

Abstract

A fructose-derived diacetate ketone has been shown to be an effective catalyst for asymmetric epoxidation. High ee’s have been obtained for a variety of trans- and trisubstituted olefins including electron-deficient α,β-unsaturated esters as well as certain cis-olefins.
Chiral ketones of various structures have been extensively investigated for asymmetric epoxidation of olefins in a number of laboratories.1 In our studies, we have found that fructose-derived ketone 1 provides high ee’s for a wide variety of trans- and trisubstituted olefins,2 and oxazolidinone ketone 2 can give high ee’s for olefins which had not been effective with ketone 1, including various cis-olefins,3a,c,d,e,g,k,l styrenes,3b,c,d,f and certain trisubstituted3h,j and tetrasubstituted olefins.3i,j In our efforts to expand the substrate scope, we have reported that ketone 3a is an effective epoxidation catalyst for a variety of electron-deficient α,β-unsaturated esters.4 Replacing the fused ketal of 1 with more electron-withdrawing diacetates significantly enhances the ketone’s reactivity and possibly reduces the Baeyer-Villiger decomposition. Herein we wish to report our detailed studies on epoxidations with ketone 3a and its related analogues.

Ketone 3a can be synthesized from ketone 1 in two steps by selective deketalization with 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ)5 and subsequent acetylation with Ac2O and catalytic amount (2 mol%) of DMAP (Scheme 1).4 Ketone 3a is highly electrophilic and can easily form a hydrate with water.6 With this method, ketone 3a can be obtained largely in ketone form by avoiding moisture. The β-acetate of ketone 3a is prone to elimination to form an enone. To minimize this elimination, it is crucial to use only a small amount of DMAP (ca. 2 mol%) and to carefully control the reaction time in the acetylation step. Nevertheless, it was found that the resulting enone was barely active and had little impact on the asymmetric epoxidation. Alternatively, ketone 3a can be readily prepared in hydrate form from ketone 1 by one pot deketalization and acetylation with ZnCl2, AcOH, and Ac2O in water (Scheme 2).7,8,9 However, if ketone 3a is desired, it can be obtained from 3a·H2O by simply dissolving 3a·H2O in solvents such as EtOAc and stirring with Na2SO4 overnight at rt, followed by filtration and concentration or by passing 3a·H2O through a short column of SiO2. In CDCl3, the hydrate gradually converted into the ketone as judged by 1H NMR (25% ketone at 10 min, 50% ketone at 30 min, 67% ketone at 1 h, 90% ketone at 2 h, 100% ketone at 11 h). In CD3CN-D2O (1.5:1, v/v), ca. 21% of the ketone was formed from the hydrate at 1 h (ca. 22% at 7 h). When the ketone was subjected to the same solvent mixture, ca. 26% of the ketone remained at 1 h (ca. 25% at 7 h). It appears that a similar amount of the ketone was present at around 1 h regardless of whether the ketone or its hydrate was used. Similar conversions and ee’s were also obtained for the epoxidation with ethyl trans-cinnamate as test substrate when either the ketone or its hydrate was used. In addition to ketone 3a, several analogues including diesters 3b–e and 6, as well as mono-ester ketones 5, 7, and 10 were also prepared for the epoxidation studies (Scheme 1 and Scheme 3).
Scheme 1.

Scheme 2.

Scheme 3.

Ethyl trans-cinnamate was used for initial epoxidations with each of these ketones (20 mol%). As shown in Table 1, diacetate ketone 3a was found to be among the most effective ketones in terms of both conversion and enantioselectivity. Various α,β-unsaturated esters were subsequently examined with ketone 3a (20–30 mol%). As shown in Table 2, high ee’s were obtained for substituted cinnamates and a variety of trisubstituted α,β-unsaturated esters (for more examples, see ref. 4). While certain enones could give good ee’s (Table 2, entry 9), ketone 3a is less effective for enones in general.10
Table 1.
