

Abstract
Earlier studies from this and other laboratories show that ethanol induces apoptotic death of fetal and neonatal neurons. One mechanism that underlies these effects is the ethanol-associated reduction in the phosphatidylinositol 3′ kinase pro-survival pathway. Another mechanism involves the oxidative stress caused by the ethanol-associated increase in reactive oxygen species (ROS).
In the present study, we used the murine HN2-5 hippocampal-derived cell line to investigate the effects of ethanol on ROS levels and apoptosis. We also investigated the potential neuroprotective effects of two structurally unrelated antioxidants: N-acetylcysteine (NAC) and melatonin. The results demonstrate that NAC blocked an ethanol-associated increase in ROS. In addition, NAC and melatonin prevented the augmentation of apoptosis in ethanol-treated neurons. Both antioxidants significantly elevated the expression of the anti-apoptotic gene XIAP in ethanol-treated and/or control neurons and melatonin increased Bcl-2 expression in ethanol-treated neurons. Thus, it is possible that the neuroprotective effects of NAC and melatonin involve their ability to augment the expression of one or more anti-apoptotic gene as well as their classical antioxidant actions. Additional studies are needed to establish the effectiveness of these antioxidants to prevent the loss of neurons which accompanies in utero exposure to ethanol.
Keywords: ethanol, antioxidant, apoptosis, neuroprotection, N-acetylcysteine, melatonin
Introduction
Although human alcohol consumption during pregnancy leads to severe physical, mental and behavioral deficits in children (Riley and McGee, 2005), there are no pharmacological therapeutic treatments available to prevent the ethanol-associated damge to the developing central nervous system. Human and animal studies demonstrate that early exposure to ethanol reduces the size of specific brain areas (Riley et al., 2004). In vitro and in vivo studies in rodent models show that ethanol decreases the number of developing cerebellar granule, Purkinje, hippocampal, and cortical neurons (Bonthius et al., 1996; Maier et al., 1999; Jacobs and Miller, 2001; Light et al., 2002; Heaton et al., 2004; Olney, 2004). Ethanol also reduces serotonin (5-HT) neurons and their projections Tajuddin and Druse, 1999; 2001; Sari and Zhou, 2004; Zhou et al., 2005). The ethanol-associated loss of neurons appears to be caused by apoptotic cell death (Ramachandran et al., 2003; Druse et al., 2004; 2005; 2007; Dikranian et al., 2005; Chen et al., 2006). Apoptotic death of neurons is preceded by increased reactive oxygen species (ROS) and mitochondrial dysfunction (Chu et al., 2007) as well as damage to DNA (Cherian et al., 2008).
One mechanism by which ethanol can damage CNS tissue and augment apoptosis is by increasing oxidative stress (Montoliu et al., 1995; Heaton et al., 2003b; Ramachandran et al., 2003; Marino et al., 2004; Pierce et al., 2006). The brain is particularly susceptible to oxidative stress because of its high oxygen consumption, high polyunsaturated fatty acid content, and low antioxidant defenses (Gruener et al., 1991; Lau et al., 2005). Moreover, in vivo studies show that ethanol reduces endogenous defenses against oxidative stress in CNS tissue. That is, ethanol treatment reduces levels of the endogenous antioxidant glutathione (GSH) (Uysal et al., 1989; Reddy et al., 1999; Calabrese et al., 2000) and alters activities of the antioxidant enzymes superoxide dismutase and catalase (Heaton et al., 2003a; 2003b).
Because of the devastating effects of in utero ethanol exposure on the developing CNS, there is considerable interest in identifying potential therapeutic agents that might prevent this damage. Although an effective therapeutic agent has yet to be used in humans, in vivo and in vitro studies in rodents show that several antioxidants, a 5-HT1A agonist, and other agents may be able to provide neuroprotection (Druse et al., 2004; 2005; Heaton et al., 2004; Marino et al., 204; Siler-Marsiglio et al., 2004; Antonio and Druse, 2008). The present investigation studied the potential neuroprotective/anti-apoptotic effects of the antioxidants N-acetyl cysteine (NAC) and melatonin in HN2-5 cells. Like the fetal rhombencephalic neurons used in our prior investigations of antioxidants and 5-HT1A agonists, the hippocampus-derived HN2-5 cells contain the 5- HT1A receptor (Lee et al., 1990; Singh et al., 1996). The present study also investigated non-antioxidant effects of NAC and melatonin that might contribute to their neurprotective effects; these studies evaluated the influence of the two antioxidants on the expression of the important anti-apoptotic genes XIAP and Bcl-2.
NAC is of potential interest as a neuroprotective agent against ethanol-induced apoptosis because it both reduces oxidative stress and prevents cell death in other models of oxidative stress (Jayalakshmi et al., 2005; Arakawa et al., 2006. Reportedly, NAC can counteract oxidative stress and promote survival by increasing GSH levels (Bosch-Morell et al., 1998; Arakawa et al., 2006) and by augmenting the activity of glutathione peroxidase (Bosch-Morell et al., 1998). The in vivo effects of NAC on CNS tissue are likely to involve astrocytes, which play an important role in GSH recycling (Watts et al., 2005). Of interest, NAC treatment can influence gene transcription. That is, NAC can reverse changes in gene expression that were originally mediated by ischemia-reperfusion (Maddika et al., 2009), As3+ (Thompson et al., 2009), or hyperglycemia (Hung et al., 2009).
Melatonin is another antioxidant of potential therapeutic interest. Melatonin is an indole, which can reduce oxidative stress in vitro (Tan et al., 2000) and in models of neurodegenerative diseases (Iacovitti et al., 1997; Cabrera et al., 2000; Feng and Zhang, 2004). It can also attenuate the increase in ROS that accompanies treatment with β-amyloid (Feng and Zhang, 2004) or hydrogen peroxide (Juknat et al., 2005). These effects of melatonin are likely mediated by its action as a free radical scavenger (Zang et al., 1998) and by its ability to increase the expression of genes that encode the endogenous antioxidant enzymes glutathione peroxidase, copper-zinc superoxide dismutase, and manganese superoxide dismutase (Rodríguez et al., 1998). This study investigated whether melatonin also increased the expression of two specific anti-apoptotic/pro-survival genes, Bcl2 and XIAP.
