

Abstract
The essential autophagy protein and haplo-insufficient tumor suppressor, Beclin 1, interacts with several cofactors (Ambra1, Bif-1, UVRAG) to activate the lipid kinase Vps34, thereby inducing autophagy. In normal conditions, Beclin 1 is bound to and inhibited by Bcl-2 or the Bcl-2 homolog Bcl-XL. This interaction involves a Bcl-2 homology 3 (BH3) domain in Beclin 1 and the BH3 binding groove of Bcl-2/Bcl-XL. Other proteins containing BH3 domains, called BH3-only proteins, can competitively disrupt the interaction between Beclin 1 and Bcl-2/Bcl-XL to induce autophagy. Nutrient starvation, which is a potent physiologic inducer of autophagy, can stimulate the dissociation of Beclin 1 from its inhibitors, either by activating BH3-only proteins (such as Bad) or by posttranslational modifications of Bcl-2 (such as phosphorylation) that may reduce its affinity for Beclin 1 and BH3-only proteins. Thus, anti-apoptotic Bcl-2 family members and pro-apoptotic BH3-only proteins may participate in the inhibition and induction of autophagy, respectively. This hitherto neglected crosstalk between the core machineries regulating autophagy and apoptosis may redefine the role of Bcl-2 family proteins in oncogenesis and tumor progression.
Keywords: Bcl-2, Bcl-xL, BH3-only proteins, Beclin 1, autophagy, apoptosis
Introduction
The death and degradation of cells or of their parts constitute a normal facet of the healthy life of eukaryotic organisms. During organismal homeostasis, extracellular and intracellular macromolecules are constantly degraded and resynthesized. Within cells, cytoplasmic organelles are continually renewed by a process involving destruction and reconstruction. Further, when one cell divides, giving rise to two daughter cells, another cell has to die to maintain the population constant. Thus, given the importance of cell death and cell degradation in tissue homeostasis, it seems likely that mechanisms have evolved to coordinately regulate these two pathways.
Two prominent mechanisms of self-destruction have captured the attention of cell biologists over the last decades: apoptosis and autophagy. Apoptosis, also called type 1 cell death, may be defined as suicidal cell death with a particular morphology involving nuclear chromatin condensation.1,2 Autophagy (sometimes associated with so-called type 2 non-apoptotic cell death) is characterized by the sequestration of cytoplasmic material in vacuoles for bulk degradation by lysosomal enzymes.3 Although a process of self-destruction, autophagy is not invariably a process of cell death; on the contrary autophagy often protects against cell death.4-7 In conditions of stress, autophagy can remove damaged organelles (and hence facilitate repair)8 or furnish additional ATP by catabolizing cellular macromolecules (and hence favor survival).9,10
The molecular mechanisms of apoptosis and autophagy are quite different and involve fundamentally distinct sets of regulatory and executioner molecules.11-13 Thus far, there is no known overlap between the proteins that are essential for autophagy in Saccharomyces cerevisiae and those that regulate programmed cell death in this species.14 However, in mammals (and likely other metazoans), there is one family of proteins, the so-called Bcl-2 protein family, that plays a dual role in the control of apoptosis and autophagy. Although Bcl-2 family proteins were initially characterized as cell death regulators, it has recently become clear that they also control autophagy. Cellular anti-apoptotic proteins such as Bcl-2,15 Bcl-XL16, Bcl-w17 and Mcl-118 can inhibit autophagy. Virus encoded Bcl-2 homologs such as KSHV vBcl-2 and murine γHV68 M11 also suppress autophagy.15,19 Conversely, pro-apoptotic BH3-only proteins from the Bcl-2 family such as BNIP3,20,21 Bad,18 Noxa, Puma, BimEL22 and Bik23 can induce autophagy.
Here, we discuss the mechanisms by which Bcl-2 family members, well-characterized regulators of apoptosis, also regulate autophagy.
