

Abstract
A new database search algorithm has been developed to identify disulfide-linked peptides in tandem MS data sets. The algorithm is included in the newly developed tandem MS database search program, MassMatrix. The algorithm exploits the probabilistic scoring model in MassMatrix to achieve identification of disulfide bonds in proteins and peptides. Proteins and peptides with disulfide bonds can be identified with high confidence without chemical reduction or other derivatization. The approach was tested on peptide and protein standards with known disulfide bonds. All disulfide bonds in the standard set were identified by MassMatrix. The algorithm was further tested on bovine pancreatic ribonuclease A (RNaseA). The 4 native disulfide bonds in RNaseA were detected by MassMatrix with multiple validated peptide matches for each disulfide bond with high statistical scores. Fifteen nonnative disulfide bonds were also observed in the protein digest under basic conditions (pH = 8.0) due to disulfide bond interchange. After minimizing the disulfide bond interchange (pH = 6.0) during digestion, only one nonnative disulfide bond was observed. The MassMatrix algorithm offers an additional approach for the discovery of disulfide bond from tandem mass spectrometry data.
Keywords: disulfide linkage, tandem mass spectrometry, database searching, proteomics, post-translational modification
Introduction
Among the 20 natural amino acids, cysteine is unique because it is involved in many biological activities through oxidation and reduction to form disulfide bonds and sulfhydryls. These modifications play an important role in protein's structure and biological function. The identification and characterization of protein disulfide bonds is an essential step to thoroughly understand their biological function. However, the determination of disulfide linkages can be a challenging task.
Several methods are used to determine the pattern of disulfide bond linkage. Among them, crystallography and NMR are excellent tools that can identify disulfide bond linkages with minimal disulfide bond interexchange.1–6 However, the application of both techniques is limited by large sample requirements and protein size. Edman Degradation can also be used for the identification of disulfide bonds.7–10 But this technique requires ultra pure samples, digestion, and further purification of the peptides for protein sequencing. Mass spectrometry has also been successfully applied to identify disulfide bonds in solution8,11–13 and from gels.14,15 Researchers have successfully demonstrated that disulfide bridge patterns can be identified by mas spectrometry (MS) analysis, following protein digestion either under partial reduction12,13,16,17 or nonreduction conditions.8,11,14,15,18–24
Partial reduction is a widely accepted approach for the determination of disulfide bonds. In this approach, the protein is digested under controlled conditions such that disulfide bonds with different reduction kinetics are reduced and alkylated gradually. Peptide containing modified cysteines are further separated and analyzed by MS and/or tandem MS. However, when a protein is highly bridged, multiple reductions and separations are necessary for complete disulfide mapping. The approach requires relatively large sample amounts compared with direct tandem MS and can provide ambiguous results when disulfide bonds have the same or similar reduction rates.16 Tandem MS analysis of protein digests under nonreducing condition has also been used frequently for the investigation of disulfide bond patterns. With appropriate enzyme(s) digestion, disulfide bonds pattern can be identified in a single run. However, data processing may be extremely complicated and time/labor-consuming for proteins with multiple unknown disulfide bonds. Also, when the sample contains large number of cysteines or multiple disulfide bonds exist in the sample, some disulfide bond linkages might go unidentified. Tools that can effectively search tandem MS data for the presence of disulfide bonds will facilitate disulfide analysis.
Several methods have been reported for peptide mass analysis for disulfide-linked peptides.25,26 Tandem MS data from disulfide-linked peptides under reduction conditions can be analyzed using traditional database search programs.27–29 However, high-throughput software for analysis of tandem MS data of disulfide-linked proteins and peptides under nonreducing condition are limited. Recently, a search program for the identification of disulfide bonds in peptides from tandem MS data was reported by Wefing et al.30 However, the program lacks a calibrated empirical scoring model or a statistical scoring model. Commonly used database search program, such as SEQUEST28 and Mascot,29 do not have options to perform analysis of tandem MS data from disulfide-linked proteins and peptides.
Herein, we report a new data search algorithm for tandem MS data to identify disulfide bonds in proteins and peptides. The algorithm was included in a newly developed tandem MS database search program, MassMatrix.31 The algorithm employs a probabilistic scoring model and fast database searching algorithm in MassMatrix to achieve reliable statistical scoring, high sensitivity, and selectivity and high-performance identification of disulfide bonds from proteins and peptides. With this approach, disulfide bonds in proteins and peptides can be identified along with other fixed and/or variable modifications in tandem mass spectrometry and without the need for reduction or derivatization of the sulfhydryls and/or disulfide bonds.
Experimental Procedures
Sample Preparation and Mass Spectrometry
All peptide and protein standards were purchased from Sigma-Aldrich (St. Louis, MO). The protein standards were digested by a combination of trypsin (Promega, Madison, WI) and chymotrypsin (Roche, Switzerland) following a protocol similar to Ressell et al.32 Briefly, protein standards were digested in 25 mM ammonium bicarbonate solution (pH = 8.0). To minimize/block the disulfide bond interchange, protein standards were also digested in 25 mM phosphate buffer (pH = 6.0) or 25 mM ammonium bicarbonate with 10 mM iodoacetamide (final concentration).24 Enzymes were used in 25:1 ratio (substrate/enzyme), and the mixture was incubated at 37 °C overnight.