Results
Ethanol increases ROS formation in HN2-5 cells NAC attenuates this effect
Figure 1 depicts data obtained from four similar experiments in which HN2-5 cells were pre-treated with NAC for 30 minutes and then co-treated with NAC and 100 mM ethanol for 1 hour. During the 1-hour interval ROS levels were measured using the fluorescent dye 2′,7′-dichlorodihydrofluorescein (DCF). The results of these studies show that ethanol treatment significantly elevates the levels of ROS. Typically, a significant increase in ROS is detected as early as 30 minutes after the addition of ethanol (∼40% increase, p < .01); by 1 hour ROS levels are ∼80% greater in ethanol-treated than control neurons (p < .01). NAC treatment reduced ROS levels in ethanol-treated cultures to values that are significantly below those in their time-matched groups (p < .01). Although this and another laboratory (Giusti et al., 1996) had technical difficulty using melatonin in DCF assays, there is clear evidence that melatonin scavenges ROS (Tan et al., 2000).
Figure 1. N-Acetylcysteine blocks the ethanol-associated increase in ROS.
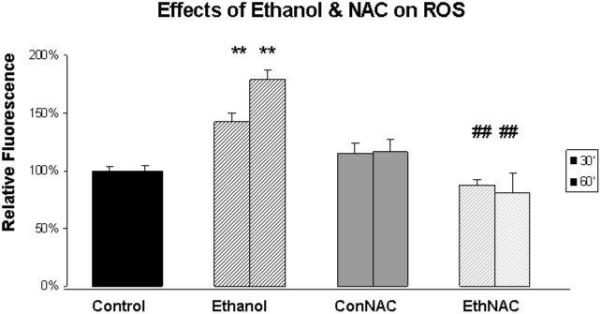
DCF was used to measure ROS in cultures of HN2-5 cells maintained in the presence of 0 or 100 mM ethanol and 0 or 1 mM NAC. DCF fluoresecence was measured over a 1-hour period in 12 replicate wells/sample. Results are presented from the 30′ and 60′ time points as the mean ± SEM of values obtained from four similar experiments. The ** identify values that are significantly different from those in the time-matched untreated control at p < .01; ## identifies values from cells treated both with ethanol and NAC that were significantly different from values obtained from ethanol-treated cells (p < .01). The abbreviations Con, Eth, and NAC are used in place of Control, Ethanol, and N-acetylcysteine, respectively.
Ethanol significantly increases the percentage of apoptotic HN2-5 cells; co-treatment with NAC and melatonin prevents this ethanol-associated damage
Figures 2A and 2B include representative images of HN2-5 cells stained with Hoechst 33342. These cells were treated with ethanol and NAC (Figure 2A) or melatonin (Figure 2B). This laboratory previously demonstrated that Hoechst 33342, which stains fragmented/apoptotic nuclei, identifies the same apoptotic cells as TUNEL (terminal deoxynucleotidyl transferase-mediated dUTP-biotin nick end-labeling); TUNEL stains fragmented DNA (Druse et al., 2004). Each value presented in the graphic representations in Figures 2C and 2D was obtained from 4-6 separate studies in which > 2000 cells were analyzed per treatment group. Ethanol treatment caused a 2.5- to 3-fold increase in the percentage of apoptotic HN2-5 cells. Importantly, co-treatment with either NAC (Figure 2C) or melatonin (Figure 2D) blocked the ethanol-associated increase in apoptosis; the percentage of apoptotic neurons in cultures treated with ethanol plus NAC or melatonin were comparable to those in untreated (no ethanol) cultures (p > .05).
Figure 2. Ethanol augments apoptosis in HN2-5 cells; both NAC and melatonin provide neuroprotection.
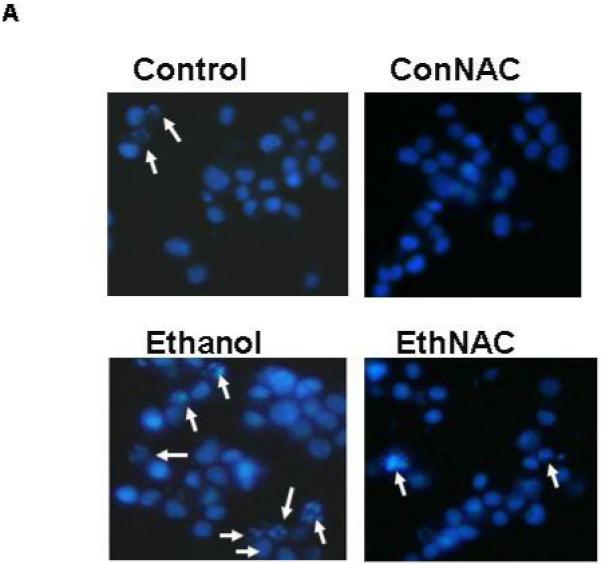
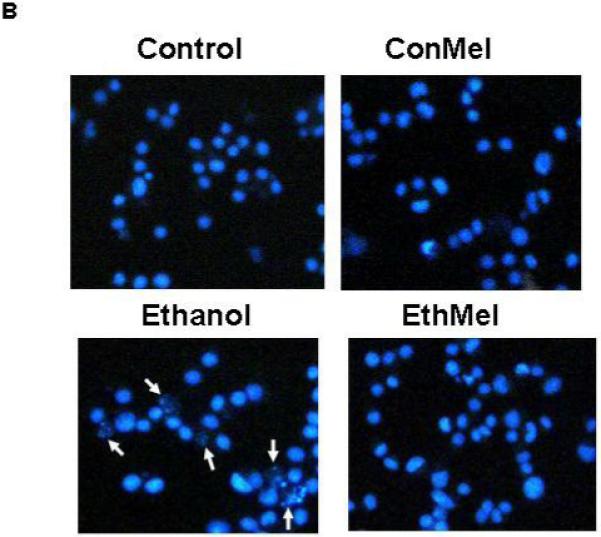
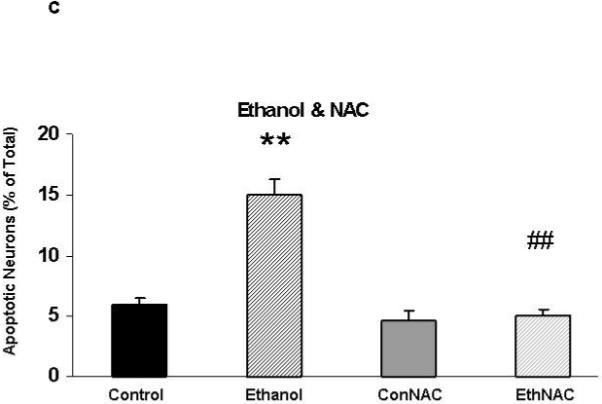
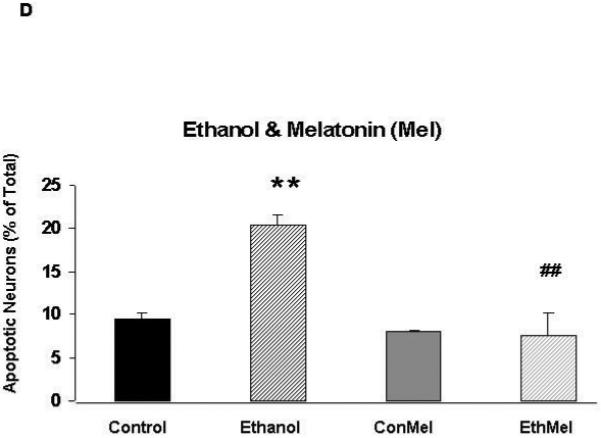