The Bcl-2 Family in Apoptosis Regulation—a Quick Guide
Bcl-2 is the prototype of a family of proteins containing at least one Bcl-2 homology (BH) region. In humans and mice, the Bcl-2 family is split into anti-apoptotic multi-domain proteins (prototypes: Bcl-2 and Bcl-XL), which contain four BH domains (numbered BH1 to BH4), pro-apoptotic multi-domain proteins (prototypes: Bax and Bak), which contain three BH domains (BH1, BH2 and BH3), and the pro-apoptotic BH3-only protein family (which has more than a dozen members).24,25 Different combinations of Bcl-2 family proteins are expressed in a cell type-, differentiation- and activation state-dependent fashion. Since they engage in multiple protein-protein interactions among each other and with other cellular proteins, the stoichiometry of Bcl-2 proteins determines the propensity of cells to succumb to different apoptosis inducers including metabolic stressors, genotoxic insults, and the absence of obligate growth factors.24,25
The molecular surface of the multidomain anti-apoptotic Bcl-2 proteins possess a hydrophobic cleft, the BH3 binding groove, which can accommodate BH3 domains from pro-apoptotic Bcl-2 protein family members. The BH3 domains from pro-apoptotic proteins are structurally defined as four-turn amphipathic α-helices, containing the sequence motif: Hy-X-X-X-Hy-X-X-X-Sm-D/E-X-Hy, where Hy represents hydrophobic residues and Sm represents small residues, typically glycine. As this motif has no invariant residues, often, poorly-conserved BH3 domains cannot be identified solely by sequence analysis.
The variability of amino acid composition of both pro-apoptotic BH3 domains as well as the residues lining the BH3-binding groove of the anti-apoptotic proteins dictate highly variable affinities for each specific pair of interactions. These variable affinities, combined with the spatial and temporal variations in the concentration of each of these proteins regulates the stoichiometry of these interactions. Thus, altering the concentration of just one BH3 domain-containing protein can influence multiple protein-protein interactions among Bcl-2 family proteins, thereby transmitting a pro-apoptotic signal throughout the cell and throughout the network of Bcl-2-like proteins.11,24,26,27
The principle site of action of apoptosis regulation by Bcl-2-like proteins is probably the mitochondrial membrane.25 As a rule, anti-apoptotic multi-domain proteins (Bcl-2, Bcl-XL, Bcl-w, Mcl-1) mainly reside in mitochondria, protecting these organelles against mitochondrial membrane permeabilization (MMP), one of the rate-limiting events of apoptosis induction. In addition, anti-apoptotic multi-domain proteins can reside in other cellular compartments and hinder the movement of pro-apoptotic proteins to mitochondria. While Bak is normally associated with the outer mitochondrial membrane, Bax resides in the cytosol of healthy cells. Either of the two principal pro-apoptotic multi-domain proteins (Bax and Bak) are required for MMP in a series of different models of apoptosis induction.28 Although the precise mechanisms of MMP are debated,25 MMP can result from a conformational change of Bax or Bak (with exposure of their N-termini), their full insertion into mitochondrial membranes as homo-oligomerized multimers and the formation of giant protein-permeable pores.29
BH3-only proteins can be activated by transcriptional induction, by post-translational modification or by liberation from endogenous inhibitors. They can exert their pro-apoptotic action by at least two different mechanisms. Some BH3-only proteins (prototypes: Bad and Noxa) preferentially interact with anti-apoptotic Bcl-2 proteins (Bad with Bcl-2 and Bcl-XL, Noxa with Mcl-1), dissociating them from BH3 or Bax/Bak-like proteins, which in turn mediate MMP. Others (prototype: t-Bid) may activate pro-apoptotic multi-domain proteins to initiate MMP, either by stimulating the translocation of Bax to mitochondrial membranes or by local effects on Bak.11,24,26
The Beclin 1 Interactome in Autophagy
Beclin 1 (the mammalian ortholog of yeast Atg6) was originally discovered as a Bcl-2-interacting protein30 and was the first human protein shown to be indispensable for autophagy.31 The overall structure of Atg6/Beclin 1, as well as its essential role in autophagosome formation, is evolutionarily conserved throughout all eukaryotic phyla. In yeast (Saccharomyces cerevisiae), which lacks Bcl-2 homologs, Atg6 acts as an obligatory allosteric activator of the class III phosphatidylinositol 3-kinase (PI3K) Vps34, the enzyme that phosphorylates phosphatidylinositol to generate phosphatidylinositol 3-phosphate (PI3P).32 Another protein present in the yeast Atg6 interactome is the myristoylated (and hence membrane-anchored) kinase, Vps15. The Atg6-dependent, Vps34-mediated generation of PI3P is important for the nucleation of the phagophore assembly complex as well as for the retrieval of additional membranes to the extending isolation membrane.3,12 In yeast, Atg6 also participates in a functionally distinct Vps34 complex, comprising Atg6, Vps34, Vps15 and Vps38, which is involved in vacuolar sorting, but not autophagy.32 An area of active research relates to the question of whether the mammalian Atg6 ortholog, Beclin 1, also partipicates in similar biochemically and functionally distinct Vps34 complexes.