Capillary-liquid chromatography-nanospray tandem mass spectrometry (Nano-LC/MS/MS) was performed on a Thermo Finnigan LTQ mass spectrometer equipped with a nanospray source operated in positive ion mode. The LC system was an UltiMate Plus (Dionex, Sunnyvale, CA) with a FAMOS autosampler and SWITCHOS column switcher. Solvent A contained water with 50 mM acetic acid, and solvent B contained acetonitrile. Five microliters of each sample was injected onto the trapping column (LC-Packings A Dionex Co, Sunnyvale, CA) and washed with solvent A. The injector port was switched to inject, and the peptides were eluted off of the trap onto a 5 cm, 75 μm i.d. ProteoPep II C18 packed nanospray tip (New Objective, Inc. Woburn, MA). Peptides were eluted into the LTQ system using a gradient of 2–80% B over 30 min, with a flow rate of 300 nL/min. The total run time was 58 min. The scan sequence of the mass spectrometer was programmed to perform a full scan followed by 10 data-dependent MS/MS scans of the most abundant peaks in the spectrum. Dynamic exclusion was used to exclude multiple MS/MS of the same peptide.
Database Searching and Searching Parameters
The .RAW data files obtained from the mass spectrometer were converted to mzData (.XML) files using BioWorks (ThermoFisher, San Jose, CA). The mzData files were searched against custom protein databases using the MassMatrix search engine (www.massmatrix.net). The custom FASTA format protein databases were composed of target protein sequences and decoy protein sequences. Target protein sequences for bovine pancreatic ribonucleaseA (RNaseA) and Insulin were reported previously.33,34 Decoy protein sequences were reversed or randomized sequence of RNaseA or Insulin. All spectra that were not determined as singly charged were searched as both doubly and triply charged ions. Peptide sequences with a length from 6 to 50 amino acid (AA) residues, missed cleavage sites of up to 4, and charges of +1, +2, and +3 were searched. For spectra with multiple matches, the highest scoring match was used. The program was tested with peptide and protein standards and with the well-characterized protein, RNaseA, which contains four known native disulfide bonds.
Results and Discussion
Classification of Disulfide Bonds and Disulfide-Linked Peptides
In MassMatrix, disulfide bonds in peptides were classified into two types: interchain disulfide bonds and intrachain disulfide bonds. Peptides with more than 2 disulfide bonds are difficult to characterize by tandem MS due to poor fragmentation and size. Therefore, only peptides with up to 2 disulfide bonds were considered in MassMatrix. However, models for peptides with more than 2 disulfide bonds may be included in MassMatrix as needed. Peptides with up to 2 disulfide bonds were classified into 4 types as shown in Figure 1. Type 1 peptides only have interchain disulfide bonds. Type 2 peptides only have intrachain disulfide bonds. Type 3 peptides are hybrids with both inter- and intrachain disulfide bonds. Type 4 peptides have circular chains. Peptides with only 1 disulfide bond fall into the following classes: 1A and 2A. Peptides with 2 disulfide bonds include types 1B, 2B, 3, and 4.
Figure 1.
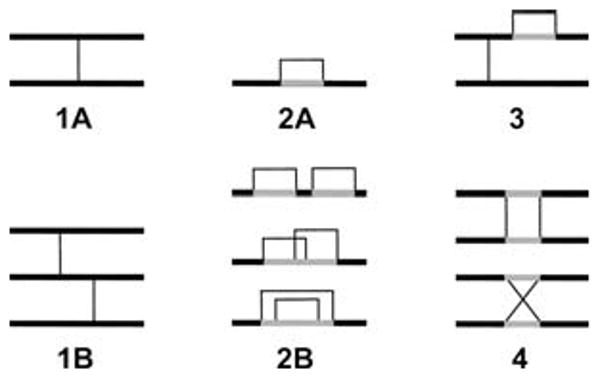
Classification of disulfide-linked peptides in MassMatrix.
Search Algorithm
A flowchart for the disulfide search algorithm in MassMatrix is shown in Figure 2. MassMatrix has two disulfide search modes: exploratory and confirmatory. There are no biological or chemical feasibility for disulfide bonds assumed in either the exploratory or confirmatory mode. In the exploratory search mode, all cysteine residues in the protein sequences are considered to be variable disulfide bonding sites, that is, all cysteine may or may not form disulfide bonds. During searching, MassMatrix will generate all possible combinations of disulfide bonds by assuming that any two cysteine residues are capable of forming a disulfide bond. Consider a protein with n cysteine residues. During exploratory search mode, MassMatrix will generate possible combinations of single disulfide bond for the protein (n = number of cysteine residues). In the confirmatory search mode, only the disulfide bonds specified in the protein database by the user will be considered and searched against experimental data. Disulfide bonds are specified in the sequence by input of a custom database. In the special .BAS MassMatrix databases, disulfide bonds are coded as “C($i)”, where i is the index number of the specified disulfide bond. Each bond has two related disulfide bonding sites. The protein is digested in silico based on the specified proteolysis reagent with consideration given to the presence of disulfide bonds generated from either the exploratory search mode or as input by the user in confirmatory search mode. These hypothetical peptides are then fragmented using the appropriate fragmentation model and scored against the experimental tandem MS data.
Figure 2.
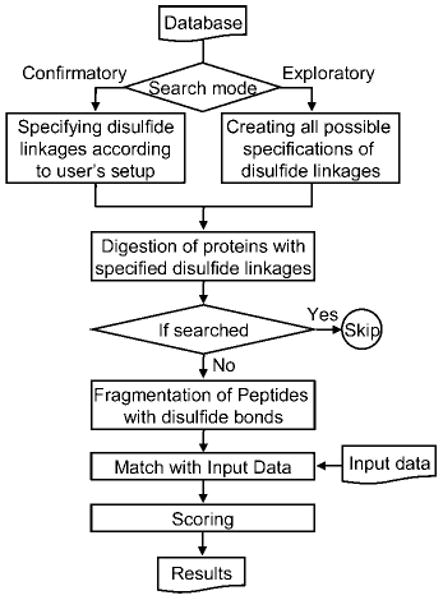
The flow diagram for the search algorithm of disulfide-linked peptides in MassMatrix.
