located at the interface of extracellular and transmembrane domains, where halothane perturbed conformations of, and widened the gap among, the Cys-loop, the β1-β2 loop, and the TM2-TM3 linker. The halothane with the highest binding affinity at the interface between the α4 and β2 subunits altered interactions between the protein and nearby lipids by competing for hydrogen bonds. Gaussian network model analyses of the α4β2 nAChR structures at the end of 20-ns simulations in the absence or presence of halothanes revealed profound changes in protein residue mobility. The concerted motions critical to protein function were also perturbed considerably. Halothane's effect on protein dynamics was not confined to the residues adjacent to the binding sites, indicating an action on a more global scale.
Keywords: anesthesia mechanism, halothane, nicotinic acetylcholine receptor, alpha4beta2, membrane protein, molecular dynamic simulation
Introduction
The α4β2 nicotinic acetylcholine receptor (nAChR) is abundant in the brain. It mediates fast synaptic transmission in the central nervous system. All subtypes of nAChRs, including α4β2, are ligand-gated, pentameric, cation-permeable channels. Binding of the endogenous neurotransmitter acetylcholine and other agonists (such as nicotine) to the extracellular (EC) domain of the receptor triggers the channel opening, allowing cations to pass across the membrane. The function of the α4β2 nAChR can be severely suppressed by general anesthetics.1-7 It has been shown that anesthetics profoundly reduce the channel opening of the α4β2 nAChR7 and inhibit the channel current.1-4 Although emerging evidence suggests that the α4β2 nAChR is one of many molecular targets for general anesthetics, there is no comprehensive understanding on how anesthetics modulate the function of the α4β2 nAChR. In fact, molecular mechanisms of anesthetic action on all putative targets remain to be determined.
Knowledge of anesthetic interaction sites is essential to understanding how anesthetics modulate protein functions. Site-directed mutagenesis, in conjunction with functional measurements, has been used widely in searching for anesthetic binding sites.8 However, it remains unclear whether the resulting sensitive mutagenesis sites were involved in direct anesthetic binding or only a part of allosteric interactions. X-ray structures of anesthetic-protein complexes are ideal for revealing anesthetic binding sites with atomic resolution, but only a few model proteins have been crystallized with anesthetics.9-11 Because of great technical challenges, there is no X-ray structure available for the α4β2 or other subtypes of nAChRs, though the recent achievement in X-ray structures of the homologous bacterial pentameric ligand-gated ion channel GLIC12,13 and ELIC14 is certainly encouraging. NMR can be used not only to determine anesthetic interaction sites,15-17 but also to divulge anesthetic-triggered changes in protein structures and dynamics.15,16,18,19 These have been successfully demonstrated for isolated domains of the α4β2 nAChR.17,20,21 Photo-affinity labeling revealed specific binding sites in the Torpedo nAChR for several different anesthetics,22-26 but there is no report in the literature about anesthetic photolabling to the α4β2 nAChR. The currently available experimental data have not generated a coherent picture about anesthetic interaction sites in the α4β2 nAChR.
Protein function is a dynamic process that can be modulated by conformational changes triggered by ligand binding. In the case of nAChRs, it is the collective motion among all five subunits, upon agonist-binding in the EC domain, that regulates channel closing and opening in the transmembrane (TM) domain.27,28 Intrinsic motions of a protein can be perturbed by anesthetics,15,29 and if a specific motion critical to function is altered, then diverse functional changes, including those observed for the α4β2 nAChR,1-4,7 are expected. Thus, in order to understand how anesthetics modulate protein function, we must first determine how they modulate protein motions.
Complementary to experimental efforts, computational studies can provide insight into how anesthetics interact with proteins and modulate their structures and dynamics.18,29-31 In the present study, we started with our published open-channel structure model of the α4β2 nAChR in a ternary lipid mixture32 and explored the nature and location of anesthetic interactions using flexible ligand docking and MD simulations. Halothane, an inhalation general anesthetic, was chosen for the study because of the availability of optimized computational parameters33,34 and photolabeling data with the Torpedo nAChR.23 In addition to revealing potential halothane binding sites in the α4β2 nAChR, we also investigated some of the following intriguing questions: since most volatile anesthetics like halothane are low affinity drugs,35 is tight binding necessary to trigger structural or dynamical changes that are essential to protein function? Can a limited number of anesthetic interaction sites produce a global effect? Is an anesthetic's effect on protein functional motion confined only to the vicinity of the binding site(s) or can it change motions on a global scale? The more than 20-ns MD simulations in the absence or presence of halothane in this study have offered some answers to these questions.
Systems and Methods
Systems
For clarity, the systems in the absence or presence of halothane molecules are denoted as control or halothane system, respectively. Our published open-channel α4β2 nAChR structure32 was used as the control in this study. Details on the system preparation and equilibration as well as a ∼11-ns MD simulation can be found in our previous publication.32 Briefly, the open-channel α4β2 nAChR was embedded in a pre-equilibrated ternary lipid bilayer36 consisting of 160 POPC, 50 POPA, and 54 cholesterols (CHOL). In addition, the system contained 34924 TIP3P water molecules, 109 Na+, and 22 Cl− to achieve a fully hydrated and neutral condition with an ion concentration of 200 mM. The protein in the control system after the previous 11-ns MD simulations was used for halothane docking. Except for the addition of halothane molecules and eight additional water molecules, the initial halothane system and the control system were virtually the same.
MD simulations
All MD simulations were performed either on BIGBEN or POPLE at the Pittsburgh Supercomputer Center using NAMD2.6.37 The CHARMM-27 force field38 was used for everything in the systems except halothane, for which we used parameters developed previously by our group.34 PME was used for long-range electrostatic interactions, and a 12-Å cutoff was used for non-bonded interactions. Both systems were energy minimized using the conjugated gradient and line search algorithm, equilibrated using gradually reduced constraint on Cα atoms (from 10 to 0 kcal/mol/Å2 over a course of 700 ps). This was followed by constraint-free MD simulations at 303 K and 1 atm (NPT) for more than 20 ns. A time step of 1 fs and a damping coefficient of 1 ps-1 were used throughout the MD simulations.
Initial halothane occupancy sites in the open-channel α4β2 nAChR
Initial halothane locations in the halothane system were determined on the basis of either experimental suggestions23 or in silico docking using Autogrid/AutoDock4.39 The Lamarckian genetic algorithm with the grid size of 0.375 Å was used, and 500 runs were typically performed. High halothane occupancy sites found from docking were included in the subsequent MD simulations. Additional halothane molecules were manually placed into the system at sites suggested by experiments,23 resulting in a total of 22 halothane molecules in the initial halothane system. After performing 19 ns of NPT simulations on this 22-halothane system, we identified six representative sites showing relatively small halothane displacements. These six halothane molecules were included in our halothane system for a more than 20-ns MD simulation.