In mammals, the interaction of Beclin 1 with Vps34 and Vps15 is conserved.33 Additional Beclin 1 interactors include UVRAG, Ambra1 and Bif-1 (also called endophilin B1) (Fig. 1).34-36 Knockdown and knockout studies performed on human and mouse cells indicate that UVRAG, Ambra1 and Bif-1 are all indispensable for the activation of autophagy, as well as for the optimal activation of Vps34. UVRAG and Ambra1 directly interact with distinct regions of the Beclin 1 molecule34,35 while Bif-1 interacts with Beclin 1 indirectly via UVRAG.36 However, the relative contribution of these molecules to autophagy (and perhaps autophagy-unrelated functions) is not equivalent. The deletion of only one beclin 1 allele, which causes a reduction of Beclin 1 protein expression by 50%, is sufficient to cause a severe defect in autophagy,37,38 a phenotype that is only observed when both alleles of ambra1 or bif-1 are deleted.35,36 Beclin 1-/- mice succumb early during embryonic development (around day e7.5),37,38 while ambra1-/- mice die from exencephaly during birth,35 and bif-1-/- mice are viable.36 A common denominator of the knockout phenotypes is that beclin 1+/- as well as bif-1-/- mice tend to develop spontaneous tumors,36-38 suggesting that a functional Beclin 1 complex (and by extension autophagy) may participate in tumor suppression.
Figure 1.
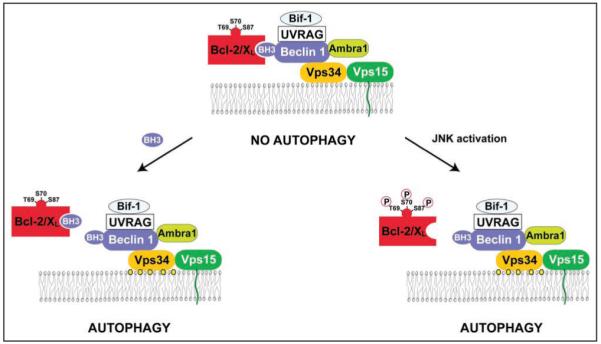
The Beclin 1 interactome in human cells and possible mechanisms underlying the regulation of its autophagy function. Beclin 1 binds to the Class III phosphatidylinositol kinase, Vps34, which is associated with the myristoylated, membrane-anchored kinase, Vps15. The lipid kinase activity of the multiprotein Beclin 1/Vps34 complex converts phosphatidylinositol into phosphatidylinositol-3-phosphate (depicted as yellow circles), which is involved in the nucleation of pre-autophagosomal structures. Beclin 1 interacts with several coactivators including UVRAG, Ambra1 and Bif-1, as well as with the inhibitors, Bcl-2 and Bcl-XL. Shown here are two mechanisms for disrupting the interaction between Beclin 1 and Bcl-2 or Bcl-XL, and thereby activating autophagy: (1) Proteins that contain BH3 domains or small molecules that mimic BH3 domains can bind to the BH3-binding groove of Bcl-2 or Bcl-XL and competitively disrupt the interaction between Bcl-2 or Bcl-XL and Beclin 1. This leads to the de-inhibition of the lipid kinase activity of the class III phosphatidylinositol-3-kinase Vps34 and autophagy induction. (2) C-Jun-N’-terminal kinase 1 (JNK1)-mediated phosphorylation of Bcl-2 during starvation can disrupt its interaction with Beclin 1, leading to autophagy induction.