In MassMatrix's Collision Induced Dissociation fragmentation model, disulfide bonds are not cleaved by collisional activation. Disulfide-linked peptides may have multiple chains that are connected to each other. Each chain may undergo fragmentation independently including neutral losses of H2O and NH3 due to residues with hydroxyl and amino groups, respectively. Only product ions created from the rupture of a single bond were considered (thus, internal fragments were not searched). Therefore, when one chain undergoes fragmentation to create product ions, other chain(s), if any, will be intact and considered as a modification to the cysteine on the first chain.
Type 1A peptides are the most preferred configuration for sequencing by tandem mass spectrometry. Type 1A peptide fragmentation produces a set of product ions for each peptide chain allowing excellent sequence coverage. The same is true for type 1B given that the peptide chains are of reasonable length so that the precursor and product ions can be detected. Other peptide types can suffer from lower sequence coverage due to blind spots in the product ion series. For example, fragmentation of type 2 and 3 peptides that occurs in the sequence connecting the two linked sulfides results in the same product ion and, thus, no useful sequence information. We refer to this region of the peptide (gray area in Figure 1) as the blind spot. Similarly, for type 4 peptides, fragmentation between the two interchain disulfide bonds on the same chain does not create any signature product ions. Resulting tandem MS spectra for type 2, 3, and 4 peptides will be missing product ions for these blind areas. At present, the best strategy is to use a protease to cut these peptide sequences and convert them to type 1A. Alternatively, we are examining the use of data-dependent MS3 to sequence these blind areas.
The scoring algorithm involved in the disulfide bond search is the same as that used for peptides without disulfide bonds.31 MassMatrix contains three independent scoring models, including two statistical models and a descriptive model. The two statistical scores, pp and pp2, are the negative common logarithm of the likelihood that the peptide match is random. The pp value is the major standard used in MassMatrix, and the pp2 value is a supplementary score. Peptide matches with either pp or pp2 value greater than 6 were considered to be statistically significant.31
Most database search programs (MassMatrix,31 SEQUEST,28 Mascot,29 etc.) assume a simple fragmentation pattern for the tandem mass spectra of theoretically calculated peptide sequences. Furthermore, there are no methods to predict relative abundances of products ions from experimental spectra of disulfide-linked peptides. Therefore, manual validation is necessary to obtain truly reliable disulfide-linked peptide identifications from shotgun proteomic experiments performed at low mass accuracy. MassMatrix provides information in the search results, such as statistical scores, graphical tandem mass spectra with highlighted matched ions, theoretical product ion tables, and so forth, to allow the user to visually validate each disulfide-linked peptide match (see Supplementary Figure 1 in Supporting Information).
Validation with Disulfide-Linked Peptide Standards
The search algorithm was first tested with CID MS/MS data from two peptide standards and an insulin digest containing known disulfide bonds. The peptide standards were type 2A and 2B peptides and analyzed directly by nanoLC—MS/MS without any digestion. The data from the peptide standards were searched without specifying any peptide cleavages due to digestion. Insulin consists of two chains connected by two interchain disulfide bonds, and one chain also has an intrachain disulfide bond. Digestion of insulin by a combination of trypsin and chymotrypsin creates two types of disulfide-linked peptides: type 1A and type 3 peptides. Each of these four types was successfully identified by searching their tandem MS data with the MassMatrix search engine. A listing of the peptide matches is provided in Table 1. Because type 1A peptides have a higher potential to return a complete set of signature product ions created from fragmentation, they normally have relatively higher pp values (Figure 3a). Type 2 peptides (Figure 3b,c) contain blind spots and are more difficult to identify in tandem MS database search especially when a large part of their sequences is contained in the blind spot. This is manifest in lower pp scores as evident in the search of the type 2B peptide (Figure 3c) listed in the Table 1. Fragmentation that occurs in the blind spot creates product ions with the same mass as the precursor ion. Therefore, we normally observe an abundant peak with the same mass-to-charge ratio (m/z) as the precursor ion in the tandem MS spectra for type 2 peptides as shown in Figure 3b,c. We are exploring the option of additional MS3 experiment to further map and identify these peptides. Type 3 peptides are hybrid where one chain has an intrachain disulfide bond and a blind area linked via an interchain disulfide to a separate peptide (Figure 3d). Because these peptides partially resemble type 1A, they can still be identified with significant scores in MassMatrix (Figure 3d).
Table 1.
Four Types of Standard Disulfide-Linked Peptides Identified in MassMatrixa
| Peptides | Type | High pp value | Source |
|---|---|---|---|
 |
1A | 17.0 | Digest of Insulin |
| 1A | 18.8 | Digest of Insulin | |
| 1A | 12.2 | Digest of Insulin | |
| 2A | 8.9 | Peptide standard 1 | |
| 2B | 3.4* | Peptide standard 2 | |
| 3 | 11.2 | Digest of Insulin |
Asterisk (*): the match for type 2B standard peptide is considered significant by MassMatrix due to its significant pp2 value which is 9.5.
Figure 3.
