

Abstract
Cognitive deficits and hippocampal atrophy, features that are shared with aging and dementia, have been described in type 2 diabetes mellitus (T2DM). T2DM is associated with obesity, hypertension, dyslipidemia, hypothalamic pituitary adrenocortical (HPA) axis abnormalities and inflammation, all of which have been shown to negatively impact the brain. However, since most reports in T2DM focused on glycemic control, the relative contribution of these modifying factors to the impairments observed in T2DM remains unclear. We contrasted 41 middle-aged dementia-free volunteers with T2DM (on average 7 years since diagnosis) with 47 age-, education-, and gender-matched non-insulin resistant controls on cognition and brain volumes. HPA axis activity and other modifiers that accompany T2DM were assessed to determine their impact on brain and cognition. Individuals with T2DM had specific verbal declarative memory deficits, reduced hippocampal and prefrontal volumes, and impaired HPA axis feedback control. Diminished cortisol suppression after dexamethasone and dyslipidemia were associated with decreased cognitive performance, whereas obesity was negatively related to hippocampal volume. Moreover, prefrontal volume was influenced by worse glycemic control. Thus, obesity and altered cortisol levels may contribute to the impact of T2DM on the hippocampal formation, resulting in decreased verbal declarative memory performance.
Keywords: Type 2 diabetes mellitus, hippocampus, cognition, HPA axis, obesity, aging
1. INTRODUCTION
The prevalence rates of type 2 diabetes mellitus (T2DM), a metabolic disorder associated with obesity and age (Morley, 2008), have risen markedly in recent years (Wild, Roglic, Green, Sicree, & King, 2004). There is a developing body of research identifying the brain as a site of possible complications in T2DM. Both cognitive impairments and structural brain abnormalities have been reported in T2DM, with several studies demonstrating that the hippocampus is specifically affected by the disease [e.g.,]. Although individuals with T2DM share memory impairments and hippocampal volume reductions with individuals at risk for dementia of the Alzheimer’s type, and the risk for developing dementia appears to be increased among diabetics, whether these two disorders are etiologically linked remains to be demonstrated. In this study, we sought to ascertain how the allostatic load that accompanies T2DM affects the brain among individuals with no evidence of dementia. Namely, what is the impact of diabetes-associated factors such as obesity, hypertension, dyslipidemia, altered cortisol control, and inflammation on cognition and brain structure in diabetes?
Decreased cognitive performance has been extensively described among individuals with T2DM, with reduced performance in verbal declarative memory and processing speed being the most consistently reported (Awad, Gagnon, & Messier, 2004). Associations between cognitive performance and diabetes-associated factors have been predominantly explored with respect to hyperglycemia, but duration of disease, age, and the presence of comorbidities, such as hypertension, have also been related to the degree of cognitive impairment in T2DM [e.g.,]. In addition to reports focusing on cognitive function, there is also a small literature on brain structural changes in T2DM. These structural changes in T2DM encompass both white and gray matter abnormalities [i.e.,] as well as specific reductions in hippocampal and amygdalar volumes, and those abnormalities have been linked to poor glycemic control. In contrast, abnormalities in prefrontal regions have been linked to the presence of cardiovascular risk factors.
Type 2 diabetes is associated with obesity, hypertension, altered lipid profiles, disturbed cortisol control, and low grade inflammation. All of these factors have individually been shown to contribute to functional and structural brain changes and may modify the impact of T2DM on brain and cognition. For example, global gray matter (GM) atrophy has been associated with increasing BMI, and abnormalities in the frontal and medial-temporal lobe (Gustafson, Lissner, Bengtsson, Bjorkelund, & Skoog, 2004) have been described among diabetes-free obese individuals. In addition, central obesity, as indicated by the waist-hip-ratio has been associated with reductions in hippocampal volumes and with an increase in white matter pathology (Jagust, Harvey, Mungas, & Haan, 2005). It should be noted, however, that not all studies of obesity have yielded consistent results [e.g.,]. Hypertension has been connected to abnormalities in the prefrontal cortex, and these abnormalities were associated with impairments in executive function, even when hypertension was successfully treated with medication (Raz, Rodrigue, & Acker, 2003). In contrast triglyceride elevations have been associated with memory dysfunction in humans [e.g., (Helkala, Niskanen, Viinamaki, Partanen, & Uusitupa, 1995)] and in rodents, and these investigators have suggested that a possible mechanism for those memory impairments may be inhibition of NMDA-mediated LTP maintenance in the hippocampus.
Cortisol dysregulation, which has been associated with T2DM, has been shown to impact brain and cognitive function in numerous studies. For example, hippocampal volume reductions and diminished cognitive function have been associated with sustained cortisol elevations [i.e., (Starkman, Gebarski, Berent, & Schteingart, 1992)]. In prior work in our laboratory we found that impairments in cortisol feedback control among individuals with T2DM, as indicated by higher cortisol levels after 1.5 mg dexamethasone administration, were related to worse cognitive performance. Inflammatory markers such as C-reactive protein (CRP) and fibrinogen, which are associated with T2DM and Metabolic Syndrome have also been associated with altered cognitive performance [e.g.,].
To the best of our knowledge, this is the first study that seeks to assess the associations between T2DM, cognition, and brain volumes while ascertaining the possible modifying role of diabetes-associated factors, such as obesity, hypertension, altered lipid profiles, cortisol, and inflammatory markers.
2. RESULTS
Demographics
The demographic variables and group descriptors are summarized in table 1. As expected, participants with T2DM had higher BMIs and greater rates of hypertension, lipid abnormalities, and inflammation than control participants.
Table 1.