This figure includes representative images of HN2-5 cells, stained with Hoechst 33342, that were co-treated with ethanol (0 or 100 mM) and either 0 or 1 mM NAC (Figure 2A) or 0 or 1 μM melatonin (Figure 2B). Apoptotic cells are identified with arrows. Figures 2C and 2D include graphic representations of the results obtained in experiments which used Hoechst 33342 to identify apoptotic HN2-5 cells in cultures co-treated with ethanol and NAC (Figure 2C) or melatonin (Figure 2D). Results are presented as the mean percent apoptotic cells ± SEM of values obtained from four to six separate experiments. Treatment of HN2-5 cells with ethanol for 24 hours results in a 2- to 2.5-fold increase in apoptosis. Co-treatment of ethanol cultures with either NAC or melatonin decreased the percentage of apoptotic neurons and prevented ethanol-associated increase apoptosis. Values from ethanol-treated cultures were significantly greater than those in control cultures (**, p < .01), and those from cultures co-treated with NAC or melatonin and ethanol were significantly less than those from the ethanol-treated cultures (##, p < .01) and comparable to control values (p > .05). The abbreviations Con, Eth, NAC and Mel are used in place of Control, Ethanol, N-acetylcysteine, and melatonin, respectively.
A 24-hour exposure of HN2-5 cells to ethanol reduced the expression of the gene that encodes the pro-survival protein XIAP by ∼50% (p < .05); co-treatment with either NAC or melatonin prevented this ethanol-associated reduction (Figure 3)
Figure 3. Ethanol reduced the expression of XIAP. Co-treatment with NAC (Figure 3A) or melatonin (Figure 3B) augmented expression of XIAP in ethanol-treated HN2-5 cells; melatonin also increased XIAP in control neurons.

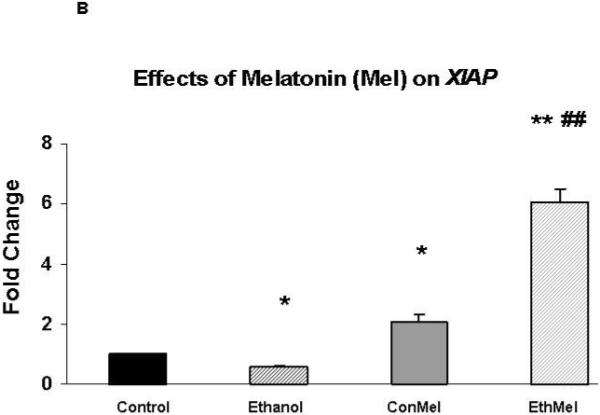
Each value represents the mean ± the SEM of values obtained from three to four separate experiments. Values are expressed as the fold change in mRNA as calculated by the 2 -ΔΔC T method [Livak and Schmittgen, 2001]. The abbreviations Con, Eth, NAC and Mel are used in place of Control, Ethanol, N-acetylcysteine, and melatonin, respectively. * and ** identify values that are significantly different from the control value at p < .05 and p< .01, respectively; the ## identifies values in the EthNAC or EthMel groups that are significantly different from those in the Ethanol group (p < .01).
In fact, both antioxidants significantly augmented XIAP in ethanol-treated neurons. In comparison with control neurons (no ethanol, no antioxidant) XIAP was increased ∼1.5 fold in HN2-5 cells that were co-treated with ethanol and NAC (p < .05) and 6-fold in cells co-treated with melatonin (p < .01). Melatonin also increased XIAP in control neurons (p < .05).
Expression of the gene that encodes the pro-survival protein Bcl-2 was augmented by melatonin (Figure 4)
Figure 4. Co-treatment of ethanol-treated HN2-5 cells with melatonin augmented expression of XIAP.
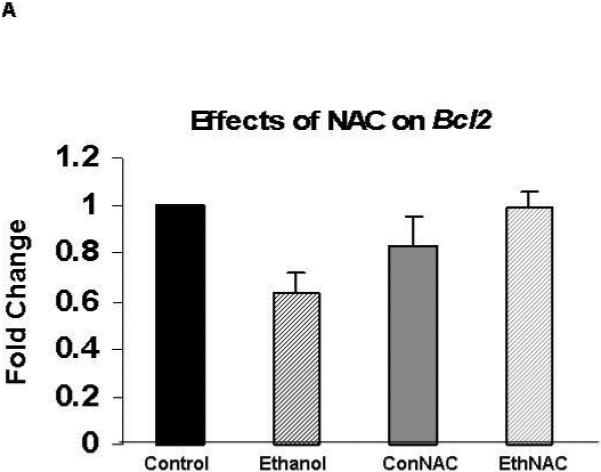
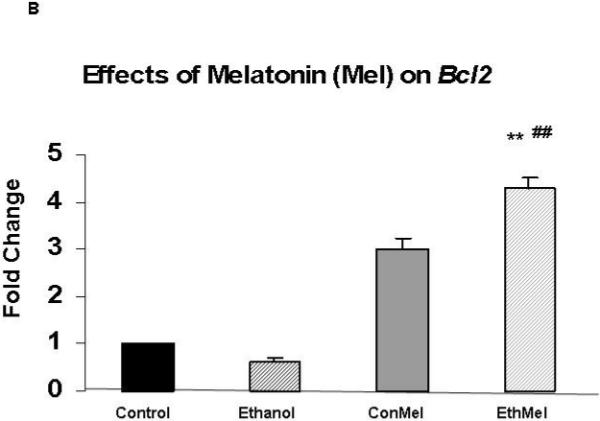
Each value represents the mean ± the SEM of values obtained from three to four separate experiments obtained from cultures of HN2-5 cells that were co-treated with NAC (Figure 4A) or melatonin (Figure 4B). Values are expressed as the fold change in mRNA as calculated by the 2 -ΔΔCT method [Livak and Schmittgen, 2001]. The abbreviations Con, Eth, NAC and Mel are used in place of Control, Ethanol, N-acetylcysteine, and melatonin, respectively. The ** identifies values that are significantly different from the control value at p< .01; the ## identifies values in the EthMel group that are significantly different from those in the Ethanol group (p <. 01).