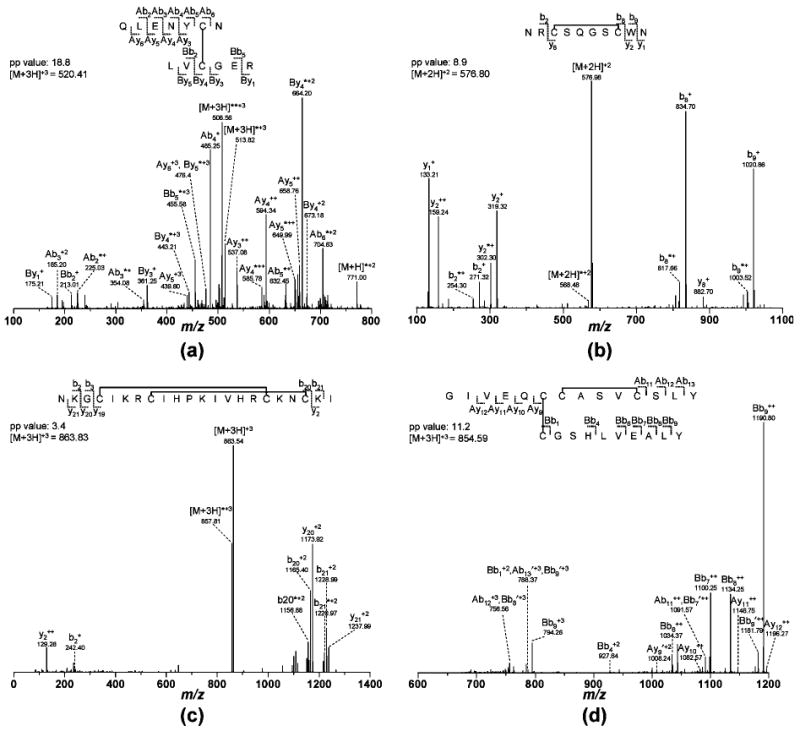
Disulfide-linked peptide matches for (a) type 1A, (b) type 2A, (c) type 2B, and (d) type 3 peptides. Ions with neutral loss of small molecules are labeled by asterisk (*, loss of ammonia) and prime (′, loss of water).
Validation with Bovine Pancreatic Ribonuclease A (RNaseA)
The tandem MS data sets from the digests of RNaseA by a combination of chymotrypsin and trypsin under basic (pH = 8.0) and acidic (pH = 6.0) conditions were searched against the RNaseA sequence using either the confirmatory or exploratory search modes. For the exploratory search, decoy sequences were added to a limited database that contained RNaseA, the reversed RNaseA sequence, and 20 randomized RNaseA sequences. The summary of the searches for the data set from the digest under pH = 8.0 are listed in Table 2. Because the search space for the decoy sequences was much larger than that for the true RNaseA, sequence peptide matches to the true sequence were considered true positives (TPs) and those from decoy sequences were considered false positives (FPs). The true positive peptide matches from RNaseA were further manually validated by two experts independently, and those that passed both manual validations were used to map the disulfide bonds of RNaseA. Manual validation of the search results was carried out in order to eliminate matches to spectra of poor quality (low S/N) or matches that did not yield any consecutive y or b ions. For RNaseA at pH = 8.0, 281 out of 302 (93.0%) TPs survived this manual validation, whereas at pH = 6.0, 102 out of 113 (90.3%) TPs survived validation. The complete lists of disulfide-linked peptide matches for the two data sets are provided in the Supplementary Tables 1 and 2 in Supporting Information. Spectra for matched peaks and a table of theoretical masses calculated from the MassMatrix fragmentation model for each validated true positive peptide match are provided in the Supplementary Figure 1 in Supporting Information.
Table 2.
Summary of the Three Searches for the Tandem MS Data Sets from RNaseA
| database | search mode | theoretical peptides calculated | search time |
|---|---|---|---|
| 1 RNaseA | Confirmatory | 971 | 23 s |
| 2 RNaseA | Exploratory | 9648 | 28 s |
| 3 RNaseA and decoys | Exploratory | 232481 | 62 s |
1. Confirmatory Search for Native Disulfide Bonds
Four nativedisulfidebondsinRNaseA,C26—C84,C40—C95,C58—C110, and C65—C72 were specified in the confirmatory search.33–35 The summary report of the four native disulfide bonds is shown in Table 3. For the RNaseA digests under both basic and acidic conditions, each native disulfide bond was confirmed by multiple validated true peptide matches with significant statistical scores. More native disulfide-linked peptides and higher total statistical scores for native disulfide bonds were observed in basic digestion because the optimal pH for trypsin digestion is pH ∼ 8.0. Therefore, all four native bonds reported by literature were confirmed with a very high confidence in our tandem MS experiment and subsequent database search performed in MassMatrix.
Table 3.
Native Disulfide-Linked Peptides in Digest of RNaseA under Basic and Acidic Conditions Identified in the Confirmatory Searcha
| digestion condition | disulfide bond | number of matches | pp value sum/high | disulfide-linked peptides |
|---|---|---|---|---|
| pH = 8.0 | C26—C84 | 41 | 554.9/21.9 | [26–29]-S-S-[80–85] |
| [26–30]-S-S-[80–85] | ||||
| [26–31]-S-S-[80–85] | ||||
| [26–31]-S-S-[77–85] | ||||
| C40—C95 | 74 | 629.3/18.9 | [40–46]-S-S-[93–97] | |
| [40–46]-S-S-[92–97] | ||||
| [40–46]-S-S-[92–98] | ||||
| C58—C110 | 10 | 112.7/17.2 | [47–61]-S-S-[105–115] | |
| C65—C72 | 58 | 807.6/32.9 | [62–73]* | |
| [62–76]* | ||||
| [62–66]-S-S-[67–73] | ||||
| [62–66]-S-S-[67–76] | ||||
| pH = 6.0 | C26—C84 | 31 | 431.6/20.5 | [26–29]-S-S-[80–85] |
| [26–30]-S-S-[80–85] | ||||
| [26–31]-S-S-[80–85] | ||||
| C40—C95 | 46 | 386.4/15.4 | [40–46]-S-S-[93–97] | |
| [40–46]-S-S-[92–97] | ||||
| [40–46]-S-S-[92–98] | ||||
| [38–46]-S-S-[92–97] | ||||
| C58—C110 | 11 | 129.8/17.5 | [52–61]-S-S-[105–115] | |
| [47–61]-S-S-[105–115] | ||||
| C65—C72 | 6 | 79.3/21.9 | [62–73]* | |
| [62–66]-S-S-[67–73] | ||||
| [62–66]-S-S-[67–76] |
Asterisk (*): peptides with intrachain disulfide bond.