Asymmetric Epoxidation of Ethyl trans-Cinnamate with Ketones 3, 5a, 6, 7, and 10a
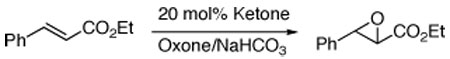 | |||
|---|---|---|---|
| entry | ketone | conv. (%) | ee (%) |
| 1b | 3a | 47 | 95 |
| 2b | 3b | 27 | 94 |
| 3b | 3c | 16 | 94 |
| 4b | 3d | 34 | 94 |
| 5b | 3e | 10 | 90 |
| 6b | 5a | 1 | 29 |
| 7b | 6 | 10 | 91 |
| 8b | 7 | 4 | 62 |
| 9c | 10 | 5 | 42 |
Method A: All reactions were carried out with substrate (0.5 mmol), ketone (0.1 mmol), Bu4NHSO4 (0.03 mmol), Oxone (2.5 mmol), and NaHCO3 (7.75 mmol) in CH3CN-aq. Na2(EDTA) (4 × 10−4 M) (6.25 mL) (v/v, 1.5/1). A mixture of Oxone and NaHCO3 was added portionwise over 4.5 h at 0 °C and stirred for 5.5 h at 0 °C. For entry 8, a mixture of Oxone and NaHCO3 was added portionwise over 4.5 h at 0 °C and stirred for 7.5 h at 0 °C and for 12 h at room temperature.
The conversion and ee were determined by chiral GC (Chiraldex G-TA).
The conversion and ee were determined by chiral GC (Chiraldex B-DM).
Table 2.
Asymmetric Epoxidation of Olefins Catalyzed by Ketone 3aa
| entry | substrate | yield (%)b |
ee (%) |
config.1 |
|---|---|---|---|---|
| 1c | X = H | 73 | 96f | (+)-(2S,3R)6c,11 |
| 2d | X = p-Me | 91 | 97g | (+) |
| 3e | X = p-OMe | 57 | 90h | (+)-(2S,3R)12 |
| 4d | 93 | 96i | (+)-(2S,3R)13 | |
| 5c | 45 | 86g | (+) | |
| 6c | 77 | 89f | (+) | |
| 7d | 77 | 93f | (+) | |
| 8d | 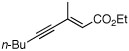 |
74 | 98h | (+) |
| 9c | 42 | 82i | (+)-(2S,3R)14 |
Method A: All reactions were carried out with substrate (0.5 mmol), ketone 3a (0.1–0.15 mmol), Bu4NHSO4 (0.03 mmol), Oxone (2.5 mmol), and NaHCO3 (7.75 mmol) in CH3CN-aq. Na2(EDTA) (4 × 10−4 M) (6.25 mL) (v/v, 1.5/1). A mixture of Oxone and NaHCO3 was added portionwise over 4.5 h at 0 °C and stirred for 7.5 h at 0 °C and for 12 h at rt. For entry 3 a mixture of Oxone and NaHCO3 was added portionwise over 4.5 h at 0 °C and stirred for 7.5 h at 0 °C.
Isolated yields.
0.15 mmol 3a used.
0.125 mmol 3a used.
0.10 mmol 3a used.
Determined by chiral GC (Chiraldex G-TA).
Determined by chiral HPLC (Chiralpak AD).
Determined by chiral HPLC (Chiralcel OD).
Determined by chiral HPLC (Chiralcel OB).
The epoxidation with other types of olefins was also investigated.15 Studies with trans-β-methylstyrene showed that less amount of ketone catalyst (10 mol%) was required when the epoxidation was carried out at a slightly higher pH (around 8.75 to 9.50) with slow addition of Oxone and K2CO3 solutions (Method B). This protocol is usually effective for relatively reactive substrates. The epoxidation with ketone 3a gave good to high ee’s for various trans- and trisubstituted olefins including less reactive trans-enimides (Table 3, entries 1–9). High ee’s were obtained for certain cis-olefins such as aromatic conjugated olefins with bulky R groups (Table 3, entries, 14–17). Low ee’s were obtained for terminal and tetrasubstituted olefins tested (Table 3, entries 19–21). Figure 1 and Figure 2 list a few possible competing spiro transition states for trans-, trisubstituted, and cis-olefins.16 Based on the determined configurations of some epoxide products in Table 2 and Table 3, the epoxidation for trans- and trisubstituted olefins likely proceeds via spiro transition state A with possible contribution from B (Figure 1) (at least in these cases). For conjugated aromatic cis-olefins, the epoxidation appears to proceed via spiro transition state E with possible contribution from F based on the configurations determined (Table 3).