Halothane binding energies
To assess halothane binding energies, free energy perturbation (FEP) calculations40,41 were performed using the October 24, 2008, concurrent versions system (CVS) snapshot of NAMD2.6.37 The revised FEP code enables the decoupling of electrostatic interactions from van der Waals interactions. The FEP calculations were started using the coordinates and velocities from the end of the 20-ns simulation of the halothane system. For each halothane site in the α4β2 nAChR, the halothane molecule was gradually annihilated as λ changed from 1 to 0. The λ decrement was 0.05 for λ values between 0.2 and 0.9, and 0.025 when λ was in the ranges of 0.0-0.2 and 0.9-1. Therefore, there were 26 separated λ windows for each halothane calculation. Within each window, an equilibration of 10 ps was followed by 100 ps of FEP data collection, all with a 2-fs time step. To avoid the so-called endpoint catastrophes resulting from annihilation of an alchemical group, a radius-shifting coefficient of 6.0 Å2 was used, and electrostatics were linearly scaled down between λ values of 1 and 0.5 to completely turn off electrostatics as the λ value was equal or smaller than 0.5. The same FEP calculation was also performed on a halothane molecule in a water box. The reported binding energies are from the difference between the FEP calculations in the α4β2 nAChR and in water.
Data analysis
The VMD program37 was used for the visualization of the simulation results. The majority of the data presented in this paper were analyzed using VMD-associated scripts, either directly from the VMD developers or with some customization. A salt bridge is defined by a cutoff distance of 5 Å between the center of oxygen atoms in an acidic residue and the center of nitrogen atoms in a basic residue. The pore radius was measured using the HOLE program.42 The Gaussian network model (GNM) (http://ignm.ccbb.pitt.edu/GNM_Online_Calculation.htm) was used for analyzing halothane's effect on mobility and concerted motion of the α4β2 nAChR.43-45 Each Cα atom in the protein was taken as a node in the GNM calculations. A cutoff distance of 10 Å was used for harmonic interactions among different nodes. Halothane molecules were not counted as additional nodes. Thus, the control and halothane systems had exactly the same number of nodes in the GNM analysis. A joint mobility profile (mean-square fluctuation, MSF) of the i-th residue corresponding to the five slowest modes is defined as
| (1) |
where λk and μk are the eigenvalue and eigenvector of the k-th mode, respectively. The concerted motion is determined from the cross-correlations between the fluctuations of residues i and j, <ΔRi · ΔRj>. Color-coded correlation maps were plotted using the Origin program (OriginLab Co., Northampton, MA).
Results and Discussion
Halothane binding in the α4β2 nAChR
Halothane molecules can occupy many sites in the α4β2 nAChR, as suggested in our initial 19-ns MD simulation with 22 halothane molecules (data are not shown). Six halothane molecules showing relatively small displacements at 6 representative sites were chosen for a new 20-ns simulation. Figure 1A depicts the halothane occupancies at the beginning and end of the 20-ns MD simulation. Halo-1 and halo-5 reside between subunits and can potentially alter the inter-subunit interactions. Halo-2, halo-3, and halo-4 are located at the interface of the EC and TM domains and are involved in the interactions with the pre-TM1 linker, the β1-β2 loop, the Cys-loop, and the TM2-TM3 linker. The coupling among these loops is believed to modulate the signal transduction from agonist binding to channel gating, and halothane molecules at the interface can potentially interrupt this coupling. Halo-6 is accommodated by residues in the main immunogenic region (MIR), which contains conserved residues in the nAChR family and plays a critical role in the pathogenesis of myasthenia gravis upon antibody binding.46
Figure 1.

(A) Halothane binding sites for the open α4β2 nAChR at the beginning and end of the 20-ns simulation. The α4 and β2 subunits of the protein are drawn in gray and white cartoon, respectively. Halothane molecules are represented by van der Waals surfaces and colored black (halo-1), magenta (halo-2), green (halo-3), red (halo-4), brown (halo-5) and blue (halo-6). (B) The displacement of the six halothane molecules over the course of the 20-ns simulation.
Halothane binding affinity to the α4β2 nAChR varies among different binding sites. Relative to the initial locations, halo-1 and halo-6 had quite small displacements over the course of the 20-ns simulation (Fig. 1B), indicating favorable interactions between halothane and the surrounding residues or lipids. Displacements of the other 4 halothanes were more substantial, but none of them was greater than 2 Å in the last 5-ns simulation. FEP calculations revealed that halothane binding energies ranged from −6.8 to −2.4 kcal/mol, consistent with the known, relatively low, binding affinity of volatile anesthetics in proteins.9,47 Table 1 summaries residue types or lipids contributing to the halothane binding pockets and the average number of water molecules within 3 Å and 5 Å radius of each halothane in the last 5-ns simulation. Hydrophobic residues are abundant in all six binding sites, but polar interactions or hydrogen bonds between halothane molecules and their surroundings exist in all cases, reflecting the amphiphilic nature of anesthetic interaction sites.48 There is no obvious correlation between binding pocket size and binding energy. The pockets for halo-4 and halo-6 are more enclosed. Their Connolly surface areas are 255 Å2 and 231 Å2, equivalent to molecular volumes of 242 Å3 and 191 Å3, respectively. None of the six halothane molecules were dehydrated. The halothane molecules associated with more water molecules, such as halo-5, tended to have lower affinity to the protein.
Table 1.
Properties of halothane binding sites in the α4β2 nAChR
| halothanea | apolarb | polarb | TYR or TRPb | POPCb | CHOLb | waterc | binding energyd | Kde | ||
|---|---|---|---|---|---|---|---|---|---|---|
| uncharged | charged | 3Å | 5Å | |||||||
| halo-1(α4-1/β2-3) | 7(78%) | 1(11%) | 1(11%) | 1.1 ± 0.8 | 5.5 ± 1.5 | -6.8 | 1.2×10-2 | |||
| halo-2(β2-3) | 4(58%) | 1(14%) | 1(14%) | 1(14%) | 1.4 ± 1.3 | 12.9 ± 2.7 | -3.4 | 3.5 | ||
| halo-3(α4-2) | 8(67%) | 2(17%) | 1(8%) | 1(8%) | 1.1 ± 1.1 | 7.0 ± 2.0 | -3.8 | 1.8 | ||
| halo-4(α4-1) | 8(58%) | 3(21%) | 3(21%) | 0.7 ± 0.6 | 2.6 ± 1.2 | -3.7 | 2.1 | |||
| halo-5(α4-1/β2-3) | 4(58%) | 1(14%) | 2(28%) | 2.9 ± 1.6 | 13.1 ± 2.7 | -2.4 | 18.5 | |||
| halo-6(β2-1) | 5(42%) | 1(8%) | 2(17%) | 4(33%) | 0.2 ± 0.5 | 2.3 ± 1.3 | -3.9 | 1.5 | ||
Specific subunit(s) involved in halothane binding are in parenthesis.
Data are presented as N (%), where N is the number of residues or lipids contributing to a binding pocket, present for more than 60% of the time over the last 5-ns of the simulation, and % is the percentage of each type of residue or lipid with respect to the total number of residues or lipids contributing to a specific binding pocket.