2. Exploratory Search for Native and Nonnative Disulfide Bonds
In the exploratory mode, MassMatrix searched the complete combination of all possible disulfide bonds in the protein provided by the model. All native disulfide-linked peptides that were seen in the confirmatory search were also seen in the exploratory search for both basic and acidic digests.
For the RNaseA digest under basic condition, 18 nonnative disulfide-linked peptides were observed with multiple validated true positive matches for each peptide in the exploratory search (Table 4). These peptides indicate that there are 15 nonnative disulfide bonds in the RNaseA. The summary of disulfide bond mapping in the RNaseA digests under basic conditions is shown in Figure 4a. Out of all 28 possible disulfide bonds formed by the eight cysteine residues in the RNaseA, 19 were identified in MassMatrix. These nonnative disulfide bonds are likely due to disulfide bond interchange during the sample processing and protein digestion.24,36
Table 4.
Nonnative Disulfide-Linked Peptides in Digest of RNaseA under Basic and Acidic Conditions Identified in the Exploratory Search
| digestion condition | disulfide bond | number of matches | pp value sum/high | disulfide-linked peptides |
|---|---|---|---|---|
| pH = 8.0 | C26–40 | 7 | 69.3/12.6 | [26–31]-S-S-[40–46] |
| C26—C95 | 8 | 89.7/13.5 | [26–31]-S-S-[92–97] | |
| C26—C110 | 8 | 32.4/6.3 | [26–31]-S-S-[105–115] | |
| C40—C58 | 2 | 13.1/6.7 | [40–46]-S-S-[47–61] | |
| C40—C65 | 8 | 107.8/18.5 | [40–46]-S-S-[62–66] | |
| C40—C84 | 3 | 27.8/11.7 | [40–46]-S-S-[80–85] | |
| C40—C110 | 10 | 51.8/6.3 | [40–46]-S-S-[105–115] | |
| C58—C65 | 2 | 10.4/5.2 | [47–61]-S-S-[62–66] | |
| C58—C95 | 2 | 25.7/13.7 | [47–61]-S-S-[92–97] | |
| C65—C84 | 3 | 48.6/16.7 | [62–66]-S-S-[80–85] | |
| C65—C95 | 5 | 56.4/13.7 | [62–66]-S-S-[92–97] | |
| C65—C110 | 4 | 36.3/11.0 | [62–66]-S-S-[105–115] | |
| C84—C95 | 28 | 286.2/16.4 | [80–85]-S-S-[93–98] | |
| [80–85]-S-S-[92–97] | ||||
| [80–85]-S-S-[92–98] | ||||
| C84—C110 | 4 | 38.1/11.4 | [80–85]-S-S-[105–115] | |
| C95—C110 | 4 | 11.6/3.5 | [92–97]-S-S-[105–115] | |
| [92–98]-S-S-[105–115] | ||||
| pH = 6.0 | C84—C95 | 8 | 121.4/20.0 | [80–85]-S-S-[92–97] |
| [80–85]-S-S-[92–98] |
Figure 4.
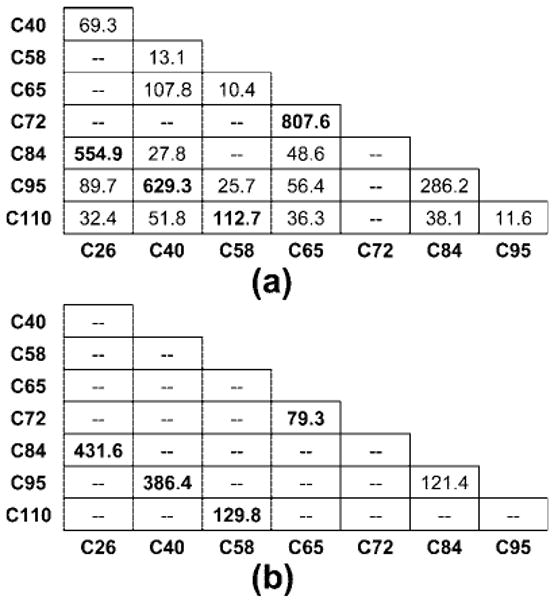
Disulfide bonds mapped by tandem MS experiment and subsequent database search in MassMatrix for digests under (a) basic and (b) acidic conditions. Confidence of the identification for each bond is indicated by the sum of pp values of peptide matches with the bond. Native disulfide bonds are bolded.
Compared with the RNaseA digest under basic condition, similar results for the four native disulfide bonds were observed in the RNaseA digest under acidic condition as shown in Table 3. However, only one validated nonnative disulfide bond (C84—C95) was observed Figure 4b and Table 4. This could be due to the fact that the disulfide bond interchange was minimized by dilute acid.24 These observations were further supported by blocking disulfide interchange by treatment with iodoacetamide and enhancing interchange by heating during digestion (see Supplementary Figure 2 in Suppporting Information).
Validation of MassMatrix Scoring Model for Disulfide-Linked Peptides
The scoring model for disulfide-linked peptides was validated by examination of the distributions of pp values for true positives and false positives (Figure 5) and receiver operating characteristic (ROC) analysis (Figure 6) for the data set from the digest under pH = 8.0. True and false positives were determined from the searches of the RNaseA data sets with decoy sequences. Because the search space for the decoy sequences was much larger than that for the true RNaseA, sequence peptide matches to the true sequence were considered true positives (TPs) and those from decoy sequences were considered false positives (FPs). True positive rate (TPR) and false positive rate (FPR) are calculated by TP/(TP + FN) and FP/(FP + TN), respectively, for a certain threshold, where FN is number of false negatives (TPs under the threshold) and TN is number of true negatives (FPs under the threshold).
Figure 5.