Table 3.
| entry | substrate | method (h) |
yieldc (ee) (%) |
config.d |
|---|---|---|---|---|
| 1 | A (24) | 75 (86e) | (+)-(R,R)2b | |
| B (8) | 81 (86e) | |||
| 2 | A (24) | 68 (92f) | (+)-(R,R)2b | |
| B (16) | 63 (93f) | |||
| 3 | B (8) | 53 (88g) | (+)-(R,R)2b | |
| 4 | B (8) | 73 (79f) | (+)-(R,R)17 | |
| 5 | A (24) | 93 (86f) | (+)-(R,R)2b | |
| B (8) | 46 (88f) | |||
| 6 | A (24) | 92 (92e) | (+)-(R,R)2b | |
| B (8) | 82 (92e) | |||
| 7 | 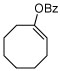 |
B (8) | 97 (95f) | (+)-(R,R)18 |
| 8 | 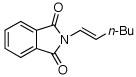 |
A (14) | 95 (75h) | (+) |
| 9 | 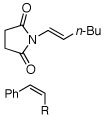 |
A (14) | 78 (86e) | (+) |
| 10 | R = Me | B (8) | 81 (49e) | (+)-(1S,2R)3c |
| 11 | R = CH2OH | B (8) | 85 (47i) | (+)-(2R,3S)19 |
| 12 | R = CH2OTBS | B (8) | 62 (70e) | (+) |
| 13 | R = CH2TMS | B (8) | 81 (65e) | (+) |
| 14 | R = TMS | B (8) | 54 (80e) | (+) |
| 15 | R = TBS | B (24) | 60 (90e) | (+) |
| 16 | B (8) | 75 (88f) | (−)-(S,S)3g | |
| 17 | 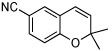 |
B (8) | 73 (90f) | (−)-(S,S)3g |
| 18 | B (8) | 50 (62e) | (+)-(2R,3S)31 | |
| 19 | B (8) | 77 (27e) | (−)-(S)2b | |
| 20 | B (8) | 72 (6f) | (+)-(S)20 | |
| 21 | 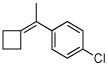 |
B (8) | 63 (67e) | (−)-(S)3i |
Method A: All reactions were carried out with olefin (0.5 mmol), ketone 3a (0.125 mmol), Bu4NHSO4 (0.03 mmol), Oxone (2.5 mmol), and NaHCO3 (7.75 mmol) in CH3CN-aq. Na2(EDTA) (4 × 10−4 M) (6.25 mL) (v/v, 1.5/1). For entries 1, 2, 5, and 6, a mixture of Oxone and NaHCO3 was added portionwise over 4.5 h at 0 °C and stirred for 7.5 h at 0 °C and for 12 h at rt. For entries 8 and 9, a mixture of Oxone and NaHCO3 was added portionwise over 3 h at 0 °C and stirred for another 11 h at 0 °C.
Method B: All epoxidations were carried out with substrate (0.5 mmol), ketone 3a·H2O (0.046 mmol) (0.1 mmol for entries 14 and 15), Oxone (1.01 mmol), and K2CO3 (2.02 mmol) in CH3CN-DMM (9 mL) (v/v, 1/2), and buffer (0.05 M Na2HPO4/0.05 M KH2PO4, pH 7.0, 3 mL) at 0 °C for 8 h, 16 h, or 24 h.
Isolated yields.
Determined by comparing the measured optical rotations with the reported ones.
Determined by chiral GC (Chiraldex B-DM).
Determined by chiral HPLC (Chiralcel OD).
The epoxide was opened (NaOMe-MeOH), the resulting alcohol was converted to its benzoate, enantioselectivity was determined by chiral HPLC (Chiralcel OD-H).
Determined by chiral HPLC (Chiralpak ADH).
Determined by chiral HPLC (Chiralcel OD-H).
Figure 1.

Possible competing spiro transition states for the epoxidation of trans- and trisubstituted olefins with ketone 3a.
Figure 2.

Possible competing spiro transition states for the epoxidation of cis-olefins with ketone 3a.
In summary, several fructose-derived diester and monoester ketones were investigated for asymmetric epoxidation. Diacetate ketone 3a has been found to be the most effective catalyst among those ketones investigated. High ee’s have been obtained for a variety of trans- and trisubstituted olefins as well as certain cis-olefins. While it is generally less enantioselective than ketone 1 for trans- and trisubstituted olefins and less enantioselective than ketone 2 for cis-olefins, ketone 3a is more effective than 1 and 2 for electron-deficient olefins, thus providing a complementary epoxidation system to ketones 1 and 2. Future efforts will be devoted to further understanding the structural effect of ketones on catalysis and developing more effective catalytic systems.