Demographics and medical data (mean ± sd) for diabetic and control groups
| Control Group (n=47) | T2DM Group (n=41) | |
|---|---|---|
| Age (yr) | 60.02±7.96 | 59.05±8.35 |
| Gender (F/M) | 23/24 | 19/22 |
| Education (yr) | 15.81±1.92 | 14.96±2.36 |
| Time from T2DM diagnosis (yr) | n.a. | 7.00±6.40 |
| BMI (Kg/m2)* | 25.49±3.79 | 32.63±6.67 |
| Hypertension (HTN)* | 11/47 | 28/41 |
| Dyslipidemia * | 20/47 | 32/41 |
| Glucose (mg/dl)* | 80.34±8.73 | 140.80±52.59 |
| Insulin (□IU/ml)* | 5.63±1.74 | 14.14±10.43 |
| HbA1c (%)* | 5.23±0.38 | 7.88±1.83 |
| Homeostasis Model Assessment * | 1.11±0.38 | 4.62±3.42 |
| Quantitative Insulin Sensitivity Check Index * | 0.38±0.02 | 0.32±0.03 |
| HDL (mg/dl)* | 57.49±14.34 | 42.39±9.55 |
| Triglycerides (mg/dl)* | 90.13±36.77 | 147±87.10 |
| current or past HTN meds* | 7/41 | 25/47 |
| Statins* | 7/41 | 25/47 |
| anti diabetic meds | n.a. | 42/47 |
| CRP (mg/l)* | 1.38±1.72 | 2.67±2.46 |
| Fibrinogen (mg/dl)* | 302.56±92.48 | 375.79±116.72 |
denotes p≤0.05 for group comparisons
Cognition
All participants were functioning in the purportedly normal range (MMSE diabetics: 29±1 vs. controls: 29.5±0.9; GDS diabetics: 1.9±0.5 vs. controls: 1.8±0.6). Both the diabetic and control group had IQ scores in the normal range, however, since the mean IQ of the diabetic group was significantly lower than the IQ of the control group (diabetics: 104.25±11.88, controls: 114.23±8.44, p≤0.001), we controlled for IQ in subsequent analyses. Impairments in the diabetic group were restricted to the verbal declarative memory domain. This was the case for immediate (F=4.161, p (Willk’s λ =0.004) and, to a lesser extent, delayed (F=2.144, p (Willk’s λ =0.084) recall. Visual declarative memory was not affected in diabetic participants; neither immediate (F=0.076, p (Willk’s λ =0.973), nor delayed (F=1.445, p (Willk’s λ =0.242) recall. Working memory (F=0.785, p (Willk’s λ =0.538), executive function (F=0.692, p (Willk’s λ =0.505) and attention (F=0.179, p (Willk’s λ =0.863) did not differ between the groups. Please see table 2.
Table 2.
IQ-adjusted scores (mean ± sem) of the verbal declarative memory tests
| Cognitive Test | Control Group | T2DM Group | F | p |
|---|---|---|---|---|
| CVLT short delay | 13.11±0.45 | 10.88±0.48 | 9.996 | 0.002 |
| Guild immediate | 7.66±0.36 | 6.03±0.39 | 6.727 | 0.011 |
| WMS-R immediate paragraph recall | 32.33±0.87 | 29.37±0.94 | 4.440 | 0.038 |
| WMS-R verbal paired associates imm. | 21.08±0.48 | 19.93±0.52 | 2.513 | 0.117 |
| CVLT long delay | 13.06±0.43 | 11.69±0.47 | 3.746 | 0.056 |
| Guild delayed | 9.03±0.42 | 7.36±0.46 | 5.962 | 0.017 |
| WMS-R delayed paragraph recall | 28.08±1.12 | 23.99±1.22 | 5.551 | 0.021 |
| WMS-R verbal paired associates del. | 7.75±0.13 | 7.44±0.14 | 2.294 | 0.134 |
Neuroimaging
White matter hyperintensities
There were very few abnormalities in either group. Therefore, we pooled participants into two groups: those with a rating of zero and those with a rating of ≥ 1. There was no difference between the diabetic and control group with respect to periventricular and deep white matter hyperintensities (X2=1.181, p=0.758 and X2=0.032, p=0.984, respectively).
Manual tracing of ROIs
We found no indication of a lateralization effect, and thus averaged left and right hemispheres. Diabetics had 12% smaller mean hippocampal volume than controls, independent of hypertension, dyslipidemia, BMI, inflammation, and scanner used. Frontal pole volume was also reduced in the diabetic group, however, when we accounted for hypertension, the frontal pole volume difference became non significant.
Groups did not differ with respect to the superior temporal gyrus, which served as a relatively small control region in the temporal lobe. Groups did not differ with respect to the superior temporal gyrus, which served as a relatively small control region in the temporal lobe. Neither global, nor cortical atrophy differed between groups. Please refer to table 3.
Table 3.
Group Comparisons of brain volumes (mean ± sd) in cubic centimeters (cc)
| Region of Interest (cc) | Control Group | T2DM Group | t | p |
|---|---|---|---|---|
| Hippocampus | 3.06 ± 0.32 | 2.68 ± 0.32 | 5.322 | ≤0.001 |
| Superior temporal gyrus | 12.01 ± 2.40 | 11.73 ± 1.80 | 0.911 | 0.365 |
| Global atrophy | 114.59 ± 50.19 | 101.05 ± 37.74 | 0.911 | 0.365 |
| Cortical atrophy | 15.52 ± 7.77 | 15.80 ± 8.93 | -.502 | 0.617 |
| Frontal pole | 32.77 ± 7.66 | 28.19 ± 6.87 | 2.721 | 0.008 |
Note: means presented here are unadjusted so as to provide meaningful volumes, but statistics are conducted adjusting for intracranial vault.
HPA axis
Cortisol data did not require logarithmic transformation; however, ACTH data were logarithmically transformed for analysis. Basal cortisol levels did not differ between groups, whereas DEX cortisol was significantly higher among diabetics (table 4). These results were independent of age, gender, or BMI, although controlling for BMI decreased the level of significance for the group differences on DEX cortisol to p=0.089.
Table 4.
Group comparison of HPA axis data (mean ± sd)
| Control Group | T2DM Group | t | p | |
|---|---|---|---|---|
| Cortisol during Oral Glucose Tolerance Test (μg/dl) | 9.65 ± 3.26 | 9.17 ± 3.29 | 0.641 | 0.524 |
| DEX cortisol (μg/dl) | 1.13 ± 0.40 | 1.78 ± 1.44 | -2.553 | 0.015 |
| Cortisol AUCG (arbitrary units) | 437.58 ± 308.13 | 517.79 ± 337.27 | -1.056 | 0.295 |
| ACTH AUCG (arbitrary units) | 2101.85 ± 2354.14 | 2007.74 ± 2351.54 | -0.067 | 0.947 |
Hypertension did not modify these associations. There were no group differences after CRH stimulation, as reflected by the cortisol or ACTH AUCG during the entire DEX/CRH.
Modifiers of cognition and brain
Because our goal was to ascertain the impact of modifiers on cognition and brain beyond the likely impact of diabetes, we ran linear regression analyses, controlling for diagnosis in the first step. Table 5 depicts the results of these analyses, showing the contribution of the modifiers after controlling for diabetes diagnosis.