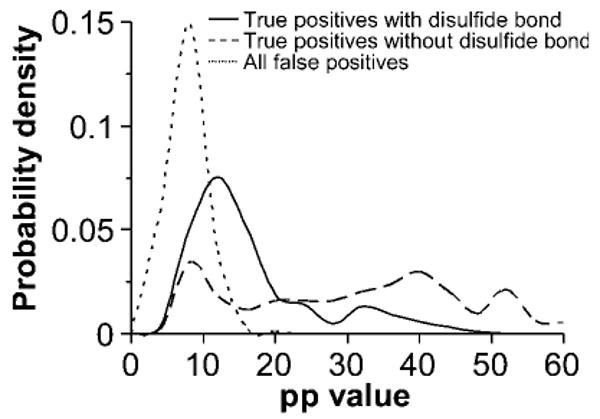
Distribution of pp values for peptides without disulfide bonds and disulfide-linked peptides, compared with false positive peptide matches.
Figure 6.
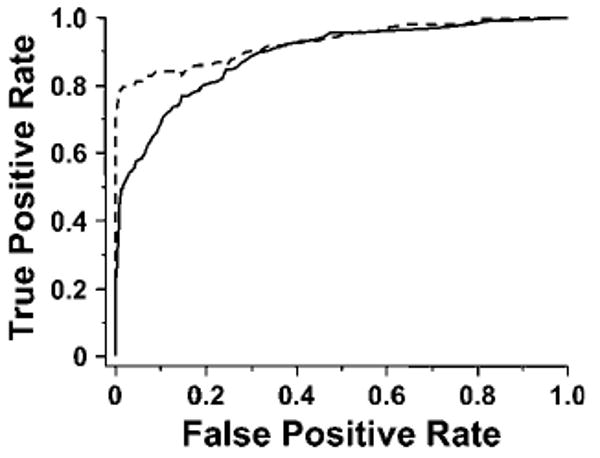
ROC curves of the search with decoy sequences. Solid line (−), true positive peptide matches with disulfide bonds vs false positives; dashed line (---), true positive peptides without disulfide bonds vs false positives. False positives include both disulfide-linked false peptides and false peptides without disulfide bonds. In ROC curves, a value to the top indicates higher sensitivity, and a value to the left indicates higher specificity.
It can be seen that the scoring model was able to discriminate TPs from FPs at a low mass accuracy of 1.0 Da, although there is some overlap between the distributions of pp value for TPs and FPs. ROC analysis also indicates that the scoring performs well for both peptides without peptide bonds and disulfide-linked peptides. However, false positives are a permanent issue for database searches at low mass accuracy.27 The appropriate threshold values in MassMatrix can be determined experimentally by using standard experimental data sets with searches against databases containing decoy sequences as described above.37 By setting up appropriate threshold scores based on ROC analysis, the algorithm achieved a TPR, that is, sensitivity, of 85% for peptides without disulfide bonds and 71% for disulfide-linked peptides with a FPR of 10%, that is, a specificity of 90%.
Disulfide-Linked Peptides versus Peptides without Disulfide Bonds
Disulfide-linked peptides tend to have smaller pp values than those without disulfide bonds in MassMatrix as shown in Figure 5. Area under the curve (AUC) for the ROC curves indicates sensitivity and specificity. An ideal algorithm with an AUC equal to 1.0 achieves 100% sensitivity and 100% specificity. It can be seen in Figure 6 that the AUCs for peptides without disulfide bonds and those with disulfide bonds are 0.93 and 0.89, respectively. Therefore, MassMatrix has better sensitivity and specificity for peptides without disulfide bonds than for disulfide-linked peptides as indicated by score distributions and ROC analysis. This is because (1) peptides with intrachain disulfide bonds often do not contain the complete set of signature product ions due to blind spots and (2) peptides with interchain disulfide have product ions clustered at low and high m/z. However, peptides without any disulfide bonds have product ions spread across the whole mass range resulting in a better signature.
Conclusions
A new database search algorithm has been developed to identify disulfide-linked peptides in tandem MS data sets. The algorithm was based on the statistical scoring model in MassMatrix and has been incorporated in MassMatrix to enable the program to map disulfide bonds form tandem MS data. The algorithm was tested on peptide and protein standards with known disulfide bonds. All disulfide bonds in the standard set were identified with high statistical scores by MassMatrix. The algorithm was further tested on bovine pancreatic ribonuclease A (RNaseA). The 4 native disulfide bonds in RNaseA were detected by MassMatrix with multiple validated peptide matches for each bond and high statistical scores. Fifteen nonnative disulfide bonds were also observed in the digest under basic conditions (pH = 8.0) which was mainly due to random disulfide bond interchange during digestion. After minimizing the disulfide bond interchange by dilute acid (pH = 6.0) during digestion, only one nonnative disulfide bond was observed. The distribution of statistical scores for true and false positives and ROC analysis indicate that the algorithm is capable to discriminate TPs with disulfide bonds from FPs.
Supplementary Material
Acknowledgments
The study was funded by the Ohio State University, the National Institutes of Health CA107106, the V Foundation/American Association for Cancer Research Translational Cancer Research Grant, and the Leukemia & Lymphoma Society. The authors thank Dr. Kari B. Green-Church and Ms. Nan Kleinholz at the Mass Spectrometry and Proteomics Facility for the assistance of mass spectrometric experiments.