Experimental Section
Representative Synthesis of Ketone 3a
To a solution of ketone 1 (6.90 g, 26.7 mmol) in CH3CN-H2O (v/v, 9/1) (90 mL) was added DDQ (0.60 g, 2.60 mmol) at rt. Upon stirring at rt for 7 h, the reaction mixture was concentrated, dissolved in EtOAc (80 mL), dried (Na2SO4), filtered, concentrated, and purified by flash chromatography (silica gel, hexane/EtOAc = 1/0 to 1/1) to give 4 as a white solid (4.00 g, 69% yield). mp 107–110 °C; [α]d 25 = −140.0 (c 0.80, MeOH); IR (film) 3469, 3402, 1747 cm−1; 1H NMR (300 MHz, CDCl3) δ 4.74 (d, J = 4.2 Hz, 1H), 4.68 (d, J = 9.6 Hz, 1H), 4.39 (m, 1H), 4.32 (d, J = 12.9 Hz, 1H), 4.00 (d, J = 9.6 Hz, 1H), 3.98 (dd, J = 12.9, 2.4 Hz, 1H), 3.29 (m, 2H), 1.54 (s, 3H), 1.39 (s, 3H); 13C NMR (75 MHz, CDCl3) δ 199.0, 113.6, 104.5, 74.4, 73.8, 69.7, 63.6, 26.6, 26.4.
To a solution of 4 (3.51 g, 16.10 mmol) and DMAP (0.039 g, 0.32 mmol) in dry DCM (150 mL) was added dropwise Ac2O (4.97 g, 48.70 mmol) at 0 °C over 20 min. Upon stirring at rt for 16 h (monitored by TLC), the reaction mixture was filtered through a short silica gel column. The filtrate was concentrated and purified by flash chromatography (silica gel, hexane/EtOAc = 1/0 to 3/1) to give ketone 3a as colorless syrup (3.84 g, 79% yield). [α]d 25 = −103.0 (c 0.98, CHCl3); IR (film) 1750 cm−1; 1H NMR (400 MHz, CDCl3) δ 5.89 (d, J = 3.9 Hz, 1H), 5.62-5.60 (m, 1H), 4.70 (d, J = 9.6 Hz, 1H), 4.44 (d, J = 13.2 Hz, 1H), 3.99 (d, J = 9.6 Hz, 1H), 3.96 (dd, J = 13.2, 2.1 Hz, 1H), 2.18 (s, 3H), 2.13 (s, 3H), 1.55 (s, 3H), 1.41 (s, 3H); 13C NMR (75 MHz, CDCl3) δ 192.0, 170.4, 169.6, 114.1, 105.2, 74.2, 72.3, 69.6, 62.7, 26.6, 26.2, 21.0, 20.6; HRMS calcd for C13H19O8 (M+1): 303.1080, Found 303.1087.
One Pot Synthesis of Ketone 3a
AcOH (17.5 mL) and deionized water (4.3 mL) were added to a mixture of ketone 1 (12.90 g, 50.0 mmol) and ZnCl2 (0.17 g, 1.25 mmol) in a 250 mL round bottom flask equipped with a Teflon-coated magnetic stir bar. After the resulting suspension was stirred at rt for 8–10 h, Ac2O (64.9 g, 635.8 mmol) was added into the reaction flask. After the resulting mixture was stirred at rt for 16 h, deionized water (30 mL) was added. Upon stirring at rt for 20 min, the reaction mixture was concentrated in vacuo (130 mmHg, 55 °C) until about 20 mL of solution remained. The resulting solution was transferred to a 100 mL beaker, and 10 mL of deionized water was used to rinse the flask and transferred to the beaker. Upon slightly shaking for 5 min, the mixture was then placed in an ice bath for 2 h, and the solid (mud-like) precipitated. The solid was filtered through a Büchner funnel, washed by ice-cold H2O (5 mL) and ice-cold hexane (20 mL), and dried under vacuum pump (10–20 mmHg) overnight to give ketone 3a·H2O as a white solid (12.2 g, 76% yield). mp 81–84 °C; [α]d 25 = −112.0 (c 1.05, CHCl3); IR (film) 3436, 1736 cm−1; 1H NMR (400 MHz, CDCl3) (ketone) δ 5.89 (d, J = 4.0 Hz, 1H), 5.60–5.62 (m, 1H), 4.70 (d, J = 9.6 Hz, 1H), 4.44 (dd, J = 13.2, 1.2 Hz, 1H), 3.99 (d, J = 9.6 Hz, 1H), 3.96 (dd, J = 13.2, 2.0 Hz, 1H), 2.17 (s, 3H), 2.12 (s, 3H), 1.55 (s, 3H), 1.40 (s, 3H); 13C NMR (75 MHz, CDCl3) (ketone) δ 192.0, 170.4, 169.6, 114.1, 105.2, 74.2, 72.3, 69.6, 62.7, 26.6, 26.2, 21.0, 20.6; Anal. Calcd for C13H20O9 (hydrate): C, 48.75; H, 6.29. Found: C, 49.14; H, 6.12.