Melatonin increased Bcl-2 expression in ethanol-treated HN2-5 cells in comparison to those maintained both under control conditions (4-fold increase, p < .01) and in the presence of ethanol (p <.01). NAC did not significantly alter Bcl-2 in either control or ethanol-treated neurons (p > .05).
Discussion
Using hippocampus-derived HN2-5 cells (Lee et al., 1990; Singh et al., 1996), which proved to be an effective model to study ethanol-associated apoptosis, the present study demonstrated the neuroprotective effects of two structurally unrelated antioxidants, NAC and melatonin. Their neuroprotective effects are likely to include their ability to counteract oxidative stress. In addition, the results of this investigation raise the possibility that the ability of NAC and melatonin to up-regulate genes that encode important anti-apoptotic proteins, i.e., Bcl-2 and XIAP, might also be important.
In HN2-5 cells, as in cultured neurons (Montoliu et al., 1995; Heaton et al., 2002; Ramachandran et al., 2003), ethanol-treatment causes a brisk rise in ROS. There is strong evidence that the ethanol-associated increase in ROS is associated with an impaired ability of the cells to detoxify the elevated levels of ROS (Uysal et al., 1989; Calabrese et al., 2000; Heaton et al., 2003; Watts et al., 2005), mitochondrial dysfunction, DNA damage and apoptotic death (Chu et al., 2007; Cherian et al., 2008). Unfortunately, neural tissue is particularly susceptible to the damage caused by oxidative stress because of its high oxygen consumption, high polyunsaturated fatty acid content, and low antioxidant defenses (Halliwell and Gutteridge, 1985; Lau et al., 2005).
Co-treatment with NAC prevents the ethanol-associated rise in ROS in HN2-5 cells. There is additional evidence that NAC reduces oxidative stress in hippocampal neurons (Jayalakshmi et al., 2005) and that melatonin scavenges ROS (Tan et al., 2000). In addition to its effects as a classical antioxidant, NAC can reduce oxidative stress by elevating intracellular levels of the endogenous antioxidant GSH (Mayer and Noble, 1994; Juknat et al., 2005) and by augmenting the activities of the antioxidant enzymes glutathione peroxidase and glutathione reductase (Jayalakshmi et al., 2005). Reportedly, melatonin’s effects also involve its ability to increase the activities of superoxide disumutase and glutathione reductase in brain (Okatani et al., 2000) and other organs (Liu and Ng, 2000).
Co-treatment with either NAC or melatonin prevents ethanol-associated apoptosis in HN2-5 cells. Protection was also found when NAC (Jayalakshmi et al., 2005; Arakawa et al., 2006) or melatonin treatment was used to counter the effects of other models of oxidative stress (Feng and Zhang, 2004; Juknat et al., 2005). Thus, it is likely that a part of the neuroprotective effects are due to the ability of these antioxidants to reduce oxidative stress. Similarly, this laboratory showed that ethanol-associated apoptosis in fetal rhombencephalic neurons is prevented by co-treatment with several different antioxidants, including phenols (EGCG, curcumin and resveratrol), an indole (melatonin) and a dithiol (α-lipoic acid) (Antonio and Druse, 2008). Others demonstrated that treatment with vitamin E (Heaton et al., 2004; Marino et al., 2004) or the bioflavonoid pycnogenol (Siler-Marsiglio et al., 2004) exert similar neuroprotective effects on ethanol-treated neurons. However, the neuroprotective effects of antioxidants are not limited to the attenuation of an ethanol-associated increase in ROS.
The results of the present study also suggest that a part of the neuroprotective effects of NAC and melatonin might include non antioxidant actions, i.e., their ability to up-regulate the expression of genes encoding one or more anti-apoptotic proteins. For example, both NAC and melatonin augmented expression of XIAP in ethanol-treated HN2-5 cells. XIAP encodes the X-inhibitor of apoptosis protein, an important inhibitor of apoptosis via its inhibition of caspase-3 (Salvesen et al., 2002). In addition, melatonin increased Bcl-2 expression. The Bcl-2 protein can block apoptosis by binding to proapoptotic Bax and preventing the formation of pores in the outer mitochondrial membrane (Tsujimoto et al., 2000). If the NAC and/or melatonin-mediated increase in the expression of XIAP and Bcl-2 in the antioxidant-treated neurons is paralleled by changes in protein, such changes could contribute to the neuroprotective effects of these molecules. In fact, another investigator (Heaton et al., 2004) reported that treatment with the antioxidant vitamin E prevented an ethanol-associated reduction in the levels of the Bcl-2 protein. That melatonin but not NAC up-regulated Bcl-2 suggests that the regulation of this gene is not limited to classical antioxidant effects.
In summary, this study demonstrates the anti-apoptotic effects of the antioxidants NAC and melatonin. While their neuroprotective effects are undoubtedly due in part to their actions as classical antioxidants, the present studies raise the possibility that these effects might also involve the up-regulation of genes that encode pro-survival/anti-apoptotic proteins. Interestingly, this laboratory demonstrated that two additional neuroprotective agents, i.e., the 5-HT1A agonist ipsapirone and S100B, both prevented ethanol-associated apoptosis and augmented expression of XIAP and either Bcl-xl or Bcl-2 (Druse et al., 2006; 2007). A common thread in the neuroprotective effects of three unrelated types of agents, i.e., antioxidants, ipsapirone, and S100B, might involve their ability to up-regulate the expression of these and possibly other neuroprotective genes. Importantly, ipsapirone was also shown to prevent the in vivo reduction in 5-HT neurons that accompanies in utero ethanol exposure (Tajuddin and Druse, 1999; 2001). Additional studies are needed to determine whether NAC and melatonin also exert neuroprotective effects against ethanol-associated neuronal loss in vivo.