Footnotes
Supporting Information Available: Supplementary Figure 1 provides tandem mass spectra for matched peaks and a table of theoretical masses calculated from the MassMatrix fragmentation model for each validated true disulfide-linked peptide match. Disulfide bonds for RNaseA mapped by tandem MS experiments and subsequent database search in MassMatrix under different conditions are provided in Supplementary Figure 2. The complete lists of disulfide-linked peptide matches for the results of RNaseA digests at pH = 8.0 and pH = 6.0 are listed in Supplementary Tables 1 and 2. This information is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Collins ES, Wirmer J, Hirai K, Tachibana H, Segawa S, Dobson CM, Schwalbe H. Characterisation of disulfide-bond dynamics in non-native states of lysozyme and its disulfide deletion mutants by NMR. ChemBioChem. 2005;6(9):1619–1627. doi: 10.1002/cbic.200500196. [DOI] [PubMed] [Google Scholar]
- 2.Meier S, Haussinger D, Pokidysheva E, Bachinger HP, Grzesiek S. Determination of a high-precision NMR structure of the minicollagen cysteine rich domain from Hydra and characterization of its disulfide bond formation. FEBS Lett. 2004;569(1–3):112–116. doi: 10.1016/j.febslet.2004.05.034. [DOI] [PubMed] [Google Scholar]
- 3.Sharma D, Rajarathnam K. 13C NMR chemical shifts can predict disulfide bond formation. J Biomol NMR. 2000;18(2):165–171. doi: 10.1023/a:1008398416292. [DOI] [PubMed] [Google Scholar]
- 4.von Ossowski L, Tossavainen H, von Ossowski I, Cai C, Aitio O, Fredriksson K, Permi P, Annila A, Keinanen K. Peptide binding and NMR analysis of the interaction between SAP97 PDZ2 and GluR-A: potential involvement of a disulfide bond. Biochemistry. 2006;45(17):5567–5575. doi: 10.1021/bi0511989. [DOI] [PubMed] [Google Scholar]
- 5.Haebel PW, Wichman S, Goldstone D, Metcalf P. Crystallization and initial crystallographic analysis of the disulfide bond isomerase DsbC in complex with the alpha domain of the electron transporter DsbD. J Struct Biol. 2001;136(2):162–166. doi: 10.1006/jsbi.2001.4430. [DOI] [PubMed] [Google Scholar]
- 6.Inaka K, Kanaya E, Kikuchi M, Miki K. Crystal structure of a mutant human lysozyme with a substituted disulfide bond. Proteins. 2001;43(4):413–419. doi: 10.1002/prot.1054. [DOI] [PubMed] [Google Scholar]
- 7.Gustafson KR, Sowder RC, Henderson LE, Cardellina JH, McMahon JB, Rajamani U, Pannell LK, Boyd MR. Isolation, primary sequence determination, and disulfide bond structure of cyanovirin-N, an anti-HIV (human immunodeficiency virus) protein from the cyanobacterium Nostoc ellipsosporum. Biochem Biophys Res Commun. 1997;238(1):223–228. doi: 10.1006/bbrc.1997.7203. [DOI] [PubMed] [Google Scholar]
- 8.John H, Forssmann WG. Determination of the disulfide bond pattern of the endogenous and recombinant angiogenesis inhibitor endostatin by mass spectrometry. Rapid Commun Mass Spectrom. 2001;15(14):1222–8122. doi: 10.1002/rcm.367. [DOI] [PubMed] [Google Scholar]
- 9.Strosberg AD, Margolies MN, Haber E. The interdomain disulfide bond of a homogeneous rabbit pneumococcal antibody light chain. J Immunol. 1975;115(5):1422–4. [PubMed] [Google Scholar]
- 10.Takemoto LJ. Beta A3/A1 Crystallin from human cataractous lens contains an intramolecular disulfide bond. Curr Eye Res. 1997;16(7):719–724. doi: 10.1076/ceyr.16.7.719.5055. [DOI] [PubMed] [Google Scholar]
- 11.Yen TY, Joshi RK, Yan H, Seto NOL, Palcic MM, Macher BA. Characterization of cysteine residues and disulfide bonds in proteins by liquid chromatography/electrospray ionization tandem mass spectrometry. J Mass Spectrom. 2000;35:990–1002. doi: 10.1002/1096-9888(200008)35:8<990::AID-JMS27>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 12.Yen TY, Yan H, Macher BA. Characterizing closely spaced, complex disulfide bond patterns in peptides and proteins by liquid chromatography/electrospray ionization tandem mass spectrometry. J Mass Spectrom. 2002;37:15–30. doi: 10.1002/jms.241. [DOI] [PubMed] [Google Scholar]
- 13.Zeng R, Xu Q, Shao XX, Wang KY, Xia QC. Determination of the disulfide bond pattern of a novel C-type lectin from snake venom by mass spectrometry. Rapid Commun Mass Spectrom. 2001;15:2213–2220. doi: 10.1002/rcm.500. [DOI] [PubMed] [Google Scholar]
- 14.Debski J, Wyslouch-Cieszynska A, Dadlez M, Grzelak K, Kludkiewicz B, Kolodziejczyk R, Lalik A, Ozyhar A, Kochmanb M. Positions of disulfide bonds and N-glycosylation site in juvenile hormone binding protein. Arch Biochem Biophys. 2004;421:260–266. doi: 10.1016/j.abb.2003.10.019. [DOI] [PubMed] [Google Scholar]
- 15.Tie JK, Mutucumarana VP, Straight DL, Carrick KL, Pope RM, Stafford DW. Determination of disulfide bond assignment of human vitamin K-dependent γ-glutamyl carboxylase by matrix-assisted laser desorption/ionization time-of-flight mass spectrometry. J Biol Chem. 2003;278(46):45468–45475. doi: 10.1074/jbc.M309164200. [DOI] [PubMed] [Google Scholar]
- 16.Jones MD, Hunt J, Liu JL, Patterson SD, Kohno T, Lu HS. Determination of tumor necrosis factor binding protein disulfide structure: deviation of the fourth domain structure from the TNFR/NGFR family cysteine-rich region signature. Biochemistry. 1997;36(48):14914–14923. doi: 10.1021/bi971696k. [DOI] [PubMed] [Google Scholar]