Average number of water molecules within 3 Å or 5 Å of a halothane in the last 5-ns of the simulation
Halothane binding energies from FEP calculations in kcal/mol
The apparent dissociation constant in mM, Kd, was calculated using the equation Δ ΔG = RTlnKd where R = 1.987 cal mol-1 K-1 and T = 303 K.
Halothane binding sites similar to the ones observed here were identified previously through the direct halothane photoaffinity labeling in the Torpedo nAChR,23 where halothane labeling was found primarily on tyrosine residues. The residues photolabeld by halothane in the Torpedo nAChR are presented in Fig. S1 (Supporting Information), where one can easily compare these binding sites with those in the α4β2 nAChR (Fig. 1). As shown in Table 1, four out of six halothane molecules were in proximity to tyrosine or tryptophan residues. Therefore, it is highly likely that our in silico halothane binding sites could be observed experimentally through photolabeling experiments.
Neither docking nor MD simulations in this study provided evidence for halothane residing inside the pore of α4β2 nAChR, even though the possibility for anesthetics acting as channel blockers has been discussed in the past. Mutations in the pore-lining residues indeed changed inhibitory potency of alcohols on channel opening,49-51 but a direct link between these residues and an anesthetic binding site could not be established because the mutations also affected the channel function in the absence of anesthetics. More recently, residues (α-E262, β-D268, γ-Q276) at the extracellular channel entry of the Torpedo nAChR were photolabeled by azietomidate,52 a photoreactive analog of the intravenous general anesthetic etomidate. Our recent computational data (to be reported elsewhere) also suggest that intravenous anesthetics can occupy the inside of the channel, but such interactions have not been observed for inhalational anesthetics like halothane. Nevertheless, it is highly likely that intravenous and inhalational anesthetics have distinctly different binding sites in nAChRs and other target proteins.
Halothane disturbance to agonist-binding site
Among all halothanes, halo-5 is the only one near nicotine (NIC1) at one of the agonist-binding sites. Experimental halothane photolabeling in the Torpedo nAChR showed labeling of γ-Y111 (homologues to β2-Y114 in Fig. 2) within the agonist-binding site.23 Agonist binding initiates the channel opening in nAChR and other Cys-loop receptors.53 It is logical to question whether anesthetics affect channel functions by disturbing agonist binding. Our data here shows that halo-5 did not compete with NIC1 for binding, but it did alter the networks of hydrogen bonds and salt bridges in the region that could affect signal propagation from agonist binding to channel opening.
Figure 2.

Nicotine (NIC1) binding site located between the α4-1 (light gray) and β2-3 (dark gray) subunits in the control (A) or halothane (B) system at the end of the 20-ns simulation. NIC1 (green) and halo-5 (brown) are shown in the ball-and-stick representation, and the neighboring residues are shown in the stick representation (white: carbon; red: oxygen; blue: nitrogen) without hydrogen for clarity. NIC1 has similar position and orientation in both systems, showing significant π-stacking with W57. Flexible loops (A-loop in orange, B-loop in cyan, and C-loop in purple) of the α4-1 subunit varied between the two systems, but the hydrogen bond between K158 of the B-loop and I201 of the C-loop remained. Y114, adjacent to halo-5, corresponds to a photoaffinity-labeled residue in the Torpedo nAChR.
The position and orientation of NIC1 relative to the third β2 subunit (β2-3 in Fig. 2) were almost the same in the absence or presence of halo-5 at the end of 20-ns simulations. The majority of the residues accommodating NIC1 binding in the β2-3 subunit were not affected significantly by halo-5. Because of its potential ability to form hydrogen bonds with residues in the C-loop, K158 of the B-loop was identified as a critical residue for enhancing nicotine affinity.54 For both systems in this study, the original π-stacking interaction between NIC1 and Y195 of the α4-1 subunit disappeared and the interaction between NIC1 and W57 of the β2-3 subunit was maintained. Our simulation data showed that the hydrogen bond between the backbone N–H of K158 and the backbone carbonyl of I201 sustained over 80% of the simulation time in both the control and halothane systems. The stability of NIC1 at the binding site was unlikely affected by halo-5.
Although halo-5 did not compete with NIC1 for binding, it clearly disturbed the region essential for signal propagation upon agonist binding. Halo-5 limited the capping motion of the C-loop in the α4-1 subunit and caused a wider gap between the C-loop and the surface of the adjacent β2-3 subunit (see Fig. S2 in Supporting Information). Hydrogen bonding between residues in the C-loop of an α subunit and the F-loop of a complementary subunit was suggested to play a key role in the gating pathway.55 As shown in Fig. 3, D171 of the β2-3 subunit formed hydrogen bonds simultaneously with K194 and Y202 of the α4-1 subunit in the control system, but only the one with Y202 survived in the presence of halo-5. The salt bridge between E200 of the α4-1 subunit and K79 of the β2-3 subunit virtually disappeared in the presence of halo-5. Consequently, the inter-subunit interactions are considerably weakened by halo-5. These inter-subunit interactions are presumably important for transmitting signals from agonist binding to channel gating.
Figure 3.
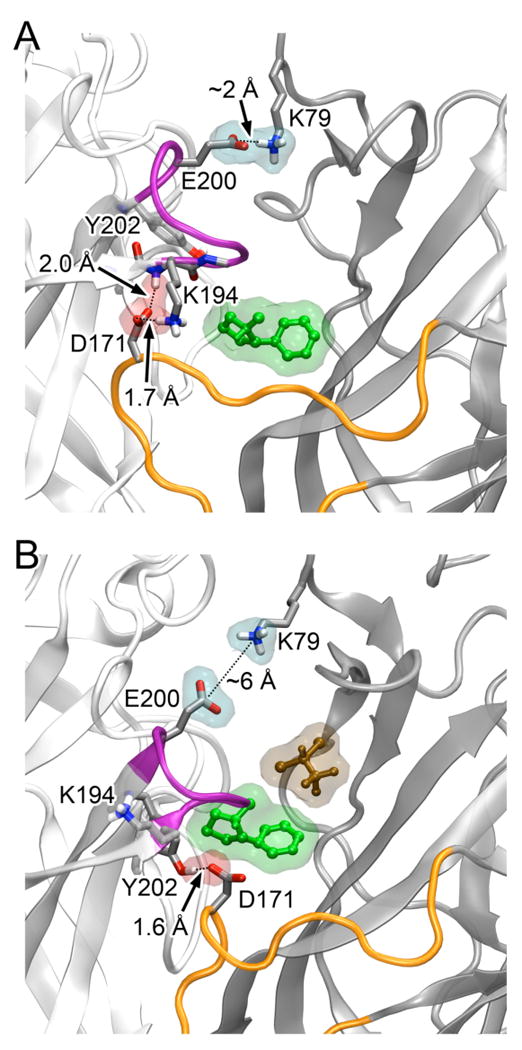
Inter-subunit salt bridges and hydrogen bonds near the NIC1 (green) binding site changed in the absence (A) or presence (B) of halo-5 (brown). The salt bridge (in a cyan surface) between K79 of the β2-3 (dark gray) and E200 of the α4-1 (light gray) disappeared in the presence of halo-5. In the absence of halo-5 in (A), D171 of the β2-3 subunit formed the salt bridge and hydrogen bond (in a pink surface) with K194 and Y202 of the α4-1 subunit, respectively. Both these salt bridge and hydrogen bond vanished in (B) and D171 formed a new hydrogen bond with Y202. The C-loop of the α4-1 subunit and the F-loop of the β2-3 subunit are highlighted in magenta and orange, respectively. A salt bridge is measured between a positive charged N atom and the center of two carboxyl oxygen atoms. A hydrogen bond is measured between the carboxyl oxygen atom and the amide or hydroxyl hydrogen.