Experimental Procedure
Cell culture of HN2-5 cells
Mouse hippocampal neuron-derived HN2-5 cells were generously donated by Dr. Probal Banerjee (SUNY, Staten Island). Cells were seeded onto 55 cm2 culture plates (6 million cells/plate) for RNA studies, 1.8 cm2 chambered slides (250,000 to 300,000 cells/chamber) for Hoechst analyses, and 96-well plates (50,000 cells/well) for DCF studies. Plates and slides were previously coated with poly-L-lysine (Sigma-Aldrich, St. Louis, MO). Cells were cultured according to established protocols (Singh et al., 1996; Adayev et al., 1999; 2003) with some modifications. After cultures reached 70% confluency, cells were maintained overnight in differentiation media [DMEM containing 5 μM retinoic acid (Sigma-Aldrich, St. Louis, MO), 1% FBS and 200 μg/ml neomycin]. Preliminary studies established that our culture conditions generated a consistent degree of differentiation (∼85%) and cell morphology. Preliminary studies also determined the optimal dose of ethanol and antioxidants and the optimal treatment time. The media concentrations of ethanol (100 mM) and serum (0.5%, v/v) were chosen because cells cultured under these conditions are sensitive to ethanol with regard to ROS levels and apoptosis. The selected neuroprotective dose of NAC (1 mM NAC, Sigma-Aldrich, St. Louis, MO) is consistent with that shown to be protective in a previous study (Mayer and Noble, 1994); the selected concentration of melatonin (1 μM melatonin, Sigma-Aldrich, Milwaukee, WI) is identical to that which reduced oxidative damage to DNA (López-Burillo et al., 2003). Differentiated cells were treated with no ethanol (control) or 100 mM ethanol (ethanol) in DMEM containing 0.5% FBS for 24 hours; they were also co-cultured in the presence or absence of NAC or melatonin. The ethanol concentration was maintained using a chamber system, previously described by this laboratory (Eriksen et al., 2002). This system maintains the ethanol concentration in the media at ≥85% of the initial concentration (Eriksen et al., 2002).
DCF determination of ROS
ROS was measured by using 2′,7′- dichlorodihydrofluorescein diacetate (DCF-DA, Sigma-Aldrich, St. Louis, MO) dye. HN2-5 cells were grown and cultured on 96-well plates as described. After the overnight differentiation period, media was removed. Cells were then washed with warmed phosphate buffered saline (PBS) and incubated with DCF-DA in PBS at 37°C for 20 minutes. The DCF-containing media was removed and cells were washed with warmed PBS to remove unincorporated DCF. Ethanol in PBS was added immediately prior to measurements of fluorescence at excitation and emission wavelengths of 485 nm and 538 nm, respectively. In each experiment, one “n” represents the mean of values obtained from 12 wells. Each experiment was repeated four times with samples generated from separate cultures.
Detection of apoptotic cells with Hoechst 33342
After the 24-hour treatments, media was removed from the cells in the 4-chambered slides, cells were washed once with PBS, and fixed with 4% formaldehyde (Sigma-Aldrich, St. Louis, MO) in PBS. Cells were then washed with PBS and incubated with a 1:500 dilution of stock solution (1.2 mg/ml in PBS) of Hoechst 33342 (Sigma-Aldrich, St. Louis, MO) in a dark humidified chamber at 37°C for 15 min. A Nikon Microphot fluorescence microscope was used in the ultraviolet range to view Hoechst-stained cells. Images were captured using a 20X objective and later analyzed at a higher computer-enhanced magnification (>100X). Neurons that were identified as apoptotic contained fragmented nuclei; non-apoptotic cells had an intact nucleus and lacked the fragmented nuclei. In earlier studies from this laboratory (Druse et al., 2004), we showed that the identical population of apoptotic fetal rhombencephalic neurons in control and ethanol-treated cultures were identified by Hoechst 33342, which stains fragmented nuclei, and TUNEL, which labels fragmented DNA in apoptotic cells. In each of 4 to 6 separate experiment 500-600 neurons were analyzed; these neurons were counted on ∼20 fields from two chambers (10 fields/chamber). Thus, a total of > 2000 were analyzed for each treatment group. Samples were analyzed methodically without consideration of treatment.
Quantitative real-time RT-PCR
As described previously (Druse et al., 2006), Trizol (Life Technology, Gaithersburg, MD) was used to extract total RNA from cultured HN2-5 cells. RNA was dissolved in 25 μl of DEPC-treated H2O, which was treated with DNA-free (Ambion) and stored at -80° until use. Using 1 to 2 μg of total RNA (DNA-free), single strand cDNA was synthesized using the First Strand cDNA synthesis kit (Pharmacia Biotech, Piscataway, NJ).
cDNA (DNA equivalent of 40 ng to 20 ng of total RNA) was used at a dilution of 1:2 or 1:4 in 20 μl of 1X Platinum Quantitative PCR Super Mix-UDG (1.5 U Platinum Taq DNA polymerase, 50 mM KCl, 3 mM MgCl2, 20 mM Tris-HCl (pH 8.4), 200 mM each of dGTP, dATP, and CTP, 400 mM dUTP, 1 U UDG) (Life Technology), 1/40,000 SYBR Green (Molecular Probes, Eugene, Oregon), 0.25 mM Rox (Life Technology), and 0.25 μM primers (Druse et al., 2006). PCR amplifications were performed in triplicate using a Perkin-Elmer Gene Amp 7300 Sequence Detector thermal cycler (Applied Biosystems, Foster City, CA). GAPDH expression was assessed to normalize sample inputs; GAPDH was not influenced by the experimental treatments used in this study. Standard curves were generated using plasmid containing target genes (Druse et al., 2006) and serial dilutions of known amounts of the input copy number of target genes. For each set of primers, a triplicate RT-PCR reaction was included that lacked cDNA or known DNA template. Specific primary sequences for XIAP and Bcl-2 (Druse et al., 2006) were selected using the Primer Express program (Applied Biosystems) and sequences available from the NCBI database. Primers were synthesized by Life Technology. Values were obtained from four to six separate experiments.
Statistical Analyses
Statistical significance of data was determined using a 2-way ANOVA and post-hoc analyses (p < 0.05). Values from real-time RT-PCR analyses were determined using the 2 -ΔΔCT method (Livak and Schmittgen, 2001), which facilitates the analysis of relative changes in gene expression from real-time quantitative RT-PCR experiments.