Table 5.
Association of modifiers with declarative memory scores and brain volumes, derived from regression analyses after controlling for diabetes diagnosis. The leftmost column contains in turn each of the neuropsychological test and brain volumes. The second column shows those modifier variables that significantly added to the diagnosis of T2DM in explaining variance of each of the brain variables
| Dependent | Modifier | Δ R2 | p (final model) | β |
|---|---|---|---|---|
| CVLT s. Delay | DEX cortisol | 0.100 | 0.004 | -1.080 |
| dyslipidemia | 0.059 | 0.023 | -2.078 | |
| CVLT l. Delay | dyslipidemia | 0.066 | 0.026 | -1.944 |
| DEX cortisol | 0.065 | 0.026 | -0.781 | |
| Guild Imm. | age | 0.063 | 0.034 | -0.087 |
| Guild Del. | DEX cortisol | 0.056 | 0.042 | -0.794 |
| Hippocampus | BMI | 0.079 | 0.014 | -0.015 |
| Frontal Pole | HbA1c | 0.102 | 0.007 | -1.560 |
| age | 0.060 | 0.046 | -0.189 | |
Associations between modifiers and verbal declarative memory
Independent of diabetes diagnosis, DEX cortisol and, to some extent, dyslipidemia were associated with performance on those declarative memory tests with more than one learning trial (CVLT list-learning and Guild delayed paragraph recall). On Guild immediate paragraph recall as well as the immediate and delayed paragraph recall of the WMS-R, the selected modifiers did not add beyond diagnosis or the confounds.
Associations between modifiers and hippocampal and frontal pole volume
In the analysis of ICV-adjusted hippocampal volume as the dependent variable, only BMI emerged as a modifier, significantly adding to the variance explained by diabetes diagnosis. The association between hippocampal volume and BMI is depicted in figure 1. Please note that although we used the ICV-adjusted hippocampal volumes in the statistical analysis, in figure 1 we display the raw hippocampal volumes. For the frontal pole volume as the dependent variable, beyond age only HbA1c significantly contributed to the variance beyond diabetes diagnosis. Although hypertension had mediated the frontal pole differences between diabetics and controls, it did not enter the model, probably because of its shared variance with other modifiers such as BMI.
Figure 1.
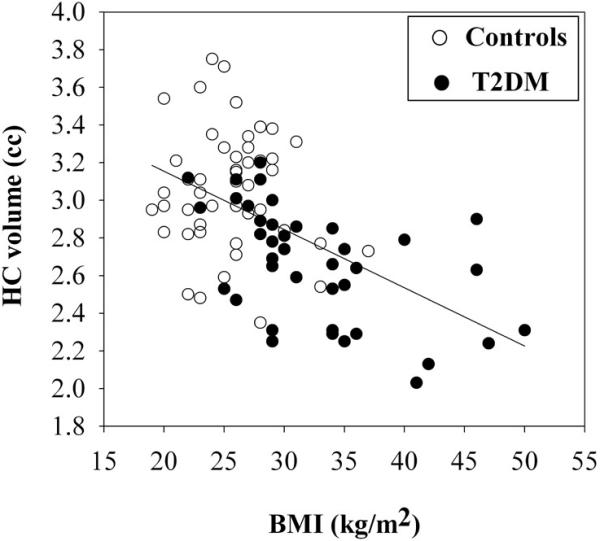
Hippocampal volume (cc) decreases with increasing BMI (kg/m2)
3. DISCUSSION
In this paper, we first sought to ascertain how T2DM impacts cognition and brain, and then establish how diabetes-associated factors further contribute or modify those associations. We found that T2DM has a deleterious effect on hippocampal integrity, demonstrating both specific verbal memory impairments and hippocampal volume reductions among individuals with T2DM. In addition, participants with diabetes had diminished suppression of cortisol after dexamethasone administration, whereas the cortisol response to CRH did not differ between groups. Moreover, they had higher rates of obesity, hypertension, altered lipid profiles, and inflammatory markers.
Our findings of specific verbal declarative memory impairments are in line with the preponderance of the literature, which has most consistently described compromised verbal memory function in T2DM . Most of our patients were relatively young (<60 years) and in reasonable glycemic control (HbA1c=7.88%), which may explain why our findings were circumscribed to reductions in declarative memory performance. Studies that have reported impairments in multiple cognitive domains have generally included much older subjects in poor glucose control.
In line with the verbal declarative memory impairments we described, the diabetic group exhibited specific hippocampal volume loss, which is consistent with our prior report and that of two other groups showing hippocampal atrophy in T2DM. Some authors have speculated that hippocampal volume loss in T2DM may be the result of hyperglycemia and the formation of toxic advanced glycation end-products. Other investigators have suggested that, given the epidemiological link between T2DM and dementia, the hippocampal atrophy in diabetes may be due to those associations. However, given the lack of postmortem evidence of increased Alzheimer’s disease lesions in the brains of individuals with T2DM, the epidemiological associations between T2DM and dementia may be more related to the reductions in brain reserve that may occur in diabetes rather than a direct etiological link to Alzheimer’s Disease (Starr & Convit, 2007). In this study all participants were screened to exclude dementia or mild cognitive impairment (MCI), which suggests that the hippocampal damage we report is not mediated by pathology associated with dementia. We propose that given the high vulnerability of the hippocampus to various metabolic insults, hippocampal-based declarative memory may be one of the first cognitive domains affected in T2DM, and that with aging and disease progression, cognitive impairments beyond hippocampal-based declarative memory may become manifest.
Verbal declarative memory performance was adversely affected by HPA axis function and lipid abnormalities. In this study, performance on tests with multiple learning trials were related to cortisol levels after dexamethasone suppression, and to a lesser extent, to lipid abnormalities, whereas we did not find such associations for recall tests without a learning component. It has been well-established that both exogenous and endogeneous cortisol elevations can specifically reduce declarative memory performance (Lupien, Maheu, Tu, Fiocco, & Schramek, 2007), and our data are in agreement with those findings. Moreover, we found evidence for that HPA axis regulation seems to partially override the diabetes effect with respect to tests with a learning component. This largely overlaps with the findings of our previous report and stresses the significance of HPA axis dysregulation for impaired verbal declarative memory in T2DM. Acute cortisol elevations can negatively impact on declarative memory function (Het, Ramlow, & Wolf, 2005) and reduce cerebral blood flow in the medial temporal lobe. Furthermore, cortisol has been shown to inhibit glucose transport in cultured hippocampal neurons (Horner, Packan, & Sapolsky, 1990) and chronically elevated corticosteroids contribute to memory and learning deficits via alterations of synaptic plasticity and decreased neurogenesis. The hippocampus, in particular during diabetes, might be more susceptible to the damaging effects of cortisol elevations. Therefore, although speculative, when a diabetic person is stressed, recurring short-term cortisol elevations may add to the allostatic burden onto the brain.