Representative Asymmetric Epoxidation Procedure using Oxone and NaHCO3 (Method A) (Table 2, Entry 2)
Aqueous Na2(EDTA) (1×10−4 M, 2.5 mL) and a catalytic amount of tetrabutylammonium hydrogen sulfate (0.010 g, 0.03 mmol) were added to a solution of ethyl trans-4-methylcinnamate (0.095 g, 0.5 mmol) in CH3CN (2.5 mL) with vigorous stirring at 0 °C. A mixture of Oxone (1.537 g, 2.5 mmol) and NaHCO3 (0.651 g, 7.75 mmol) was pulverized, and a small portion of this mixture was added to the reaction mixture to bring pH to >7.0. Then a solution of ketone 3a (0.038 g, 0.125 mmol) in CH3CN (1.25 mL) was added. The rest of the Oxone and NaHCO3 was added to the reaction mixture portionwise over a period of 4.5 h. Upon stirring for an additional 7.5 h at 0 °C and 12 h at rt, the resulting mixture was diluted with water, and extracted with ethyl acetate. The combined extracts were washed with brine, dried over Na2SO4, filtered, concentrated, and purified by flash chromatography (silica gel, hexane/EtOAc = 1/0 to 95/5) to give the epoxide as a colorless oil (0.094 g, 91% yield, 97% ee).
[For Table 2 entry 3, 7.5 mL of CH3CN and 5.0 mL of aqueous Na2(EDTA) (1×10−4 M) were used due to the poorer solubility of the substrate. For Table 2 entry 3 as well as Table 3, the silica gel was buffered with 1% Et3N in hexane.]
Representative Asymmetric Epoxidation Procedure using Oxone and K2CO3 (Method B) (Table 3, Entry 1)
To a solution of olefin (0.059 g, 0.5 mmol), ketone 3a (hydrate form) (0.015 g, 0.046 mmol), and tetrabutylammonium hydrogen sulfate (0.01 g, 0.03 mmol) in MeCN-DMM (v/v, 1/2) (9 mL) was added buffer (0.05 M aq Na2HPO4-0.05 M aq KH2PO4, pH 7.0) (3 mL) with stirring. Upon cooling to 0 °C, a solution of Oxone (0.212 M in 4 × 10−4 M aq EDTA, 4.8 mL) and a solution of K2CO3 (0.42 M in 4 × 10−4 M aq EDTA, 4.8 mL) were added dropwise simultaneously and separately over 8 h via syringe pump. The reaction was quenched by addition of pentane and extracted with pentane. The combined organic layers were dried over Na2SO4, filtered, concentrated, and purified by flash chromatography (silica gel was buffered with 1% Et3N in organic solvent, first pentane, then pentane/Et2O = 20/1) to give the epoxide as a colorless oil (0.054 g, 81% yield, 86% ee).
Supplementary Material
The synthesis and characterization of ketones 3, 4, 5, 6, 9, and 10; the characterization of epoxides, the X-ray structure of ketone 5a, the NMR spectra of ketones and epoxides, and the data for the determination of the enantiomeric excess of the epoxides obtained with ketone 3a (136 pages). This material is available free of charge via the Internet at http://pubs.acs.org.
Acknowledgements
We are grateful to the generous financial support from the General Medical Sciences of the National Institutes of Health (GM59705-08).
References
- 1.For leading reviews, seeDenmark SE, Wu Z. Synlett. 1999;847Frohn M, Shi Y. Synthesis. 2000:1979.Shi Y. Acc. Chem. Res. 2004;37:488. doi: 10.1021/ar030063x.Yang D. Acc. Chem. Res. 2004;37:497. doi: 10.1021/ar030065h.Wong OA, Shi Y. Chem. Rev. 2008;108:3958. doi: 10.1021/cr068367v.