Glossary
Abbreviations
- NAC
N-acetylcysteine
- Mel
Melatonin
- ROS
reactive oxygen species
- DCF-2′
7′-dichlorodihydrofluorescein
- GSH
glutathione
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Literature references
- Adayev T, El-Sherif Y, Barua M, Penington NJ, Banerjee P. Agonist stimulation of the serotonin1A receptor causes suppression of anoxia-induced apoptosis via mitogen-activated protein kinase in neuronal HN2-5 cells. J. Neurochem. 1999;72:1489–1496. doi: 10.1046/j.1471-4159.1999.721489.x. [DOI] [PubMed] [Google Scholar]
- Adayev T, Ray I, Sondhi R, Sobocki T, Banerjee P. The G-protein-coupled 5-HT1A receptor causes suppression of caspase-3 through MAPK and protein kinase Cα. Biochim. Biophys. Acta. 2003;1640:85–96. doi: 10.1016/s0167-4889(03)00023-5. [DOI] [PubMed] [Google Scholar]
- Antonio AM, Druse MJ. Antioxidants prevent ethanol-associated apoptosis in fetal rhombencephalic neurons. Dev. Brain Res. 2008;1204:16–23. doi: 10.1016/j.brainres.2008.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arakawa M, Ishimura A, Arai Y, Kawabe K, Suzuki S, Ishige K, Ito Y. N-acetylcysteine selectively protects cerebellar granule cells from 4-hydroxynonenal-induced cell death. Neurosci. Res. 2006;55:255–263. doi: 10.1016/j.neures.2006.03.008. [DOI] [PubMed] [Google Scholar]
- Bonthius DJ, Bonthius NE, Napper RM, Astley SJ, Clarren SK. Purkinje cell deficits in nonhuman primates following weekly exposure to ethanol during gestation. Teratology. 1996;53:230–236. doi: 10.1002/(SICI)1096-9926(199604)53:4<230::AID-TERA5>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- Bosch-Morell F, Martinez-Soriano F, Colell A, Fenandez-Checa JC, Romero FJ. Chronic ethanol feeding induces cellular antioxidants decrease and oxidative stress in rat peripheral nerves. Effect of S-adenosyl-L-methionine and N-acetyl-L-cysteine. Free Radic. Biol. Med. 1998;25:365–368. doi: 10.1016/s0891-5849(98)00036-7. [DOI] [PubMed] [Google Scholar]
- Cabrera J, Reiter RJ, Tan DX, Qi W, Sainz RM, May JC, Garcia JJ, Kim SJ, El-Sokkary G. Melatonin reduces oxidative neurotoxicity due to quinolinic acid: in vitro and in vivo findings. Neuropharmacology. 2000;39:507–514. doi: 10.1016/s0028-3908(99)00128-8. [DOI] [PubMed] [Google Scholar]
- Calabrese V, Testa G, Ravagna A, Bates TE, Stella AMG. HSP70 induction in the brain following ethanol administration in the rat: regulation by glutathione redox state. Biochem, Biophys. Res. Commun. 2000;269:397–400. doi: 10.1006/bbrc.2000.2311. [DOI] [PubMed] [Google Scholar]
- Cherian PP, Schenker S, Henderson GI. Ethanol-mediated DNA damage and PARP-1 apoptotic responses in cultured fetal cortical neurons. Alcohol. Clin. Exp. Res. 2008;32:1884–1892. doi: 10.1111/j.1530-0277.2008.00769.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu J, Tong M, de la Monte SM. Chronic ethanol exposure causes mitochondrial dysfunction and oxidative stress in immature central nervous system neurons. Acta. Neuropathol. 2007;113:659–673. doi: 10.1007/s00401-007-0199-4. [DOI] [PubMed] [Google Scholar]
- Dikranian K, Qin YQ, Labruyere J, Nemmers B, Olney JW. Ethanol-induced neuroapoptosis in the developing rodent cerebellum and related brain stem structures. Brain Res. Dev. Brain Res. 2005;155:1–13. doi: 10.1016/j.devbrainres.2004.11.005. [DOI] [PubMed] [Google Scholar]
- Druse MJ, Tajuddin NF, Gillespie RA, Le PT. The serotonin-1A agonist ipsapirone prevents ethanol-associated death of total rhombencephalic neurons and prevents the reduction of fetal serotonin neurons. Brain Res. Dev. Brain Res. 2004;150:79–88. doi: 10.1016/j.devbrainres.2004.02.009. [DOI] [PubMed] [Google Scholar]
- Druse MJ, Tajuddin NF, Gillespie RA, Dickson E, Atieh M, Pietrzak CA, Le PT. Signaling pathways involved with serotonin1A agonist-mediated neuroprotection against ethanol-induced apoptosis of fetal rhombencephalic neurons. Brain Res. Dev. Brain Res. 2005;159:18–28. doi: 10.1016/j.devbrainres.2005.06.015. [DOI] [PubMed] [Google Scholar]
- Druse MJ, Tajuddin NF, Gillespie RG, Le T. The effects of ethanol and the serotonin1A agonist ipsapirone on the expression of the serotonin1A receptor and several antiapoptotic proteins in fetal rhombencephalic neurons. Brain Res. 2006;1092:79–86. doi: 10.1016/j.brainres.2006.02.065. [DOI] [PubMed] [Google Scholar]
- Druse M, Gillespie RA, Tajuddin NF, Rich M. S100B-mediated protection against the pro-apoptotic effects of ethanol on fetal rhombencephalic neurons. Brain Res. 2007;1150:46–54. doi: 10.1016/j.brainres.2007.02.092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eriksen JL, Gillespie R, Druse MJ. Effects of ethanol and 5-HT1A agonists on astroglial S100B. Brain Res. Dev. Brain Res. 2002;139:97–105. doi: 10.1016/s0165-3806(02)00510-2. [DOI] [PubMed] [Google Scholar]
- Feng Z, Zhang J. Melatonin reduces amyloid beta-induced apoptosis in pheochromocytoma (PC12) cells. J. Pineal Res. 2004;37:257–66. doi: 10.1111/j.1600-079X.2004.00164.x. [DOI] [PubMed] [Google Scholar]
- Giusti P, Franceshchini D, Petrone M, Manev H, Floreani M. In vitro and in vivo protection against kainate-induced excitotoxicity by melatonin. J. Pineal Res. 1996;20:226–231. doi: 10.1111/j.1600-079x.1996.tb00263.x. [DOI] [PubMed] [Google Scholar]
- Gruener N, Gozlan O, Goldstein T, Davis J, Besner I, Iancu TC. Iron, transferrin, and ferritin in cerebrospinal fluid of children. Clin. Chem. 1991;37:263–265. [PubMed] [Google Scholar]