In mice and humans, Beclin 1 constitutively interacts with the anti-apoptotic proteins of the Bcl-2 family, Bcl-2,15,31 Bcl-XL,18 Bcl-w17 and Mcl-1.18 In contrast to the previously mentioned proteins contained in the Beclin 1 interactome that activate autophagy (e.g., Vps34, UVRAG, Ambra1, Bif-1), Bcl-2 and Bcl-XL act as inhibitors of autophagy. Distinct regions of Beclin 1 are involved in binding to Vps34 and in binding to Bcl-2/Bcl-XL.30,39 Although in some cell types, Bcl-2 binding to Beclin 1 disrupts its interaction with Vps34, such effects are not universally required for the autophagy inhibitory effects of Bcl-Bcl-XL.15,18 Rather, it appears that the Bcl-2/Bcl-XL—Beclin 1—Vps34 multiprotein complex (compared to Beclin 1-Vps34 complexes lacking Bcl-2/Bcl-XL) possess decreased Vps34 phosphatidylinositol 3-kinase activity (Fig. 1).15 Induction of autophagy by a variety of different inducers including nutrient starvation or treatment with the inositol trisphoshate receptor (IP3R) antagonist xestospongin B, is accompanied by the dissociation of Beclin 1 from Bcl-2 or Bcl-XL.15,18,40
In short, Beclin 1 acts as an essential activator of an autophagy-inducing lipid kinase, Vps34. In conditions in which autophagy is inhibited, Beclin 1 is inactivated by its interaction with multi-domain proteins of the Bcl-2 family. Thus, the targeting of Beclin 1 by Bcl-2 family members represents an essential mechanism of autophagy regulation.
Beclin 1 as a Novel BH3-Only Protein
A region within Beclin 1 that binds to Bcl-2 or Bcl-XL was initially identified by performing truncation mutations of Beclin 1 (aa 112-159).18,30,41 Subsequently, sequence analysis suggested that this region of Beclin 1 may contain a putative BH3 domain (aa 112-123). Indeed, residues important for binding the BH3 domain of Bak, Bad and Bim to Bcl-XL are highly conserved in the Beclin 1 BH3 domain. These amino acids include L112, L116, G120 and F123 which are buried in the hydrophobic groove of Bcl-XL, as well as D121 which forms ion pairs with a conserved arginine from Bcl-XL (Fig. 2).41
Figure 2.
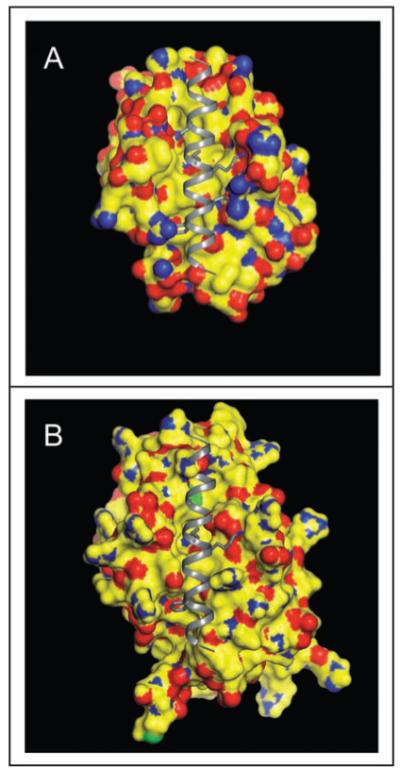
Beclin 1 BH3 domain bound to the hydrophobic surface groove of Bcl-XL (A) or Bcl-2 (B). The molecular surface of Bcl-2/Bcl-XL is color-coded by atom type: yellow represents carbon, blue represents nitrogen, red represents oxygen, and green represents sulfur. The Beclin 1 BH3 helix is shown in gray ribbon. Conserved residues within the BH3 helix, L112, L116, D121 and F123, that are involved in binding to the hydrophophic groove of Bcl-2 homologs are represented in atomic detail with atoms colored as above, except that carbon is depicted in grey. This figure demonstrates that conformational changes are required to accommodate a BH3 domain in the hydrophobic grooves of Bcl-2/Bcl-XL. The crystallographic structure of the Beclin 1 BH3 domain bound to Bcl-XL has been previously determined41 whereas in (B), the Beclin 1 BH3 domain is modeled into a molecular surface representation of Bcl-2, based on a structural superposition of Bcl-2 and Bcl-XL.