- 2.(a) Tu Y, Wang Z-X, Shi Y. J. Am. Chem. Soc. 1996;118:9806. [Google Scholar]; (b) Wang Z-X, Tu Y, Frohn M, Zhang J-R, Shi Y. J. Am. Chem. Soc. 1997;119:11224. [Google Scholar]; (c) Shu L, Shi Y. Tetrahedron. 2001;57:5213. [Google Scholar]
- 3.(a) Tian H, She X, Shu L, Yu H, Shi Y. J. Am. Chem. Soc. 2000;122:11551. [Google Scholar]; (b) Tian H, She X, Xu J, Shi Y. Org. Lett. 2001;3:1929. doi: 10.1021/ol010066e. [DOI] [PubMed] [Google Scholar]; (c) Tian H, She X, Yu H, Shu L, Shi Y. J. Org. Chem. 2002;67:2435. doi: 10.1021/jo010838k. [DOI] [PubMed] [Google Scholar]; (d) Shu L, Wang P, Gan Y, Shi Y. Org. Lett. 2003;5:293. doi: 10.1021/ol020229e. [DOI] [PubMed] [Google Scholar]; (e) Shu L, Shi Y. Tetrahedron Lett. 2004;45:8115. [Google Scholar]; (f) Goeddel D, Shu L, Yuan Y, Wong OA, Wang B, Shi Y. J. Org. Chem. 2006;71:1715. doi: 10.1021/jo0520285. [DOI] [PubMed] [Google Scholar]; (g) Wong OA, Shi Y. J. Org. Chem. 2006;71:3973. doi: 10.1021/jo0604179. [DOI] [PubMed] [Google Scholar]; (h) Shen Y-M, Wang B, Shi Y. Angew. Chem. Int. Ed. 2006;45:1429. doi: 10.1002/anie.200501520. [DOI] [PubMed] [Google Scholar]; (i) Shen Y-M, Wang B, Shi Y. Tetrahedron Lett. 2006;47:5455. [Google Scholar]; (j) Wang B, Shen Y-M, Shi Y. J. Org. Chem. 2006;71:9519. doi: 10.1021/jo061341j. [DOI] [PubMed] [Google Scholar]; (k) Burke CP, Shi Y. Angew. Chem. Int. Ed. 2006;45:4475. doi: 10.1002/anie.200600840. [DOI] [PubMed] [Google Scholar]; (l) Burke CP, Shi Y. J. Org. Chem. 2007;72:4093. doi: 10.1021/jo070205r. [DOI] [PubMed] [Google Scholar]
- 4.Wu X-Y, She X, Shi Y. J. Am. Chem. Soc. 2002;124:8792. doi: 10.1021/ja020478y. [DOI] [PubMed] [Google Scholar]
- 5.For leading references, seeFernandez JMG, Mellet CO, Marin AM, Fuentes J. Carbohydr. Res. 1995;274:263. doi: 10.1016/0008-6215(95)00065-2.Tu Y, Wang Z-X, Frohn M, He M, Yu H, Tang Y, Shi Y. J. Org. Chem. 1998;63:8475.
- 6.For other examples on ketone/hydrate, seeDenmark SE, Forbes DC, Hays DS, DePue JS, Wilde RG. J. Org. Chem. 1995;60:1391.Denmark SE, Wu Z, Crudden CM, Matsuhashi H. J. Org. Chem. 1997;62:8288. doi: 10.1021/jo971781y.Wang Z-X, Shi Y. J. Org. Chem. 1997;62:8622. doi: 10.1021/jo962392r.Song CE, Kim YH, Lee KC, Lee S-G, Jin BW. Tetrahedron: Asymmetry. 1997;8:2921.Denmark SE, Wu Z. J. Org. Chem. 1998;63:2810.Yang D, Yip Y-C, Tang M-W, Wong M-K, Cheung K-K. J. Org. Chem. 1998;63:9888.Yang D, Yip Y-C, Jiao G-S, Wong M-K. J. Org. Chem. 1998;63:8952.(h) ref. 5b.Armstrong A, Hayter BR. Tetrahedron. 1999;55:11119.Carnell AJ, Johnstone RAW, Parsy CC, Sanderson WR. Tetrahedron Lett. 1999;40:8029.(k) ref. 1a.Wang Z-X, Miller SM, Anderson OP, Shi Y. J. Org. Chem. 1999;64:6443. doi: 10.1021/jo001343i.Armstrong A, Washington I, Houk KN. J. Am. Chem. Soc. 2000;122:6297.Tian H, She X, Shi Y. Org. Lett. 2001;3:715. doi: 10.1021/ol000385q.Stearman CJ, Behar V. Tetrahedron Lett. 2002;43:1943.(p) ref. 3c.Shu L, Shen Y-M, Burke C, Goeddel D, Shi Y. J. Org. Chem. 2003;68:4963. doi: 10.1021/jo0206770.Chan W-K, Yu W-Y, Che C-M, Wong M-K. J. Org. Chem. 2003;68:6576. doi: 10.1021/jo034296d.Crane Z, Goeddel D, Gan Y, Shi Y. Tetrahedron. 2005;61:6409.