Halothane impact on the EC-TM interface and beyond
Interactions between the Cys-loop and the β1-β2 loop in the EC domain with the TM2-TM3 linker in the TM domain are widely regarded as a pathway to couple agonist binding to channel opening.56,57 Halo-2, halo-3, and halo-4, at the interface of EC and TM domains (Fig. 1), disrupted these interactions and could affect normal channel function. These three halothanes initially occupied analogous structural regions, but their final locations at the end of the 20-ns simulation varied depending on subunit types with which they were associated. Halo-2 in the β2-3 subunit moved ∼8 Å downward during the simulation and was later flanked by the Cys-loop and the TM2-TM3 linker. An analogous site in the Torpedo nAChR corresponding to halo-2 binding was identified in the halothane-photolabling experiment.23 As shown in Fig. 4, A and B, the gap between the Cys-loop and the TM2-TM3 linker was significantly widened due to the occupancy of halo-2. A previous mutagenesis study suggested that cis-trans isomerization of proline in the TM2-TM3 linker of the Torpedo nAChR accompanied the cascade from agonist binding to channel gating.58 However, recent X-ray structures showed that homologous prolines in both open-channel GLIC12,13 and closed-channel ELIC14 were in trans conformation. Homologous proline residues P271 and P270 in the TM2-TM3 linkers of the α4 and β2 subunits, respectively, were also in the trans conformation in both control and halothane systems. Interestingly, the dihedral angle (O–C–CA–N) of P138 in the β2-3 subunit changed from 0 to −100° in the presence of halo-2, as illustrated in Fig. 4C.
Figure 4.
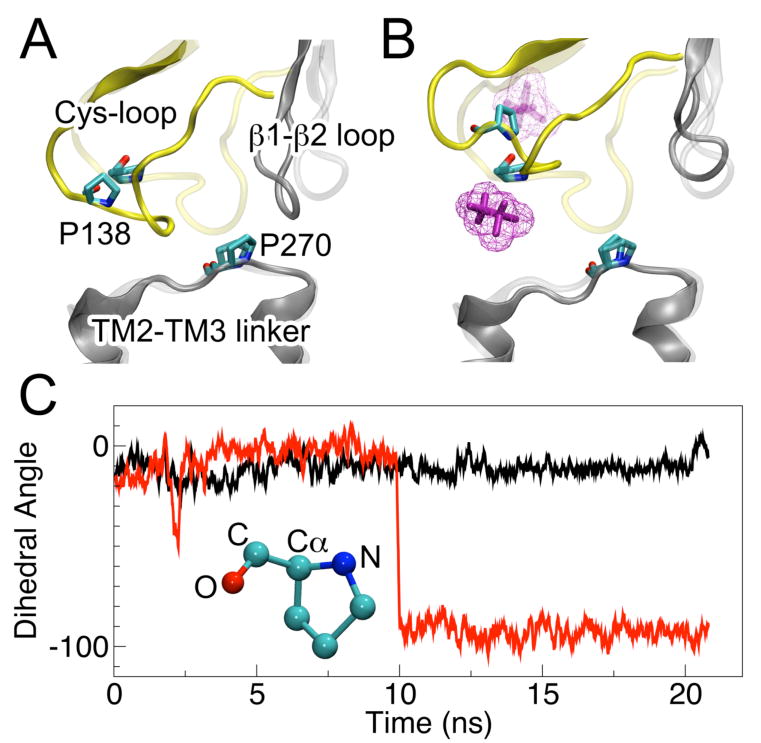
Conformation of the loops at the EC-TM interface of the β2-3 subunit at the beginning (transparent) and end (solid) of the 20-ns simulations in (A) the control system or (B) the halothane system. Halo-2 (magenta) moved from above to below the Cys-loop. The gap between the Cyc-loop and TM2-TM3 linker became wider in the halothane system. (C) Accompanying the movement of halo-2, the dihedral angle of P138 (O–C–Cα–N) in the control system (black) and halothane system (red) behaved differently.
Halo-3 and halo-4 were both located in α4 subunits (α4-2 and α4-1, respectively) and moved in a similar fashion over the course of the MD simulation, close to the tip of the β1-β2 loop. Compared to the control system, the gaps between the β1-β2 loop and the Cys-loop of these two subunits were enlarged by ∼5 Å and ∼2 Å by halo-3 and halo-4, respectively. The Cys-loop, at the interface of EC and TM domains, was proposed as the pivot point in the rotational motion of the TM domains during the channel opening.59 Allosteric effects due to halothane disturbance to the β1-β2 loop and the Cys-loop could alter salt bridges in distant regions of the protein. We found that the probability to form an inter-subunit E200-K79 salt bridge near the agonist binding site was an order of magnitude smaller in the halothane system than in the control system (Fig. S3). The loss of the E200-K79 salt bridge between the α4-1 and β2-3 subunits could result from not only the local halo-5, but also the remote halo-2 and halo-4. A stable E200-K79 salt bridge between the α4-2 and β2-2 subunits virtually disappeared in the presence of halo-3, even though no halothane was found near the salt bridge.
Halothane effect on lipid-protein interactions
Among all six halothanes, halo-1 had the lowest binding energy (−6.8 kcal/mol) in the α4β2 nAChR. Three main factors contribute to this high affinity: First, halo-1 was almost desolvated in its binding site in the TM domains between the α4-1 and β2-3 subunits. Second, its methyl fluoride formed a favorable electrostatic interaction with the side chain of S272. Third, its hydrogen, having a partial positive charge,34 formed a hydrogen bond with the phosphate oxygen of a nearby POPC lipid. In addition, there are other favorable electrostatic interactions between halo-1, the POPC, and the adjacent cholesterol (Fig. 5).
Figure 5.