Two complementary approaches, structural analyses and functional analyses in the background of appropriate mutations, have conclusively proven that Beclin 1 possesses a functional BH3 domain. The structural evidence obtained from molecular modeling,16 crystal structures of Bcl-XL complexed to a peptide corresponding to the BH3 domain of Beclin 1,41 and HSQC NMR spectra of 15N-labeled Bcl-XL measured in the presence and absence of saturating amounts of Beclin 1 BH3 domain peptides42 demonstrated that the amphipathic BH3 helix of Beclin 1 binds to a conserved hydrophobic groove of Bcl-XL. These findings are similar to previously determined interactions between Bcl-XL -and other BH3 domains.41
A complementary approach to demonstrate that Beclin 1 interacts with Bcl-XL through a BH3 domain has relied upon mutational analyses. Replacements of several critical amino acids within the putative BH3 domain of Beclin 1 (L112A, L116A, L116E, L116Q, G120E, D121A, F123A) reduce or abrogate the interaction between Beclin 1 and Bcl-2/Bcl-XL in GST pulldown, analytical gel-filtration and/or coimmunoprecipitation assays.15,18,41,42 These data were confirmed by fluorescence anisotropy measurements involving recombinant Bcl-XL protein and BH3 peptides derived from Beclin 1, showing that the Beclin 1 BH3 mutants L116A and F123A lose their affinity for Bcl-XL.18 Accordingly, these Beclin 1 mutants (L116A and F123A) exhibit a gain-of-function phenotype in that they are more potent than wild type Beclin 1 in inducing autophagy when transfected into cells.15,18 This gain-of-function phenotype correlates with the failure of the pro-autophagic activity of these mutants to be inhibited by Bcl-2 or Bcl-XL.15,18 Conversely, mutations of Bcl-2 (G145A) or Bcl-XL (G138A) that abolish their capacity to interact with BH3 peptides, also disrupt their interactions with Beclin 1 in co-immunoprecipitation assays and in vitro binding assays. As predicted, these Bcl-2 G145A and Bcl-XL G138A mutants are defective in autophagy inhibition in cellular assays.15,18
Taken together, these results demonstrate that Beclin 1 possesses a BH3-only domain that dictates its physical and functional interaction with the BH3 binding groove of multi-domain proteins of the Bcl-2 family.
Mechanisms of Regulation of Beclin 1-Bcl-2/Bcl-XL Binding
As mentioned above, autophagy induction correlates with the dissociation of Beclin 1 from Bcl-2 or Bcl-XL. Recent data suggest that multiple distinct mechanisms may account for the release of Beclin 1 from its inhibitory interaction with Bcl-2 or Bcl-XL.
Since the Beclin 1 BH3 domain interacts with the BH3-binding groove of the multi-domain, anti-apoptotic Bcl-2 proteins (see above), one plausible model is that other proteins BH3 domain-containing proteins may competitively displace the Beclin 1 BH3 domain, disrupting the interaction of Beclin 1 and Bcl-2/Bcl-XL. This would release Beclin 1 from the inhibitory effects of Bcl-2/Bcl-XL. The first evidence in support of this model was provided by studies using the BH3-mimetic compound ABT-737, an agent designed to bind to the BH3 binding groove of Bcl-2 or Bcl-XL (but not Mcl-1).43 ABT-737 inhibits the binding of Beclin 1 BH3 peptide to Bcl-XL in a competitive manner, with an IC50 in the micromolar range, as determined by measuring the binding of synthetic peptides to purified recombinant Bcl-XL in vitro.18 Similarly, ABT-737 pretreatment abolishes the immunoprecipitation between Beclin 1 and Bcl-2 or Bcl-XL (but not Mcl-1) in cells that are resistant to the pro-apoptotic action of ABT-737. This effect correlates with the induction of high levels of autophagy. ABT-737-induced autophagy cannot be inhibited by Bcl-2 or Bcl-XL overexpression, yet it is abolished by transfection with Mcl-1 or by the siRNA-mediated knockdown of Beclin 1.18 These results clearly demonstrate that competitive disruption of the Beclin 1 interaction with Bcl-2 or Bcl-XL suffices to induce autophagy.