In the data we are presenting here, hippocampal volume was strongly associated with obesity (BMI), beyond diabetes diagnosis. The negative impact of obesity on hippocampal volume is consistent with the findings of another study that reported a moderate negative association between central obesity and hippocampal volume in a group that consisted of both non-diabetic and diabetic participants of comparable age to ours. The mechanism for obesity causing damage to the brain is likely multifactorial and this is an area in clear need of development.
One possible mechanism for the association between obesity and brain impairments is obesity-associated inflammation. Pro-inflammatory cytokines such as IL-6, which are elevated in obesity have been linked to cognitive decline [e.g.,]. In addition, elevated triglyceride levels, as found in obesity, adversely affect memory performance, although we did not find direct associations between triglyceride levels and hippocampal volume. Another possible mechanism could be reduced glucose availability to the hippocampus, as the glucose transporter GLUT-4 has been found to be reduced in the hippocampus of obese Zucker rats. Moreover, in a recent report, Selim and coworkers described decreased cerebral blood flow velocities with increasing BMI in participants with T2DM (Selim, Jones, Novak, Zhao, & Novak, 2008), and a high BMI has been associated with endothelial dysfunction, albeit in the peripheral vascular bed. Taken together, these findings suggest that multiple co-occurring factors may contribute to the loss of hippocampal integrity in T2DM, maybe exerting their effects via decreased substrate supply to the hippocampus.
The group with diabetes also had reduced frontal pole volume; and this volume reduction was explained by higher rates of hypertension in that group. This is in agreement with the literature, which shows an association between decreased frontal lobe integrity and hypertension, likely due to the decreased blood flow in prefrontal areas of the brain seen in hypertension (Beason-Held, Moghekar, Zonderman, Kraut, & Resnick, 2007). Moreover, we found that frontal pole volume, in addition to being related to age, was also related to long-term glucose control as indicated by HbA1c levels. The association to HbA1c was present even after accounting for a diabetes diagnosis. Decreased cerebral blood flow in the frontal lobe has been described in T2DM and age is accompanied by decreased vascular reserve (O’Rourke, 2007). Therefore, our findings might reflect the impact of poor glucose control in T2DM and hypertension on the cerebral vasculature in the prefrontal cortex. It is conceivable that, in T2DM, damage to the hippocampus and prefrontal cortex could affect HPA axis feedback regulation, which in turn further exacerbates this damage. However, longitudinal studies are needed to better characterize the order of these effects.
In extrapolating from our findings and the current literature, we hypothesize that obesity, hypertension and impaired long-term glucose control may negatively impact brain metabolic substrate delivery by affecting endothelial function, vascular reactivity and cerebral blood flow in T2DM. This may in turn lead to a loss of neuronal integrity in the hippocampus and prefrontal cortex. The negative impact on hippocampal and prefrontal integrity may then lead to HPA axis dysregulation, which again might further aggravate hippocampal and prefrontal cortex grey matter pathology. The decreased hippocampal integrity paired with cortisol elevations might then result in compromised memory function. This view of an indirect effect of hippocampal damage on memory function via a dysregulated HPA axis has been supported by studies in rodents.
Our study has many strengths, however due to its cross-sectional design it does not permit us to draw conclusions about cause and effect. Also, the relatively broad age distribution, despite making our sample more representative of the age group that typically develops T2DM, adds variability to the data. In addition, the measure we used as an estimate of “basal” cortisol, namely a single blood value, is not a reliable measure of basal cortisol levels. In future studies multiple samples, collected throughout the day, should be utilized. Furthermore, although our analyses were designed based on the selection of a priori factors, our sample size is limited and there is the possibility of some adventitious findings. However, giving the lack of comprehensive data in this area, we felt that the exploratory analyses that we presented here are justified. There is also the possibility of type 2 error in that some of the factors we chose to represent certain domains (such as dexamethasone suppression of cortisol for the HPA axis or CRP for the domain of inflammation) may not be optimal. Future research should employ a longitudinal design and include other physiologically relevant measures of cortisol, such as diurnal cortisol, as well as inflammatory markers that have been shown to directly signal to the brain, such as IL-beta, IL-6 and TNF-alpha. In addition future studies should also include direct assessment of endothelial function, which may aid in understanding the mechanisms behind the harmful effects of T2DM on the brain, and provide rationale for new prevention and treatment efforts.
In conclusion, we demonstrated that middle-aged individuals with T2DM with typical levels of glycemic control exhibit loss of hippocampal integrity, reductions in prefrontal cortex volume and diminished feedback regulation of the HPA axis. Obesity, hypertension and cortisol regulation act as modifiers of brain and cognition and possibly exert their negative impact on the brain by reducing metabolite substrate delivery, perhaps thorough a loss of endothelial function based vascular reactivity. We propose that in order to successfully address the negative impact on T2DM on the brain, treatment should go beyond keeping patients in good glycemic control and include better blood pressure control, weight reduction, and stress management.
4. EXPERIMENTAL PROCEDURE
4.1 Participants
Eighty-eight volunteers, 41 with T2DM and 47 matched non-insulin resistant controls participated in the study. The participants were consecutive cases evaluated as part of an NIH-sponsored study. All participants were functioning in the normal range. Diabetic participants were referred to us by collaborating endocrinologists, or responded to advertisements on the internet. Control participants were selected from our ongoing studies of normal aging. All participants had a minimum of a high school education. Evidence of neurological, medical (other than T2DM, dyslipidemia, or hypertension), or active psychiatric conditions such as depression or substance abuse were exclusion criteria. The study protocol was approved by the NYU School of Medicine Institutional Board of Research Associates. Participants gave informed written consent and were compensated for their participation. A subset of these participants were included in two previously published related reports .
4.1.1. Participants with T2DM
Diabetic participants met one or more of the following criteria: (1) fasting blood glucose >125 mg/dl on two separate occasions, (2) 2-hour blood glucose level >200 mg/dl during a 75-gram oral glucose tolerance test, or (3) had received a prior diagnosis of T2DM. Diabetic individuals were being treated with hypoglycemic agents and/or by lifestyle modification but not insulin or insulin secretagogues.