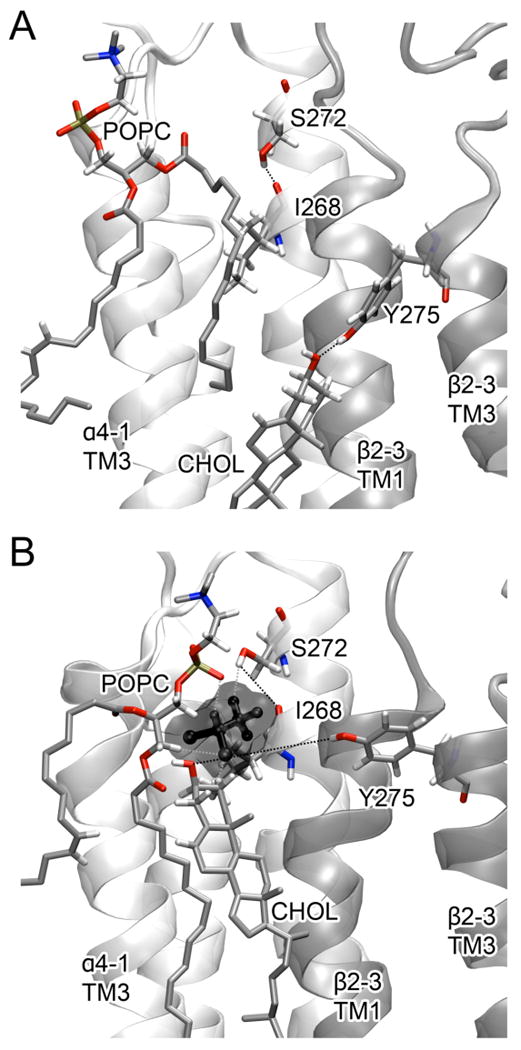
Effect of halo-1 (black) on lipid-protein interactions. (A) A cholesterol molecule (CHOL) formed a hydrogen bond with Y275 of the β2-3 subunit in the control system. (B) The hydrogen bond between CHOL and Y275 was broken in the halothane system. In addition to the interactions with POPC and CHOL, halo-1 interacts with S272 of the α4-1 subunit and weakens the hydrogen bond between S272 and I268.
The presence of halo-1 altered interactions within the protein and between the protein and lipids. The hydroxyl hydrogen atom of S272 formed a hydrogen bond with the carboxyl oxygen atom of I268 in the control system, but it interacted with halo-1 in the halothane system (Fig. 5). Y275 and a cholesterol molecule formed a hydrogen bond in the control system (Fig. 5A), but this hydrogen bond disappeared in the presence of halo-1 and the cholesterol presented a favorable electrostatic interaction with the nearby POPC (Fig. 5B). The interaction between Y275 and cholesterol molecules in the vicinity could contribute to anchoring the protein in the membrane and affect protein functions, considering that the absence of cholesterol impaired the function of the reconstituted nAChRs.60 The disruption to protein-lipid interactions by anesthetic molecules was also observed in previous experimental and computational studies.18,30 These data suggest the possibility that anesthetics could modulate protein functions by altering protein-lipid interactions.
Halothane effect on the α4β2 nAChR channel in the open conformation
Halothane had no apparent effect on overall structural stability of the α4β2 nAChR when measured by root mean-square deviation (RMSD) (Fig. S4). The hydrophobic girdle inside the channel, formed by residues from α4-V259 (β2-V253) to α4-L263 (β2-L257), remained to be the constriction region to water.32 The average minimum pore radius reduced from ∼3.7 Å to ∼ 3.2 Å over the course of the simulations in both the control and halothane systems (Fig. S5). A closer inspection, however, revealed differences in water distribution along the channel axis in the presence or absence of halothane. Figure 6 shows that, as the simulations proceeded, the water population increased inside the channel above the hydrophobic girdle by α4-L263 (β2-L257) at Z ∼ –27.5 Å, but reduced below the girdle in the halothane system, in comparison with that in the control system. It is interesting to note that the turning point of the positive and negative differences in water population between the two systems is located at the entrance of the hydrophobic girdle. The overall pattern of the difference in pore radius matches well with the difference in water distribution profile (Fig. 6), albeit some exceptions exist such as those at Z ∼ −50 Å. It is well known that dynamic and electrostatic factors, in addition to geometric factors, contribute to the channel hydration.32,61,62 Without significantly altering the pore radius, the change of local hydrophobicity due to side-chain reorientation can affect the water population in the pore.32,63 The water population was reduced by 17% at the hydrophobic girdle of the α4β2 nAChR in the halothane system. Although the observed lowering of water population should not be translated directly to the same degree of reduction in ion conductance, the association between water density in the pore and ion transport through channels has been well documented.61,64,65 It is conceivable that water depletion in the pore will affect ion permeation.
Figure 6.
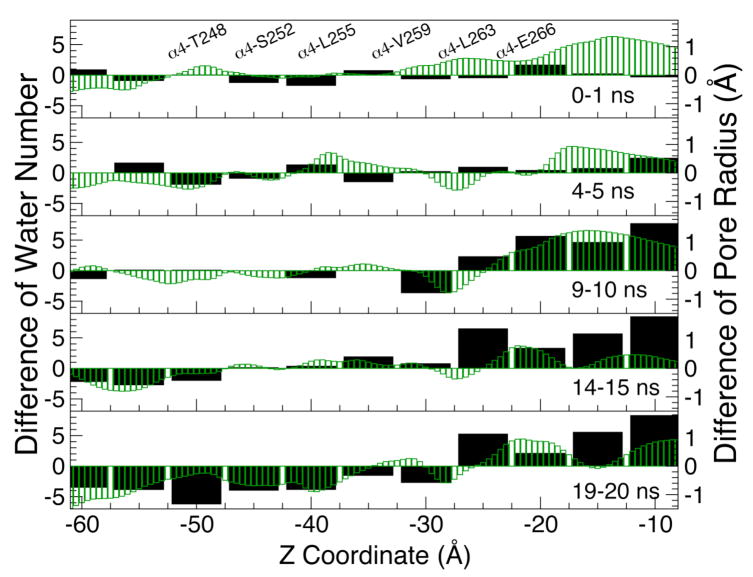
The difference between the halothane and control systems for the number of water molecules counted in 5-Å steps in the pore (wide black bars and the left vertical axis) and the pore radius profiles (thin green bars and the right vertical axis) along the channel axis (Z-coordinate. Z ∼ -10 Å: the EC/TM domain interface; Z ∼ −24 Å: the Cα atom of α4-E266). Each panel represents the average over 1 ns during the simulation. A positive value on the chart indicates a greater number of water molecules or larger pore radius in the halothane system than in the control system. The Cα positions of several pore lining residues are labeled in the top panel. The details of pore profiles are provided in Fig. S4 (Supporting Information).
Halothane modulation on mobility of the α4β2 nAChR
The modulation of α4β2 nAChR's mobility by halothane was evaluated on two different frequency scales using Cα root mean-square fluctuation (RMSF) calculations and GNM analysis.
We compared the Cα RMSF of the α4β2 nAChR in the control and halothane systems to understand how halothane affected the protein flexibility in various regions on a ps to ns timescale. The loop regions are intrinsically more flexible and likely more susceptible to halothane modulation. These loops are either involved in agonist binding (i.e., the C-loop), or required for signal transduction of channel gating (i.e., the β1-β2 loop, the F-loop, the Cys-loop, and the TM2-TM3 linker). Figure 7 shows the color-coded Cα RMSF difference between the control and halothane systems, for two neighboring subunits, α4-1 and β2-3, along with 4 halothane molecules. While the F-loop of the β2-3 subunit showed a depression in RMSF in the presence of halothane, the β1-β2 loop, the TM2-TM3 linker, and the Cys-loop showed an increased flexibility. More detailed analysis of the α4β2 nAChR RMSF data, in the presence or absence of halothane, is shown in Fig. S6.