To date, EGL-1 is the only pro-apoptotic, BH3-only protein identified in the nematode, Caenorhabditis elegans. Transgenic expression of EGL-1 causes an increase in baseline autophagy, as measured using an LGG-1::DsRED reporter construct (LGG-1 is the C. elegans ortholog of Atg8/LC3). In wild-type nematodes, starvation strongly induces autophagy, and this induction is blunted in egl-1-deficient nematode embryos. This suggests that BH3-only proteins may act as obligate inducers of autophagy, at least in C. elegans.18 In human cells, upon nutrient starvation, the amount of endogenous Beclin 1 that co-immunoprecipitates with Bcl-XL or Bcl-2 declines15,18 while the amount of the BH3 protein Bad (whose activation is known to be triggered by serum withdrawal) that co-immunoprecipitates with Bcl-XL or Bcl-2 increases.18 The knockdown of Bad (in human HeLa cells) or its knockout (in mouse embryonic fibroblasts) results in a partial defect in starvation-induced autophagy that can be restored by the addition of excess ABT-737.18 Enforced overexpression of Bad (but not a Bad mutant with a disruption in the BH3 domain) is sufficient to induce autophagy both in normal conditions and in the setting of caspase inhibition.18 Together, these results indicate that BH3-only proteins may play an evolutionarily conserved role in the activation of starvation-induced autophagy.
A second mechanism that may link starvation to the dissociation of Beclin 1 from its inhibitory interaction with Bcl-2 involves the phosphorylation of Bcl-2 by the stress-activated c-Jun N-terminal protein kinase 1 (JNK1) (Fig. 1).44 Upon nutrient withdrawal, JNK1 is activated and induces phosphorylation of serine and threonine residues (T69, S70 and S87) in the non-structured loop of Bcl-2 that is located between the N-terminal BH4 and BH1 domain. JNK1 inhibition or replacement of wild type Bcl-2 by a non-phosphorylatable Bcl-2 mutant (T69A/S70A/S87A) abolishes the starvation-induced dissociation of Bcl-2 and Beclin 1 and inhibits autophagy. Expression of constitutively active JNK1 leads to Bcl-2 phosphorylation, dissociation of Bcl-2 from Beclin 1, and stimulation of autophagy; however, active JNK1 fails to stimulate autophagy when Bcl-2 is replaced by its non-phosphorylatable mutant. In jnk1-/- MEFs, starvation fails to induce Bcl-2 multi-site phosphorylation, dissociation of the Bcl-2/Beclin 1 complex, or autophagy. Furthermore, a multi-site Bcl-2 phosphomimetic mutant (T69E/S70E/S87E) fails to co-immunoprecipitate with Beclin 1 and has no autophagy-inhibitory activity. Together, these findings indicate that JNK1-mediated phosphorylation of Bcl-2 causes its dissociation from Beclin 1, thereby inducing autophagy.
Although the two mechanisms for the dissociation of Beclin 1 from Bcl-2/Bcl-XL described above (competitive disruption by BH3-only proteins or phosphorylation of Bcl-2) appear distinct, they may be functionally linked. Phosphorylation of recombinant Bcl-2 reduces its affinity for Bax as well as for the BH3-only protein Bid, while the non-phosphorylatable Bcl-2 mutant (T69A/S70A/S87A) exhibits enhanced binding to three different the BH3-only proteins (Bid, Bad and Bim) in co-immunoprecipitation assays.45 These observations indicate that multi-site phosphorylation in the nonstructured loop reduces the affinity of Bcl-2 for BH3 domains in general, including that of Beclin 1. Another plausible, but as-of-yet unexplored, possibility is that phosphorylated Bcl-2 might change its profile of selectivity for distinct BH3 domains in a way that Bcl-2-bound Beclin 1 is replaced by other BH3-only proteins, leading to the selective liberation of Beclin 1.
A competing hypothesis is that there are indeed two independent signaling routes that link starvation to the dissociation of Beclin 1 and Bcl-2-like proteins. For example, the dissociation of Beclin 1 and Bcl-XL could occur preferentially as a result of Bad activation (presumably because Akt becomes inactive, resulting in dephosphorylation of Bad and hence its dissociation from cytosolic 14-3-3σ).11 In contrast, disruption of the Beclin 1/Bcl-2 interaction could be preferentially regulated by the phosphorylation level of Bcl-2 (which increases as a result of JNK1 activation). It is not yet known whether Beclin 1/Bcl-XL binding, like Beclin 1/Bcl-2 binding, is regulated by JNK1-mediated phosphorylation, although the potential phosphorylation sites in the nonstructured loop are conserved between Bcl-2 and Bcl-XL.