4.1.2. Control participants
The control group were age-, gender-, and education-matched to the diabetic group. Participant selection was made blind to neuropsychological, MRI, and HPA axis results. Control participants did not have evidence of overt insulin resistance, as reflected by a quantitative insulin sensitivity check index (QUICKI) ≥ 0.35(Chen, Sullivan, & Quon, 2005). The QUICKI has been validated against clamp assessments of insulin sensitivity [i.e.].
4.2. Procedures
All participants underwent neuropsychological testing, medical (including bloods for fibrinogen and CRP) and HPA axis assessment, and an MRI scan of the brain. Ten participants (7 diabetics, 3 controls) did not participate in the HPA axis protocol.
All of the evaluations have been described in detail elsewhere.
Briefly, hypertension and dyslipidemia were defined based on NCEP guidelines, which include medication use and/or meeting certain threshold values, were handled as dychotomous variables in our analyses. Fasting glucose and insulin levels and HbA1c plasma levels were assessed once during the study.
4.2.1 Neuropsychological and Psychiatric assessment
All cognitive assessments used are standardized neuropsychological tests described in detail elsewhere. Briefly, declarative memory was assessed with the California Verbal Learning Test (CVLT), short and long free recall, the individual scores from the Wechsler Memory Scale -Revised (WMS-R), which includes tests of both verbal and visual memory, and the Guild immediate and delayed paragraphs. Working memory was tested using the Digit Span Backward and the tapping backward subtest from the WMS-R. Executive Function was measured with the Controlled Word Association Test, the Stroop task, and the Tower of London. Attention was assessed with the Perceptual Speed Test and the Digit Symbol Substitution Test from the WMS-R. General Intellectual Functioning was assessed using the Shipley Institute of Living Scale and WAIS-R full scale IQ score estimates were derived from the Shipley score. To ensure that all participants were functioning in the purportedly normal range and to allow comparison to other studies, overall functional assessment was done using the Mini Mental State Examination (MMSE) (Cockrell & Folstein, 1988) and Global Deterioration Scale (GDS) (Reisberg, Ferris, de Leon, & Crook, 1988).
4.2.2 Magnetic resonance Imaging data acquisition
4.2.2.1.Image acquisition
MRI scans were acquired on either a 1.5 T GE Vision (43 controls, 27 diabetics) or 1.5 T Siemens Avanto system (4 controls, 13 diabetics), using equivalent sequences. Please see for imaging parameters of the GE Vision System.
For the Siemens system, we used a magnetization-prepared rapid gradient echo acquisition (TR 1300 ms, TE 4.38 ms, 192 slices, slice thickness 1.2 mm, no gap; FOV 250×250 mm, matrix 256×128, flip angle 15 degrees) for the region of interest (ROI) based volume measurements. Fast fluid-attenuated inversion recovery (FLAIR) was used for white matter ratings. TR 9000 ms, TE 97 ms, acquisition matrix 154 × 256, FOV 210×210, slice thickness 3 mm; 50 slices, no gaps.
4.2.2.2. Quantification of white matter hyperintensities
FLAIR images were used to quantify white matter disease and rule out primary neurological disease. The white matter hyperintensities were rated according to the modified Fazekas scale.
4.2.2.3. Manual tracing of ROIs
Drawings were made blind to participants’ identity and diagnosis. The hippocampus, superior temporal gyrus, which served as the control region, and prefrontal region and frontal pole were outlined on coronal images, and intracranial vault volume (ICV) was outlined on sagittal images. Please see for details. A threshold procedure was used to estimate the CSF portion of the ICV (global atrophy) and total frontal region (cortical atrophy). To adjust for individual differences in brain size, we residualized all volumes to the ICV by means of regression analysis and then used the residualized volumes in the statistical analyses. Correspondence of the volumes obtained on the two scanners was ascertained by obtaining images for 10 subjects on both scanners and then assessing the intracranial vault volume obtained with the two sets of images. The volumes obtained were indistinguishable (paired t-test: t=0.029, p=0.977).
4.2.3 HPA axis assessment
After a 10-hour overnight fast we collected two independent blood samples for cortisol assessment as part of the baseline bloods during a standardized glucose tolerance test. Those two samples were averaged to yield a measure of unstimulated standardized basal morning cortisol secretion. Even though this measure is not representative of diurnal cortisol, we call it “basal” in order to clearly distinguish it from cortisol levels we obtain after Dexamethasone suppression (DEX cortisol). HPA axis feedback was assessed on a different day, using the short version of the DEX/CRH test. Participants took 1.5 mg dexamethasone at 11 pm. On the next day subjects arrived at 1 pm and received a standardized lunch. Afterward, an intravenous catheter was placed in the forearm and kept patent with a heparin lock. No blood sample was drawn for at least one hour after catheter insertion to allow sufficient time for cortisol to return to baseline. Then, two samples were drawn to measure cortisol and ACTH levels. At 3 pm, 100 μg of ovine CRH was injected intravenously. Blood samples were subsequently drawn at 3.30 pm, 3.45 pm, 4 pm, and 4.15 pm. The two samples drawn before CRH administration were averaged to obtain a measure of the suppressed HPA axis (DEX cortisol). The samples drawn after CRH injection, together with those two samples were used to compute an area under the curve (AUC), using the trapezoidal rule, please see for details. The main variable of interest was cortisol after dexamethasone suppression (DEX cortisol), which was used as an index for HPA axis feedback control. We did not assess cortisol binding globulin (CBG) levels.
4.3. Assays
Glucose was measured using a glucose oxidase method (VITROS 950 AT, Amersham, England), insulin by chemiluminescence (Advia Centaur, Bayer Corporation), and HbA1c using an automated HPLC method (Tosoh Corporation, Kanagawa, Japan) certified by the National Glycohemoglobin Standardization Program. Total cortisol was measured with an enzyme immunoassay (EIA; IBL, Hamburg, Germany) with a sensitivity of 0.1 μg/dL. Intact adrenocorticotropin (ACTH) was measured with a radioimmunoassay (Nichols Institute, Bad Nauheim, Germany) with a sensitivity of 2 pg/ml. CRP was measured using an enzymatic immunoassay (Vitros CRP slide, Ortho Clin. Diagnostics) and fibrinogen was determined by photometry (Beckman-Coulter advance system).