Figure 7.
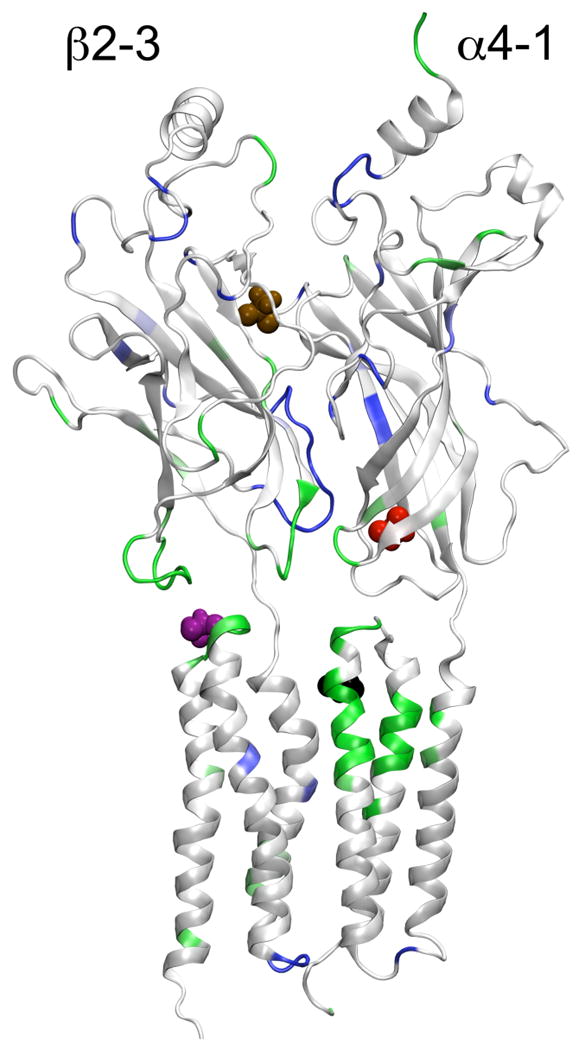
The side view from inside the channel of the α4-1 and β2-3 subunits in the presence of halo-1 (black), halo-2 (magenta), halo-4 (red), and hao-5 (brown) at the end of the 20-ns simulation. The structure of the α4-1 and β2-3 subunits is color-coded based on the Cα RMSF difference between the halothane and control systems: residues with reduced RMSF are blue, those with increased RMSF are green, and those with no significant change are white. The presence of halothane at the EC-TM interface increased the regional flexibility.
We captured halothane-induced changes in low frequency motions of the α4β2 nAChR via GNM analysis, which dissects the potential motions into a set of normal modes. The slowest GNM modes usually describe the collective motions relevant to the protein's biological functions.44,66-68 In this study, we chose the five lowest frequency modes to evaluate halothane's effects on the motions of the α4β2 nAChR. The input structures used for GNM analysis were taken from the end of the 20-ns MD simulations of the halothane and control systems. When GNM analysis was performed on individual subunits, halothane-induced changes in protein mobility were confined only within the subunits that contained halothane-binding sites. For example, the mobility of the β2-3 subunit, evaluated by the joint effect of lowest five modes, [(ΔRi)2]1-5, was affected significantly near the halothane binding sites. In contrast, the halothane-free β2-2 subunit had nearly identical [(ΔRi)2]1-5 in the control and the halothane systems (Fig. S7). The distinct differences between the halothane-bound β2-3 subunit and the halothane-free β2-2 subunit are reflected in the correlation maps in Fig. 8. The cross-correlations between fluctuations of residues i and j were calculated for individual β2-2 and β2-3 subunits in the absence or presence of halothane. As can be seen in Fig. 8, after adding halothane to the β2-3 subunit, the concerted motion was enhanced in the EC domain but reduced at the interface of the EC and TM domains, where the Cys-loop, the β1-β2 loop, and the TM2-TM3 linker meet. In contrast, the concerted motions of the β2-2 subunit were the same in the control and halothane systems.
Figure 8.

Cross correlation maps of the β2-2 (upper left corner) and β2-3 (lower right corner) subunits in the absence (A) or presence (B) of haltohane. The maps were generated based on GNM analysis of structures at the end of the 20-ns MD simulations. (C) The differences of cross correlation between the halothane and control systems.
Two generalizations can be made on the basis of GNM results of individual subunits. First, the halothane effects were encoded in the structures near the halothane binding sites after only 20-ns of MD simulation, as in the case of the β2-3 subunit. We would not be able to observe GNM differences if the structures were identical in the control and halothane systems, as in the case of the β2-2 subunit. Second, halothane altered the mobility of the protein near the halothane binding sites.
We also asked the question whether the halothane's modulation to protein motion is limited only to the residues adjacent to the binding sites, or it can extend to a global scale and affect the entire protein assembly. Our GNM analysis on the pentameric structures of the α4β2 nAChR in the control and halothane systems revealed that halothane's effect on protein mobility could extend to all of the subunits. Figure 9 shows the [(ΔRi)2]1-5 of all subunits in the α4β2 nAChR pentamer. It is clear that the β2-2 subunit experienced a substantial amount of changes in mobility even though there was no halothane directly bound to it. Apparently, halothane binding can modulate protein mobility not only locally but also globally.
Figure 9.

Mean-square fluctuations (MSF) of individual subunit in the control (black) or halothane (red) system. The data were generated using the five slowest normal modes of the GNM analysis of the pentameric structures at the end of the 20-ns simulations. The key structural features are labeled in the first and third panels.
Conclusions
More than one plausible halothane interaction site exists in the α4β2 nAChR. The majority of the halothane binding sites are located at the EC and TM interface. The apparent halothane disassociation constant, Kd, can be as low as 12 μM, but most halothane binding sites have a Kd in the low mM range. Despite the fact that halothane molecules bind at most sites with low affinity, they can nevertheless perturb the structure and motion of the α4β2 nAChR. In particular, the disturbance by these halothane molecules to the Cys-loop, the β1-β2 loop, and the TM2-TM3 linker at the EC and TM interface triggered changes in the correlated motions between the EC and TM domains, impeding the signal propagation from agonist binding in the EC domain to channel gating in the TM domain. Therefore, anesthetic binding with Kd values in the low mM range can modulate protein functions through altering motions critical to these functions.
It has been suggested28 that cooperative movement among the five subunits plays a crucial role in the function of the nAChR and other related proteins in the same superfamily. It is unknown until this study that all five subunits are required to transmit the effect of low-affinity halothane binding to a global scale, presumably via cooperative motion among these subunits. Our data on the α4β2 nAChR provides compelling evidence that a limited number of anesthetic interaction sites can produce a global effect on protein motion and consequently on protein function.