- 17.Jones MD, Patterson SD, Lu HS. Determination of disulfide bonds in highly bridged disulfide-linked peptides by matrix-assisted laser desorption/ionization mass spectrometry with post-source decay. Anal Chem. 1998;70:136–143. doi: 10.1021/ac9707693. [DOI] [PubMed] [Google Scholar]
- 18.Cole AR, Hall NE, Treutlein HR, Eddes JS, Reid GE, Moritz RL, Simpson RJ. Disulfide bond structure and N-glycosylation sites of the extracellular domain of the human interleukin-6 receptor. J Biol Chem. 1999;274(11):7207–7215. doi: 10.1074/jbc.274.11.7207. [DOI] [PubMed] [Google Scholar]
- 19.Krokhin OV, Cheng K, Sousa SL, Ens W, Standing KG, Wilkins JA. Mass spectrometric based mapping of the disulfide bonding patterns of integrin α chains. Biochem. 2003;42:12950–12959. doi: 10.1021/bi034726u. [DOI] [PubMed] [Google Scholar]
- 20.Merewether LA, Le J, Jones MD, Lee R, Shimamoto G, Lu HS. Development of disulfide peptide mapping and determination of disulfide structure of recombinant human osteoprotegerin chimera produced in Escherichia coli. Arch Biochem Biophys. 2000;375(1):101–110. doi: 10.1006/abbi.1999.1636. [DOI] [PubMed] [Google Scholar]
- 21.Patterson SD, Katta V. Prompt fragmentation of disulfide-linked peptides during matrix-assisted laser desorption ionization mass spectrometry. Anal Chem. 1994;66(21):3727–3732. doi: 10.1021/ac00093a030. [DOI] [PubMed] [Google Scholar]
- 22.Pitt JJ, Da Silva E, Gorman JJ. Determination of the disulfide bond arrangement of Newcastle disease virus hemagglutinin neuraminidase. Correlation with a beta-sheet propeller structural fold predicted for paramyxoviridae attachment proteins. J Biol Chem. 2000;275(9):6469–6478. doi: 10.1074/jbc.275.9.6469. [DOI] [PubMed] [Google Scholar]
- 23.Wallis TP, Huang CY, Nimkar SB, Young PR, Gorman JJ. Determination of the disulfide bond arrangement of dengue virus NS1 protein. J Biol Chem. 2004;279(20):20729–20741. doi: 10.1074/jbc.M312907200. [DOI] [PubMed] [Google Scholar]
- 24.Gorman JJ, Wallis TP, Pitt JJ. Protein disulfide bond determination by mass spectrometry. Mass Spectrom Rev. 2002;21:183–216. doi: 10.1002/mas.10025. [DOI] [PubMed] [Google Scholar]
- 25.Caporale C, Sepe C, Caruso C, Pucci P, Buonocore V. Assignment of protein disulfides by a computer method using mass spectrometric data. FEBS Lett. 1996;393:241–247. doi: 10.1016/0014-5793(96)00894-0. [DOI] [PubMed] [Google Scholar]
- 26.Fenyo D. A software tool for the analysis of mass spectrometric disulfide mapping experiments. Comput Appl Biosci. 1997;13(6):617–618. doi: 10.1093/bioinformatics/13.6.617. [DOI] [PubMed] [Google Scholar]
- 27.Sadygov RG, Cociorva DC, Yates JR. Large-scale database searching using tandem mass spectra: Looking up the answer in the back of the book. Nat Methods. 2004;1(3):195–202. doi: 10.1038/nmeth725. [DOI] [PubMed] [Google Scholar]
- 28.Eng JK, McCormack AL, Yates JR. An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. J Am Soc Mass Spectrom. 1994;5:976–989. doi: 10.1016/1044-0305(94)80016-2. [DOI] [PubMed] [Google Scholar]
- 29.Perkins DN, Pappin DJC, Creasy DM, Cottrell JS. Probability-based protein identification by searching sequence database using mass spectrometry data. Electrophoresis. 1999;20:3551–3567. doi: 10.1002/(SICI)1522-2683(19991201)20:18<3551::AID-ELPS3551>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 30.Wefing S, Schnaible V, Hoffmann D. SearchXLinks. a program for the identification of disulfide bonds in proteins from mass spectra. Anal Chem. 2006;78:1235–1241. doi: 10.1021/ac051634x. [DOI] [PubMed] [Google Scholar]
- 31.Xu H, Freitas AF. A high mass accuracy sensitive probability based scoring algorithm for database searching of tandem mass spectrometry data. BMC Bioinf. 2007;8:133. doi: 10.1186/1471-2105-8-133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Russell WK, Park ZY, Russell DH. Proteolysis in mixed organic-aqueous solvent systems: applications for peptide mass mapping using mass spectrometry. Anal Chem. 2001;73(11):2682–2685. doi: 10.1021/ac001332p. [DOI] [PubMed] [Google Scholar]
- 33.Smyth DG, Stein WH, Moore S. The sequence of amino acid residues in bovine pancreatic ribonuclease: revision and confirmations. J Biol Chem. 1963;238(1):227–234. [PubMed] [Google Scholar]
- 34.Yazdanparast R, Andrews PC, Smith DL, Dixon JE. Assignment of disulfide bonds in proteins by fast atom bombardment mass spectrometry. J Biol Chem. 1987;262(6):2507–2513. [PubMed] [Google Scholar]
- 35.Raines RT. Ribonuclease A. Chem Rev. 1998;98:1045–1065. doi: 10.1021/cr960427h. [DOI] [PubMed] [Google Scholar]
- 36.Vinci F, Ruoppolo M, Pucci P, Freedman RB, Maino G. Early intermediates in the PDI-assisted folding of ribonuclease A. Protein Sci. 2006;9:525–535. doi: 10.1110/ps.9.3.525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Elias JE, Haas W, Faherty BK, P GS. Comparative evaluation of mass spectrometry platforms used in large-scale proteomics investigations. Nat Methods. 2005;2(9):647–648. doi: 10.1038/nmeth785. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.