4.4. Statistical analyses
Demographic variables and group descriptors were tested using Student’s t-tests for independent samples and X2 tests where appropriate. MANOVA was used to compare groups on cognitive variables grouped by domain, with IQ as a covariate. From those analyses, we produced IQ-adjusted means, which were then used to contrast the groups. ANOVA was used to analyze brain variables and covariates were used when required.
The contributions of modifiers to brain and cognitive variables were ascertained using linear regression analyses; T2DM diagnosis (0 vs. 1) was forced in as the first step and a priori selected modifiers were entered in a stepwise fashion with a probability of F set to 0.05. Briefly, the following confounds and modifiers were selected: Age and gender have been shown to have an impact on cognition and were thus considered as potential confounds. HbA1c was selected because it is directly associated with diabetes and because of the well-documented associations to brain and cognition. BMI, hypertension, markers of inflammation (CRP, fibrinogen), and dyslipidemia were considered as modifiers because of their established influence on brain and cognition. Out of the cortisol variables measured, the cortisol value post dexamethasone suppression (DEX cortisol), which was the only HPA axis variable that separated the groups, was used.
Individual variable values that deviated more than 2 standard deviations from the group mean were considered outliers and excluded from analyses. CRP levels above 10 mg/l, which likely indicates acute inflammation, were also excluded from the analyses that included CRP.
5. ACKNOWLEDGEMENTS
We thank Prof. Clemens Kirschbaum for conducting the cortisol and ACTH assays. This study was supported by grants DK 064087, P30-AG-08051 and NCRR M01 RR00096.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
6. REFERENCES
- Allen KV, Frier BM, Strachan MWJ. The relationship between type 2 diabetes and cognitive dysfunction: longitudinal studies and their methodological limitations. European Journal of Pharmacology. 2004;490:169–175. doi: 10.1016/j.ejphar.2004.02.054. [DOI] [PubMed] [Google Scholar]
- Arkin JM, Alsdorf R, Bigornia S, Palmisano J, Beal R, Istfan N, Hess D, Apovian CM, Gokce N. Relation of Cumulative Weight Burden to Vascular Endothelial Dysfunction in Obesity. The American Journal of Cardiology. 2008;101:98–101. doi: 10.1016/j.amjcard.2007.07.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arvanitakis Z, Schneider JA, Wilson RS, Li Y, Arnold SE, Wang Z, Bennett DA. Diabetes is related to cerebral infarction but not to AD pathology in older persons. Neurology. 2006;67:1960–1965. doi: 10.1212/01.wnl.0000247053.45483.4e. [DOI] [PubMed] [Google Scholar]
- Awad N, Gagnon M, Messier C. The relationship between impaired glucose tolerance, type 2 diabetes, and cognitive function. Journal Of Clinical And Experimental Neuropsychology: Official Journal Of The International Neuropsychological Society. 2004;26:1044–1080. doi: 10.1080/13803390490514875. [DOI] [PubMed] [Google Scholar]
- Beason-Held LL, Moghekar A, Zonderman AB, Kraut MA, Resnick SM. Longitudinal Changes in Cerebral Blood Flow in the Older Hypertensive Brain. Stroke. 2007;38:1766–1773. doi: 10.1161/STROKEAHA.106.477109. [DOI] [PubMed] [Google Scholar]
- Beeri MS, Silverman JM, Davis KL, Marin D, Grossman HZ, Schmeidler J, Purohit DP, Perl DP, Davidson M. Type 2 diabetes is negatively associated with Alzheimer’s disease neuropathology. The Journals of Gerontology.Series A, Biological Sciences and Medical Sciences. 2005;60:471–475. doi: 10.1093/gerona/60.4.471. Mohs et, a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruehl H, Rueger M, Dziobek I, Sweat V, Tirsi A, Javier E, Arentoft A, Wolf OT, Convit A. Hypothalamic-Pituitary-Adrenal Axis Dysregulation and Memory Impairments in Type 2 Diabetes. Journal of Clinical Endocrinology Metabolism. 2007;92:2439–2445. doi: 10.1210/jc.2006-2540. [DOI] [PubMed] [Google Scholar]
- Cartier A, Lemieux I, Almeras N, Tremblay A, Bergeron J, Despres JP. Visceral Obesity and Plasma Glucose-Insulin Homeostasis: Contributions of Interleukin-6 and Tumor Necrosis Factor-{alpha} in Men. Journal of Clinical Endocrinology Metabolism. 2008;93:1931–1938. doi: 10.1210/jc.2007-2191. [DOI] [PubMed] [Google Scholar]
- Cervos-Navarro J, Diemer NH. Selective vulnerability in brain hypoxia. Critical Reviews in Neurobiology. 1991;6:149–182. [PubMed] [Google Scholar]
- Chalmers J, Risk MTA, Kean DM, Grant R, Ashworth B, Campbell IW. Severe Amnesia After Hypoglycemia. Diabetes Care. 1991;14:922–925. doi: 10.2337/diacare.14.10.922. [DOI] [PubMed] [Google Scholar]
- Chen H, Sullivan G, Quon MJ. Assessing the predictive accuracy of QUICKI as a surrogate index for insulin sensitivity using a calibration model. Diabetes. 2005;54:1914–1925. doi: 10.2337/diabetes.54.7.1914. [DOI] [PubMed] [Google Scholar]
- Cockrell JR, Folstein MF. Mini-mental state examination (MMSE) Psychopharmacology Bulletin. 1988;24:689–692. [PubMed] [Google Scholar]
- Conrad CD. What Is the Functional Significance of Chronic Stress-Induced CA3 Dendritic Retraction Within the Hippocampus? Behavioral And Cognitive Neuroscience Reviews. 2006;5:41–60. doi: 10.1177/1534582306289043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Convit A, de Leon MJ, Tarshish C, De Santi S, Tsui W, Rusinek H, George AE. Specific hippocampal volume reductions in individuals at risk for Alzheimer’s disease. Neurobiology of Aging. 1997;18:131–138. doi: 10.1016/s0197-4580(97)00001-8. [DOI] [PubMed] [Google Scholar]