Halothane or other inhalation anesthetics might interact differently with the α4β2 nAChR in different conformations. The present study focused only on the α4β2 nAChR in the open-channel conformation. The investigation of halothane's effect on the α4β2 nAChR in the closed conformation is ongoing and will be reported elsewhere.
Supplementary Material
Acknowledgments
The authors thank Dr. Esmael J. Haddadian for helpful discussions, Dr. Jerome Henin and Dr. Troy Wymore for assisting with the FEP calculations, and Dr. Phil Blood for implementing the new FEP code at the Pittsburgh Supercomputing Center. This research is supported in part by the National Science Foundation through TeraGrid resources provided by the Pittsburgh Supercomputing Center. TeraGrid systems are hosted by Indiana University, LONI, NCAR, NCSA, NICS, ORNL, PSC, Purdue University, SDSC, TACC, and UC/ANL. This research is also supported by grants from the National Institutes of Health (R01GM066358, R01GM056257, and R37GM049202, and T32GM075770).
Footnotes
Supporting Information Available: Seven figures demonstrating the influence of halothane on the structure and dynamics of the α4β2 nAChR are included in the on-line supporting information. This information is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Flood P, Ramirez-Latorre J, Role L. Anesthesiology. 1997;86:859. doi: 10.1097/00000542-199704000-00016. [DOI] [PubMed] [Google Scholar]
- 2.Violet JM, Downie DL, Nakisa RC, Lieb WR, Franks NP. Anesthesiology. 1997;86:866. doi: 10.1097/00000542-199704000-00017. [DOI] [PubMed] [Google Scholar]
- 3.Cardoso RA, Yamakura T, Brozowski SJ, Chavez-Noriega LE, Harris RA. Anesthesiology. 1999;91:1370. doi: 10.1097/00000542-199911000-00029. [DOI] [PubMed] [Google Scholar]
- 4.Yamakura T, Harris RA. Anesthesiology. 2000;93:1095. doi: 10.1097/00000542-200010000-00034. [DOI] [PubMed] [Google Scholar]
- 5.Mori T, Zhao X, Zuo Y, Aistrup GL, Nishikawa K, Marszalec W, Yeh JZ, Narahashi T. Mol Pharmacol. 2001;59:732. doi: 10.1124/mol.59.4.732. [DOI] [PubMed] [Google Scholar]
- 6.Downie DL, Vicente-Agullo F, Campos-Caro A, Bushell TJ, Lieb WR, Franks NP. J Biol Chem. 2002;277:10367. doi: 10.1074/jbc.M107847200. [DOI] [PubMed] [Google Scholar]
- 7.Yamashita M, Mori T, Nagata K, Yeh JZ, Narahashi T. Anesthesiology. 2005;102:76. doi: 10.1097/00000542-200501000-00015. [DOI] [PubMed] [Google Scholar]
- 8.Yamakura T, Borghese C, Harris RA. J Biol Chem. 2000;275:40879. doi: 10.1074/jbc.M005771200. [DOI] [PubMed] [Google Scholar]
- 9.Liu R, Loll PJ, Eckenhoff RG. Faseb J. 2005;19:567. doi: 10.1096/fj.04-3171com. [DOI] [PubMed] [Google Scholar]
- 10.Bhattacharya AA, Curry S, Franks NP. J Biol Chem. 2000;275:38731. doi: 10.1074/jbc.M005460200. [DOI] [PubMed] [Google Scholar]
- 11.Franks NP, Jenkins A, Conti E, Lieb WR, Brick P. Biophys J. 1998;75:2205. doi: 10.1016/S0006-3495(98)77664-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hilf RJ, Dutzler R. Nature. 2009;457:115. doi: 10.1038/nature07461. [DOI] [PubMed] [Google Scholar]
- 13.Bocquet N, Nury H, Baaden M, Le Poupon C, Changeux JP, Delarue M, Corringer PJ. Nature. 2009;457:111. doi: 10.1038/nature07462. [DOI] [PubMed] [Google Scholar]
- 14.Hilf RJ, Dutzler R. Nature. 2008;452:375. doi: 10.1038/nature06717. [DOI] [PubMed] [Google Scholar]
- 15.Canlas CG, Cui T, Li L, Xu Y, Tang P. J Phys Chem B. 2008;112:14312. doi: 10.1021/jp805952w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cui T, Bondarenko V, Ma D, Canlas C, Brandon NR, Johansson JS, Xu Y, Tang P. Biophys J. 2008;94:4464. doi: 10.1529/biophysj.107.117853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bondarenko V, Yushmanov VE, Xu Y, Tang P. Biophys J. 2008;94:1681. doi: 10.1529/biophysj.107.116772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tang P, Simplaceanu V, Xu Y. Biophys J. 1999;76:2346. doi: 10.1016/S0006-3495(99)77391-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Xu Y, Seto T, Tang P, Firestone L. Biophys J. 2000;78:746. doi: 10.1016/S0006-3495(00)76632-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yushmanov VE, Xu Y, Tang P. Biochemistry. 2003;42:13058. doi: 10.1021/bi0350396. [DOI] [PubMed] [Google Scholar]
- 21.Bondarenko V, Xu Y, Tang P. Biophys J. 2007;92:1616. doi: 10.1529/biophysj.106.095364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Eckenhoff RG. Proc Natl Acad Sci U S A. 1996;93:2807. doi: 10.1073/pnas.93.7.2807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chiara DC, Dangott LJ, Eckenhoff RG, Cohen JB. Biochemistry. 2003;42:13457. doi: 10.1021/bi0351561. [DOI] [PubMed] [Google Scholar]
- 24.Ziebell MR, Nirthanan S, Husain SS, Miller KW, Cohen JB. J Biol Chem. 2004;279:17640. doi: 10.1074/jbc.M313886200. [DOI] [PubMed] [Google Scholar]
- 25.Nirthanan S, Garcia G, 3rd, Chiara DC, Husain SS, Cohen JB. J Biol Chem. 2008;283:22051. doi: 10.1074/jbc.M801332200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chiara DC, Hong FH, Arevalo E, Husain SS, Miller KW, Forman SA, Cohen JB. Mol Pharmacol. 2009;75:1084. doi: 10.1124/mol.108.054353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Szarecka A, Xu Y, Tang P. Proteins. 2007;68:948. doi: 10.1002/prot.21462. [DOI] [PubMed] [Google Scholar]
- 28.Purohit P, Mitra A, Auerbach A. Nature. 2007;446:930. doi: 10.1038/nature05721. [DOI] [PubMed] [Google Scholar]