- Halliwell B, Gutteridge JMC. Oxygen Radicals and the Nervous System. 1985. [Google Scholar]
- Heaton MD, Paiva M, Mayer J, Miller R. Ethanol-mediated generation of reactive oxygen species in developing rat cerebellum. Neurosci. Lett. 2002;334:83–86. doi: 10.1016/s0304-3940(02)01123-0. [DOI] [PubMed] [Google Scholar]
- Heaton MB, Paiva M, Madorsky I, Mayer J, Moore DB. Effects of ethanol on neurotrophic factors, apoptosis-related proteins, endogenous antioxidants, and reactive oxygen species in neonatal striatum: relationship to periods of vulnerability. Dev. Brain Res. 2003a;140:237–252. doi: 10.1016/s0165-3806(02)00610-7. [DOI] [PubMed] [Google Scholar]
- Heaton MB, Paiva M, Madorsky I, Shaw G. Ethanol effects on neonatal rat cortex: comparative analyses of neurotrophic factors, apoptosis-related proteins, and oxidative processes during vulnerable and resistant periods. Dev. Brain Res. 2003b;145:249–262. doi: 10.1016/j.devbrainres.2003.08.005. [DOI] [PubMed] [Google Scholar]
- Heaton MB, Madorsky I, Paiva M, Siler-Marsiglio KI. Vitamin E amelioration of ethanol neurotoxicity involves modulation of apoptosis protein levels in neonatal rat cerebellar granule cells. Dev. Brain Res. 2004;150:117–124. doi: 10.1016/j.devbrainres.2004.03.010. [DOI] [PubMed] [Google Scholar]
- Hung KY, Liu SY, Kao SH, Huang JW, Chiang CK, Tsai TJ. N-acetylcysteine-mediated antioxidation prevents hyperglycemia-induced apoptosis and collagen synthesis in rat mesangial cells. Am. J.Nephrol. 2009;29:192–202. doi: 10.1159/000155657. [DOI] [PubMed] [Google Scholar]
- Iacovitti L, Stull ND, Johnston K. Melatonin rescues dopamine neurons from cell death in tissue culture models of oxidative stress. Brain Res. 1997;768:317–26. doi: 10.1016/s0006-8993(97)00668-9. [DOI] [PubMed] [Google Scholar]
- Jacobs JS, Miller MW. Proliferation and death of cultured fetal neocortical neurons: effects of ethanol on the dynamics of cell growth. J. Neurocytology. 2001;30:391–401. doi: 10.1023/a:1015013609424. [DOI] [PubMed] [Google Scholar]
- Jayalakshmi J, Sairam M, Singh SB, Sharma SK, Ilavazhagan G, Banerjee PK. Neuroprotective effect of N-acetyl cysteine on hypoxia-induced oxidative stress in primary hippocampal culture. Brain Res. 2005;1046:97–104. doi: 10.1016/j.brainres.2005.03.054. [DOI] [PubMed] [Google Scholar]
- Juknat AA, Mendez M, Quaglino A, Fameli CI, Mena M, Kotler ML. Melatonin prevents hydrogen peroxide-induced Bax expression in cultured rat astrocytes. J. Pineal Res. 2005;38:84–92. doi: 10.1111/j.1600-079X.2004.00166.x. [DOI] [PubMed] [Google Scholar]
- Lau FC, Shukitt-Hale B, Joseph JA. The beneficial effects of fruit polyphenols on brain aging. Neurobiol. Aging. 2005;26(Suppl 1):128–132. doi: 10.1016/j.neurobiolaging.2005.08.007. [DOI] [PubMed] [Google Scholar]
- Lee HJ, Hammond D, Large TH, Roback J, Sim J, Brown D, Otten U, Wainer B. Neuronal properties and trophic activities of immortalized hippocampal cells from embryonic and young adult mice. J. Neurosci. 1990;10:1779–1787. doi: 10.1523/JNEUROSCI.10-06-01779.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Light KE, Belcher SM, Pierce DR. Time course and manner of Purkinje neuron death following a single ethanol exposure on postnatal day 4 in the developing rat. Neurosci. 2002;114:327–337. doi: 10.1016/s0306-4522(02)00344-5. [DOI] [PubMed] [Google Scholar]
- Liu F, Ng TB. Effect of pineal indoles on activities of antioxidant defense enzymes superoxide dismutase, catalase, and glutathione reductase, and levels of reduced and oxidized glutathione in rat tissues. Biochem. Cell. Bio. 2000;78:447–453. doi: 10.1139/o00-018. [DOI] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2-ΔΔCT method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- López-burillo S, Tan D-X, Mayo JC, Sainz RM, Manchester LC, Reiter RJ. Melatonin, xanthurenic acid, resveratrol, EGCG, vitamin C and α-lipoic aid differentially reduce oxidative DNA damage induced by Fenton reagents: a study of their individual and synergistic actions. J. Pineal Res. 2003;34:269–277. doi: 10.1034/j.1600-079x.2003.00041.x. [DOI] [PubMed] [Google Scholar]
- Maddika S, Elimban V, Chapman D, Dhalla NS. Role of oxidative stress in ischemia-reperfusion-induced alterations in myofibrillar ATPase activities and gene expression in the heart. Can. J. Pharmacol. 2009;87:120–129. doi: 10.1139/Y08-105. [DOI] [PubMed] [Google Scholar]
- Maier SE, Miller JA, Blackwell JM, West JR. Fetal alcohol exposure and temporal vulnerability: regional differences in cell loss as a function of the timing of binge-like alcohol exposure during brain development. Alcohol. Clin. Exp. Res. 1999;23:726–734. doi: 10.1111/j.1530-0277.1999.tb04176.x. [DOI] [PubMed] [Google Scholar]
- Marino MD, Aksenov MY, Kelly SJ. Vitamin E protects against alcohol-induced cell loss and oxidative stress in the neonatal rat hippocampus. Int J. Devl. Neuroscience. 2004;22:363–377. doi: 10.1016/j.ijdevneu.2004.04.005. [DOI] [PubMed] [Google Scholar]
- Mayer M, Noble M. N-Acetyl-L-cysteine is a pluripotent protector against cell death and enhancer of trophic factor-mediated cell survival in vitro. Proc. Natl. Acad. Sci. 1994;91:7496–7500. doi: 10.1073/pnas.91.16.7496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montoliu C, Sancho-Tello M, Azorin I, Burgal M, Valles S, Renau-Piqueras J, Guerri C. Ethanol increases cytochrome P4502E1 and induces oxidative stress in astrocytes. J. Neurochem. 1995;65:2561–2570. doi: 10.1046/j.1471-4159.1995.65062561.x. [DOI] [PubMed] [Google Scholar]