- de Quervain DJF, Henke K, Aerni A, Treyer V, McGaugh JL, Berthold T, Nitsch RM, Buck A, Roozendaal B, Hock C. Glucocorticoid-induced impairment of declarative memory retrieval is associated with reduced blood flow in the medial temporal lobe. European Journal of Neuroscience. 2003;17:1296–1302. doi: 10.1046/j.1460-9568.2003.02542.x. [DOI] [PubMed] [Google Scholar]
- den Heijer T, Vermeer SE, van Dijk EJ, Prins ND, Koudstaal PJ, Hofman A, Breteler MM. Type 2 diabetes and atrophy of the medial temporal lobe structures on brain MRI. Diabetologia. 2003;46:1604–1610. doi: 10.1007/s00125-003-1235-0. [DOI] [PubMed] [Google Scholar]
- Farr SA, Yamada KA, Butterfield DA, Abdul HM, Xu L, Miller NE, Banks WA, Morley JE. Obesity and Hypertriglyceridemia Produce Cognitive Impairment. Endocrinology. 2008 doi: 10.1210/en.2007-1722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Real JM, Pickup JC. Innate immunity, insulin resistance and type 2 diabetes. Trends Endocrinol.Metab. 2008;19:10–16. doi: 10.1016/j.tem.2007.10.004. [DOI] [PubMed] [Google Scholar]
- Ferrannini E, Natali A, Bell P, Cavallo-Perin P, Lalic N, Mingrone G. Insulin Resistance and Hypersecretion in Obesity. Journal of Clinical Investigation. 1997;100:1166–1173. doi: 10.1172/JCI119628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gold S, Dziobek I, Sweat V, Tirsi A, Rogers K, Bruehl H, Tsui W, Richardson S, Javier E, Convit A. Hippocampal damage and memory impairments as possible early brain complications of type 2 diabetes. Diabetologia. 2007;50:711–719. doi: 10.1007/s00125-007-0602-7. [DOI] [PubMed] [Google Scholar]
- Gold SM, Dziobek I, Rogers K, Bayoumy A, McHugh PF, Convit A. Hypertension and hypothalamo-pituitary-adrenal axis hyperactivity affect frontal lobe integrity. The Journal of Clinical Endocrinology and Metabolism. 2005;90:3262–3267. doi: 10.1210/jc.2004-2181. [DOI] [PubMed] [Google Scholar]
- Gustafson D, Lissner L, Bengtsson C, Bjorkelund C, Skoog I. A 24-year follow-up of body mass index and cerebral atrophy. Neurology. 2004;63:1876–1881. doi: 10.1212/01.wnl.0000141850.47773.5f. [DOI] [PubMed] [Google Scholar]
- Haltia LT, Viljanen A, Parkkola R, Kemppainen N, Rinne JO, Nuutila P, Kaasinen V. Brain white matter expansion in human obesity and the recovering effect of dieting. J Clin Endocrinol.Metab. 2007;92:3278–3284. doi: 10.1210/jc.2006-2495. [DOI] [PubMed] [Google Scholar]
- Hassing LB, Grant MD, Hofer SM, Pedersen NL, Nilsson SE, Berg S, McClearn GE, Johansson B. Type 2 diabetes mellitus contributes to cognitive decline in old age: A longitudinal population-based study. Journal of the International Neuropsychological Society. 2004;10:299–607. doi: 10.1017/S1355617704104165. [DOI] [PubMed] [Google Scholar]
- Heitner J, Dickson D. Diabetics do not have increased Alzheimer-type pathology compared with age-matched control subjects. A retrospective postmortem immunocytochemical and histofluorescent study. Neurology. 1949 Nov;:1306–1311. doi: 10.1212/wnl.49.5.1306. 1997. [DOI] [PubMed] [Google Scholar]
- Helkala EL, Niskanen L, Viinamaki H, Partanen J, Uusitupa M. Short-term and long-term memory in elderly patients with NIDDM. Diabetes Care. 1995;18:681–685. doi: 10.2337/diacare.18.5.681. [DOI] [PubMed] [Google Scholar]
- Het S, Ramlow G, Wolf OT. A meta-analytic review of the effects of acute cortisol administration on human memory. Psychoneuroendocrinology. 2005;30:771–784. doi: 10.1016/j.psyneuen.2005.03.005. [DOI] [PubMed] [Google Scholar]
- Horner HC, Packan DR, Sapolsky RM. Glucocorticoids inhibit glucose transport in cultured hippocampal neurons and glia. Neuroendocrinology. 1990;52:57–64. doi: 10.1159/000125539. [DOI] [PubMed] [Google Scholar]
- Jagust W, Harvey D, Mungas D, Haan M. Central obesity and the aging brain. Arch Neurol. 2005;62:1545–1548. doi: 10.1001/archneur.62.10.1545. [DOI] [PubMed] [Google Scholar]
- Jongen C, van der Ground J, ppelle LJ, iessels GJ, iergever JP, luim JPW. Automated measurement of brain and white matter lesion volume in type 2 diabetes mellitus. Diabetologia. 2007;50:1509–1516. doi: 10.1007/s00125-007-0688-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katz A, Nambi SS, Mather K, Baron AD, Follmann DA, Sullivan G, Quon MJ. Quantitative insulin sensitivity check index: a simple, accurate method for assessing insulin sensitivity in humans. The Journal of Clinical Endocrinology and Metabolism. 2000;85:2402–2410. doi: 10.1210/jcem.85.7.6661. [DOI] [PubMed] [Google Scholar]
- Korf ESC, White LR, Scheltens P, Launer LJ. Brain aging in very old men with type 2 diabetes: the honolulu-Asia aging study. Diabetes Care. 2006;29:2268–2274. doi: 10.2337/dc06-0243. [DOI] [PubMed] [Google Scholar]
- Kumar A, Haroon E, Darwin C, Pham D, Ajilore O, Rodriguez G, Mintz J. Gray matter prefrontal changes in type 2 diabetes detected using MRI. J Magn Reson.Imaging. 2008;27:14–19. doi: 10.1002/jmri.21224. [DOI] [PubMed] [Google Scholar]
- Last D, de Bazelaire C, Alsop DC, Hu K, Abduljalil AM, Cavallerano J, Marquis RP, Novak V. Global and Regional Effects of Type 2 Diabetes on Brain Tissue Volumes and Cerebral Vasoreactivity. Diabetes Care. 2007;30:1193–1199. doi: 10.2337/dc06-2052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leibson CL, Rocca WA, Hanson VA, Cha R, Kokmen E, O’Brien PC, Palumbo PJ. Risk of Dementia among Persons with Diabetes Mellitus: A Population-based Cohort Study. American Journal of Epidemiology. 1997;145:301–308. doi: 10.1093/oxfordjournals.aje.a009106. [DOI] [PubMed] [Google Scholar]