- 7.For a leading reference on selective deketalization with aqueous AcOH, seeKang J, Lim GJ, Yoon SK, Kim MY. J. Org. Chem. 1995;60:564.
- 8.For a leading reference on Lewis acid-catalyzed acetylation, seeOrita A, Tanahashi C, Kakuda A, Otera J. J. Org. Chem. 2001;66:8926. doi: 10.1021/jo0107453.
- 9.For a related two-step procedure with AcOH-H2O, Ac2O-ZnCl2 from ketone 1, seeNieto N, Molas P, Benet-Buchholz J, Vidal-Ferran A. J. Org. Chem. 2005;70:10143. doi: 10.1021/jo051682h.
- 10.For leading reviews on various effective asymmetric epoxidation of electron-deficient olefins seePorter MJ, Skidmore J. Chem. Commun. 2000:1215.Lauret C, Roberts SM. Aldrichimica Acta. 2002;35:47.Kelly DR, Roberts SM. Biopolymers. 2006;84:74. doi: 10.1002/bip.20373.Shibasaki M, Kanai M, Matsunaga S. Aldrichimica Acta. 2006;39:31.Díez D, Núñez MG, Antón AB, García P, Moro RF, Garrido NM, Marcos IS, Basabe P, Urones JG. Curr. Org. Synth. 2008;5:186.Lattanzi A. Curr. Org. Synth. 2008;5:117.
- 11.Cabon O, Buisson D, Larcheveque M, Azerad R. Tetrahedron: Asymmetry. 1995;6:2211. [Google Scholar]
- 12.Yamamoto M, Hayashi M, Masaki M, Nohira H. Tetrahedron: Asymmetry. 1991;2:403. [Google Scholar]
- 13.Abidi SL, Wolfhagen JL. J. Org. Chem. 1979;44:433. [Google Scholar]
- 14.Bougauchi M, Watanabe S, Arai T, Sasai H, Shibasaki M. J. Am. Chem. Soc. 1997;119:2329. [Google Scholar]
- 15.(a) For recent epoxidation examples catalyzed by ketone 3a, see ref. 9 Nieto N, Munslow IJ, Fernández-Pérez H, Vida-Ferran A. Synlett. 2008:2856.Hirooka Y, Nitta M, Furuta T, Kan T. Synlett. 2008:3234.
- 16.Some planar transition states may also be competing. However, the extent of their involvement awaits further studies.
- 17.Wang Z-X, Shi Y. J. Org. Chem. 1998;63:3099. doi: 10.1021/jo980604+. [DOI] [PubMed] [Google Scholar]
- 18.Zhu Y, Shu L, Tu Y, Shi Y. J. Org. Chem. 2001;66:1818. doi: 10.1021/jo001593z. [DOI] [PubMed] [Google Scholar]
- 19.Denis JN, Greene AE, Serra AA, Luche MJ. J. Org. Chem. 1986;51:46. [Google Scholar]
- 20.Capriati V, Florio S, Luisi R, Salomone A. Org. Lett. 2002;4:2445. doi: 10.1021/ol026212d. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The synthesis and characterization of ketones 3, 4, 5, 6, 9, and 10; the characterization of epoxides, the X-ray structure of ketone 5a, the NMR spectra of ketones and epoxides, and the data for the determination of the enantiomeric excess of the epoxides obtained with ketone 3a (136 pages). This material is available free of charge via the Internet at http://pubs.acs.org.