- Okatani Y, Wakatsuki A, Kaneda C. Melatonin increases activities of glutathione peroxidase and superoxide dismutase in fetal rat brain. J. Pineal Res. 2000;28:89–96. doi: 10.1034/j.1600-079x.2001.280204.x. [DOI] [PubMed] [Google Scholar]
- Olney JW. Fetal alcohol syndrome at the cellular level. Addiction Biol. 2004;9:137–149. doi: 10.1080/13556210410001717006. [DOI] [PubMed] [Google Scholar]
- Pierce DR, Cook CC, Hinson JA, Light KE. Are oxidative mechanisms primary in ethanol induced Purkinje neuron death of the neonatal rat? Neuroscience Lett. 2006;400:30–34. doi: 10.1016/j.neulet.2006.02.025. [DOI] [PubMed] [Google Scholar]
- Ramachandran V, Perez A, Chen J, Senthil D, Schenker S, Henderson G. In utero ethanol exposure causes mitochondrial dysfunction, which can result in apoptotic cell death in fetal brain: a potential role for 4-hydroxynonenal. Alcohol. Clin. Exp. Res. 2001;25:862–871. [PubMed] [Google Scholar]
- Ramachandran V, Watts LT, Maffi SK, Chen J, Schenker S, Henderson G. Ethanol-induced oxidative stress precedes mitochondrially mediated apoptotic death of cultured fetal cortical neurons. J. Neurosci. Res. 2003;74:577–588. doi: 10.1002/jnr.10767. [DOI] [PubMed] [Google Scholar]
- Reddy SK, Husain K, Schlorff EC, Scott RB, Somani SM. Dose response of ethanol ingestion on antioxidant defense system in rat brain subcellular fractions. Neurotoxicology. 1999;20:977–987. [PubMed] [Google Scholar]
- Riley EP, McGee CL, Sowell ER. Teratogenic effects of alcohol: a decade of brain imaging. Am. J. Med. Genet. C. Semin. Med. Genet. 2004;127:35–41. doi: 10.1002/ajmg.c.30014. [DOI] [PubMed] [Google Scholar]
- Riley EP, McGee CL. Fetal alcohol spectrum disorders: and overview with emphasis on changes in brain and behavior. Exp. Biol. Med. 2005;230:357–365. doi: 10.1177/15353702-0323006-03. [DOI] [PubMed] [Google Scholar]
- Rodriguez AB, Nogales G, Ortega E, Barriga C. Melatonin controls superoxide anion level: modulation of superoxide dismutase activity in ring dove heterophils. J. Pineal Res. 1998;24:9–14. doi: 10.1111/j.1600-079x.1998.tb00360.x. [DOI] [PubMed] [Google Scholar]
- Salvesen GS, Duckett CS. IAP proteins: blocking the road to deathߣs door. Nat. Rev. Mol. Cell. Biol. 2002;3:401–410. doi: 10.1038/nrm830. [DOI] [PubMed] [Google Scholar]
- Sari Y, Zhou FC. Prenatal alcohol exposure causes long-term serotonin neuron deficit in mice. Alcohol. Clin. Exp. Res. 2004;28:941–948. doi: 10.1097/01.alc.0000128228.08472.39. [DOI] [PubMed] [Google Scholar]
- Siler-Marsiglio KI, Paiva M, Madorsky I, Serrano Y, Neeley A, Heaton MB. Protective mechanisms of pycnogenol in ethanol-insulted cerebellar granule cells. Int. J. Neurobiol. 2004;61:267–276. doi: 10.1002/neu.20057. [DOI] [PubMed] [Google Scholar]
- Singh JK, Chromy BA, Boyers MJ, Dawson G, Banerjee P. Induction of the serotonin1A receptor in neuronal cells during prolonged stress and degeneration. J. Neurochem. 1996;66:2361–2372. doi: 10.1046/j.1471-4159.1996.66062361.x. [DOI] [PubMed] [Google Scholar]
- Tajuddin NF, Druse MJ. In utero ethanol exposure decreased the density of serotonin neurons. Maternal ipsapirone treatment exerted a protective effect. Brain Res. Dev. Brain Res. 1999;117:91–97. doi: 10.1016/s0165-3806(99)00102-9. [DOI] [PubMed] [Google Scholar]
- Tajuddin NF, Druse MJ. A persistent deficit of serotonin neurons in the offspring of ethanol-fed dams: protective effects of maternal ipsapirone treatment. Brain Res. Dev. Brain Res. 2001;129:181–188. doi: 10.1016/s0165-3806(01)00199-7. [DOI] [PubMed] [Google Scholar]
- Tan DX, Manchester LC, Reiter RJ, Plummer BF, Limson JSTW, Qi W. Melatonin directly scavenges hydrogen peroxide: a potentially new metabolic pathway of melatonin biotransformation. Free Rad. Biol. Med. 2000;29:1177–1185. doi: 10.1016/s0891-5849(00)00435-4. [DOI] [PubMed] [Google Scholar]
- Thompson JA, White CC, Cox DP, Chan JY, Kavanagh TJ, Fausto N, Franklin CC. Distinct Nrf1/2-independent mechanisms mediate As3+-induced glutamate-cysteine ligase subunit gene expression in murine hepatocytes. Free Radic. Biol. Med. 2009;46:1614–1625. doi: 10.1016/j.freeradbiomed.2009.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsujimoto Y, Shimizu S. Bcl-2 family: Life or death switch. FEBS Lett. 2000;466:6–10. doi: 10.1016/s0014-5793(99)01761-5. [DOI] [PubMed] [Google Scholar]
- Uysal M, Kutalp G, Ozdemirler G, Aykac G. Ethanol-induced changes in lipid peroxidation and glutathione content in rat brain. Drug Alcohol Depend. 1989;23:227–230. doi: 10.1016/0376-8716(89)90085-9. [DOI] [PubMed] [Google Scholar]
- Watts LT, Rathinam ML, Schenker S, Henderson G. Astrocytes protect neurons from ethanol-induced oxidative stress and apoptotic death. J. Neurosci. Res. 2005;80:655–666. doi: 10.1002/jnr.20502. [DOI] [PubMed] [Google Scholar]
- Zang LY, Cosma G, Gardner H, Vallyathan V. Scavenging of reactive oxygen species by melatonin. Biochim. Biophys. Acta. 1998;1425:469–477. doi: 10.1016/s0304-4165(98)00099-3. [DOI] [PubMed] [Google Scholar]
- Zhou FC, Sari Y, Powrozek TA. Fetal alcohol exposure reduces serotonin innervation and compromises development of the forebrain along the serotonergic pathway. Alcohol. Clin. Exp. Res. 2005;29:141–149. doi: 10.1097/01.alc.0000150636.19677.6f. [DOI] [PubMed] [Google Scholar]