- Lezak MD. Neuropsychological Assessment. 3 ed. Oxford University Press; New York: 1995. [Google Scholar]
- Lupien SJ, Maheu F, Tu M, Fiocco A, Schramek TE. The effects of stress and stress hormones on human cognition: Implications for the field of brain and cognition. Brain and Cognition. 2007;65:209–237. doi: 10.1016/j.bandc.2007.02.007. [DOI] [PubMed] [Google Scholar]
- Mattson MP, Gurthrie PB, Kater SB. Intrinsic factors in the selective vulnerability of hippocampal pyramidal neurons. Progress in Clinical & Biological Research. 1989;317:333–351. [PubMed] [Google Scholar]
- McEwen BS. Possible mechanisms for atrophy of the human hippocampus. Molecular Psychiatry. 1997;2:255–263. doi: 10.1038/sj.mp.4000254. [DOI] [PubMed] [Google Scholar]
- Morley JE. Diabetes and Aging: Epidemiologic Overview. Clinics in Geriatric Medicine. 2008;24:395–405. doi: 10.1016/j.cger.2008.03.005. [DOI] [PubMed] [Google Scholar]
- O’Rourke MF. Arterial aging: pathophysiological principles. Vascular Medicine. 2007;12:329–341. doi: 10.1177/1358863X07083392. [DOI] [PubMed] [Google Scholar]
- Pannacciulli N, Del Parigi A, Chen K, Le DS, Reiman EM, Tataranni PA. Brain abnormalities in human obesity: A voxel-based morphometric study. Neuroimage. 2006;31:1419–1425. doi: 10.1016/j.neuroimage.2006.01.047. [DOI] [PubMed] [Google Scholar]
- Peila R, Rodriguez BL, Launer LJ, Honolulu-Asia Aging S. Type 2 diabetes, APOE gene, and the risk for dementia and related pathologies: The Honolulu-Asia Aging Study. Diabetes. 2002;51:1256–1262. doi: 10.2337/diabetes.51.4.1256. [DOI] [PubMed] [Google Scholar]
- Raz N, Rodrigue KM, Acker JD. Hypertension and the brain: vulnerability of the prefrontal regions and executive functions. Behavioral Neuroscience. 2003;117:1169–1180. doi: 10.1037/0735-7044.117.6.1169. [DOI] [PubMed] [Google Scholar]
- Reisberg B, Ferris SH, de Leon MJ, Crook T. The Global Deterioration Scale (GDS) Psychopharmacology Bulletin. 1988;24:661–663. [PubMed] [Google Scholar]
- Roozendaal B, Phillips RG, Power AE, Brooke SM, Sapolsky RM, McGaugh JL. Memory retrieval impairment induced by hippocampal CA3 lesions is blocked by adrenocortical suppression. Nature Neuroscience. 2001;4:1169–1171. doi: 10.1038/nn766. [DOI] [PubMed] [Google Scholar]
- Scheltens P, Barkhof F, Leys D, Pruvo JP, Nauta JJ, Vermersch P, Steinling M, Valk J. A semiquantative rating scale for the assessment of signal hyperintensities on magnetic resonance imaging. Journal of the Neurological Sciences. 1993;114:7–12. doi: 10.1016/0022-510x(93)90041-v. [DOI] [PubMed] [Google Scholar]
- Selim M, Jones R, Novak P, Zhao P, Novak V. The effects of body mass index on cerebral blood flow velocity. Clin Auton.Res. 2008;18:331–338. doi: 10.1007/s10286-008-0490-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skalicky J, Muzakova V, Kandar R, Meloun M, Rousar T, Palicka V. Evaluation of oxidative stress and inflammation in obese adults with metabolic syndrome. Clin Chem Lab Med. 2008 doi: 10.1515/CCLM.2008.096. [DOI] [PubMed] [Google Scholar]
- Starkman MN, Gebarski SS, Berent S, Schteingart DE. Hippocampal formation volume, memory dysfunction, and cortisol levels in patients with Cushing’s syndrome. Biological Psychiatry. 1992;32:756–765. doi: 10.1016/0006-3223(92)90079-f. [DOI] [PubMed] [Google Scholar]
- Starr VL, Convit A. Diabetes, sugar-coated but harmful to the brain. Current Opinion in Pharmacology. 2007;7:638–642. doi: 10.1016/j.coph.2007.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stranahan AM, Arumugam TV, Cutler RG, Lee K, Egan JM, Mattson MP. Diabetes impairs hippocampal function through glucocorticoid-mediated effects on new and mature neurons. Nat Neurosci. 2008;11:309–317. doi: 10.1038/nn2055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sweat V, Starr V, Bruehl H, Arentoft A, Tirsi A, Javier E, Convit A. C-Reactive Protein is Linked to Lower Cognitive Performance in Overweight and Obese Women. Inflammation. 2008 doi: 10.1007/s10753-008-9065-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taki Y, Kinomura S, Sato K, Inoue K, Goto R, Okada K, Uchida S, Kawashima R, Fukuda H. Relationship between body mass index and gray matter volume in 1,428 healthy individuals. Obesity.(Silver.Spring) 2008;16:119–124. doi: 10.1038/oby.2007.4. [DOI] [PubMed] [Google Scholar]
- van Harten B, Oosterman J, van Loon B, Scheltens P, Weinstein HC. Brain Lesions on MRI in Elderly Patients with Type 2 Diabetes Mellitus. European Neurology. 2006;57:70. doi: 10.1159/000098054. [DOI] [PubMed] [Google Scholar]
- Weaver JD, Huang MH, Albert M, Harris T, Rowe JW, Seeman TE. Interleukin-6 and risk of cognitive decline: MacArthur Studies of Successful Aging. Neurology. 2002;59:371–378. doi: 10.1212/wnl.59.3.371. [DOI] [PubMed] [Google Scholar]
- Wild S, Roglic G, Green A, Sicree R, King H. Global Prevalence of Diabetes: Estimates for the year 2000 and projections for 2030. Diabetes Care. 2004;27:1047–1053. doi: 10.2337/diacare.27.5.1047. [DOI] [PubMed] [Google Scholar]
- Winocur G, Greenwood CE, Piroli GG, Grillo CA, Reznikov LR, Reagan LP, McEwen BS. Memory impairment in obese Zucker rats: An investigation of cognitive function in an animal model of insulin resistance and obesity. Behavioral Neuroscience. 2005;119:1389–1395. doi: 10.1037/0735-7044.119.5.1389. [DOI] [PubMed] [Google Scholar]