- 29.Szarecka A, Xu Y, Tang P. Biophys J. 2007;93:1895. doi: 10.1529/biophysj.106.102780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tang P, Xu Y. Proc Natl Acad Sci U S A. 2002;99:16035. doi: 10.1073/pnas.252522299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yonkunas MJ, Xu Y, Tang P. Biophys J. 2005;89:2350. doi: 10.1529/biophysj.105.063396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Haddadian EJ, Cheng MH, Coalson RD, Xu Y, Tang P. J Phys Chem B. 2008;112:13981. doi: 10.1021/jp804868s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tang P, Zubryzcki I, Xu Y. J Comput Chem. 2001;22:436. [Google Scholar]
- 34.Liu Z, Xu Y, Saladino AC, Wymore T, Tang P. J Phys Chem A. 2004;108:781. [Google Scholar]
- 35.Campagna JA, Miller KW, Forman SA. N Engl J Med. 2003;348:2110. doi: 10.1056/NEJMra021261. [DOI] [PubMed] [Google Scholar]
- 36.Cheng MH, Liu LT, Saladino AC, Xu Y, Tang P. J Phys Chem B. 2007;111:14186. doi: 10.1021/jp075467b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, Chipot C, Skeel RD, Kale L, Schulten K. J Comput Chem. 2005;26:1781. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.MacKerell AD, Bashford D, Bellott, Dunbrack RL, Evanseck JD, Field MJ, Fischer S, Gao J, Guo H, Ha S, Joseph-McCarthy D, Kuchnir L, Kuczera K, Lau FTK, Mattos C, Michnick S, Ngo T, Nguyen DT, Prodhom B, Reiher WE, Roux B, Schlenkrich M, Smith JC, Stote R, Straub J, Watanabe M, Wiorkiewicz-Kuczera J, Yin D, Karplus M. J Phys Chem B. 1998;102:3586. doi: 10.1021/jp973084f. [DOI] [PubMed] [Google Scholar]
- 39.Morris GM, Goodsell DS, Halliday RS, Huey R, Hart WE, Belew RK, Olson AJ. J Comput Chem. 1998;19:1639. [Google Scholar]
- 40.Zacharias M, Straatsma TP, McCammon JA. The Journal of Chemical Physics. 1994;100:9025. [Google Scholar]
- 41.Chipot C, Pearlman DA. Molecular Simulation. 2002;28:1. [Google Scholar]
- 42.Smart OS, Neduvelil JG, Wang X, Wallace BA, Sansom MS. J Mol Graph. 1996;14:354. doi: 10.1016/s0263-7855(97)00009-x. [DOI] [PubMed] [Google Scholar]
- 43.Bahar I, Atilgan AR, Erman B. Fold Des. 1997;2:173. doi: 10.1016/S1359-0278(97)00024-2. [DOI] [PubMed] [Google Scholar]
- 44.Yang LW, Liu X, Jursa CJ, Holliman M, Rader AJ, Karimi HA, Bahar I. Bioinformatics. 2005;21:2978. doi: 10.1093/bioinformatics/bti469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yang LW, Rader AJ, Liu X, Jursa CJ, Chen SC, Karimi HA, Bahar I. Nucleic Acids Res. 2006;34:W24. doi: 10.1093/nar/gkl084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bellone M, Tang F, Milius R, Conti-Tronconi BM. J Immunol. 1989;143:3568. [PubMed] [Google Scholar]
- 47.Johansson JS, Manderson GA, Ramoni R, Grolli S, Eckenhoff RG. Febs J. 2005;272:573. doi: 10.1111/j.1742-4658.2004.04500.x. [DOI] [PubMed] [Google Scholar]
- 48.Xu Y, Tang P. Biochim Biophys Acta. 1997;1323:154. doi: 10.1016/s0005-2736(96)00184-8. [DOI] [PubMed] [Google Scholar]
- 49.Borghese CM, Ali DN, Bleck V, Harris RA. Alcohol Clin Exp Res. 2002;26:1764. doi: 10.1097/01.ALC.0000042012.58231.D9. [DOI] [PubMed] [Google Scholar]
- 50.Wenningmann I, Barann M, Vidal AM, Dilger JP. Mol Pharmacol. 2001;60:584. [PubMed] [Google Scholar]
- 51.Zhou QL, Zhou Q, Forman SA. Biochemistry. 2000;39:14920. doi: 10.1021/bi001281q. [DOI] [PubMed] [Google Scholar]
- 52.Chiara DC, Hong FH, Arevalo E, Husain SS, Miller KW, Forman SA, Cohen JB. Mol Pharmacol. 2009;75:1084. doi: 10.1124/mol.108.054353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sine SM, Engel AG. Nature. 2006;440:448. doi: 10.1038/nature04708. [DOI] [PubMed] [Google Scholar]
- 54.Xiu X, Puskar NL, Shanata JA, Lester HA, Dougherty DA. Nature. 2009;458:534. doi: 10.1038/nature07768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gleitsman KR, Kedrowski SM, Lester HA, Dougherty DA. J Biol Chem. 2008;283:35638. doi: 10.1074/jbc.M807226200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Xiu X, Hanek AP, Wang J, Lester HA, Dougherty DA. J Biol Chem. 2005;280:41655. doi: 10.1074/jbc.M508635200. [DOI] [PubMed] [Google Scholar]
- 57.Bouzat C, Gumilar F, Spitzmaul G, Wang HL, Rayes D, Hansen SB, Taylor P, Sine SM. Nature. 2004;430:896. doi: 10.1038/nature02753. [DOI] [PubMed] [Google Scholar]
- 58.Lummis SC, Beene DL, Lee LW, Lester HA, Broadhurst RW, Dougherty DA. Nature. 2005;438:248. doi: 10.1038/nature04130. [DOI] [PubMed] [Google Scholar]
- 59.Law RJ, Henchman RH, McCammon JA. Proc Natl Acad Sci U S A. 2005;102:6813. doi: 10.1073/pnas.0407739102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Dalziel AW, Rollins ES, McNamee MG. FEBS Lett. 1980;122:193. doi: 10.1016/0014-5793(80)80435-2. [DOI] [PubMed] [Google Scholar]
- 61.Hummer G, Rasaiah JC, Noworyta JP. Nature. 2001;414:188. doi: 10.1038/35102535. [DOI] [PubMed] [Google Scholar]
- 62.Ivanov I, Cheng X, Sine SM, McCammon JA. J Am Chem Soc. 2007;129:8217. doi: 10.1021/ja070778l. [DOI] [PubMed] [Google Scholar]
- 63.Saladino AC, Xu Y, Tang P. Biophys J. 2005;88:1009. doi: 10.1529/biophysj.104.053421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lynden-Bell RM, Jayendran CR. J Chem Phys. 1996;105:9266. [Google Scholar]
- 65.Beckstein O, Sansom MS. Phys Biol. 2004;1:42. doi: 10.1088/1478-3967/1/1/005. [DOI] [PubMed] [Google Scholar]
- 66.Konrad H, Gerald RK. J Chem Phys. 1999;111:10766. [Google Scholar]
- 67.Tama F, Sanejouand YH. Protein Eng. 2001;14:1. doi: 10.1093/protein/14.1.1. [DOI] [PubMed] [Google Scholar]
- 68.Ma J. Curr Protein Pept Sci. 2004;5:119. doi: 10.2174/1389203043486892. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.