Apoptosis versus Autophagy Regulation by Bcl-2 Proteins
As discussed above, Bcl-2 proteins are best known for their capacity to inhibit or to induce apoptosis, with BH3-only proteins functioning in apoptosis induction. Beclin 1 is a novel member of the BH3-only branch of the Bcl-2 family; yet, overexpression of Beclin 1 solely induces autophagy and has no detectable apoptogenic effects (Levine B and Kroemer G, unpublished results). How is it possible that proteins that share structural domains and a common molecular target have such different physiological functions? The microinjection of a peptide encompassing the BH3 domain of Beclin 1 can induce Bax-dependent apoptosis.18 However, enforced expression of full-length Beclin 1 fails to trigger MMP and apoptosis, suggesting that some regions of Beclin 1 outside of the BH3 domain may exert an inhibitory, anti-apoptotic function. How such an inhibitory effect would be accomplished remains an open question. One possibility is that other domains of Beclin 1 could neutralize potential “apoptogenic” effects of the BH3 domain by targeting Beclin 1 to the ER (or other non-mitochondrial sites), thereby hindering Beclin 1 from interacting with the apoptosis-regulatory mitochondrial pool of Bcl-2 or Bcl-XL. Another possibility is that the Beclin 1 BH3 domain may bind with lower affinity to the Bcl-XL/Bcl-2 BH3-binding groove than do BH3 domains from pro-apoptotic proteins. Therefore, at physiologically relevant concentrations, the Beclin 1 BH3 domain would not displace the pro-apoptotic proteins from the Bcl-2/Bcl-XL BH3 binding groove to induce apoptosis. In addition, the induction of apoptosis by microinjection of a Beclin 1 BH3 peptide may simply represent an artifact of non-physiological high concentrations, and may not be biologically relevant to the function of the full-length protein endogenously expressed in cells. Thus, BH3 domains may represent yet another example of divergent evolution wherein a common structural motif, the BH3 domain, is utilized by diverse groups of functional proteins, including not only apoptotic family members but also autophagy proteins, to mediate their interactions with Bcl-2/ Bcl-XL molecules to regulate diverse cellular functions.
One particularly intriguing aspect about the regulation of autophagy by BH3 domains relates to concepts of spatio-temporal organization and organelle specificity. ABT-737 induces the colocalization of GFP-LC3 with the mitochondrial marker HSP60 to a greater degree than with the endoplasmic reticulum (ER) marker calreticulin,18,46 indicating that it may stimulate “mitophagy” (autophagy of mitochondria) 47 more efficiently than “reticulophagy” (autophagy of the ER).48 However, a substantial part of the negative regulation of Beclin 1 appears to occur at the ER. This has been demonstrated by comparative studies of Bcl-2 and Bcl-XL mutants whose expression is restricted to the ER or mitochondria, by replacing the Bcl-2/Bcl-XL carboxy-terminal insertion sequence with an equivalent sequence from cytochrome b5 or ActA, respectively.49 Only ER-targeted, but not mitochondrion-targeted Bcl-2 and Bcl-XL suppress autophagy.9,15,50,51 Furthermore, the treatment of cells with ABT-737 decreases the fraction of Beclin 1 that co-immunoprecipitates with ER-targeted Bcl-2 but does not affect the amount of Beclin 1 that co-immunoprecipitates with mitochondrion-targeted Bcl-2.18 A similar result is obtained when the interaction of Beclin 1 with wild type Bcl-2 (which interacts both with ER membranes and with mitochondria) is evaluated after subcellular separation into the microsomal fraction (that is enriched in ER) and the heavy membrane fraction (that is enriched in mitochondria); ABT-737 diminishes the interaction between wild type Bcl-2 and Beclin 1 in microsomes but not in the heavy membrane fraction.18 An analogous approach led to the conclusion that Bcl-2 phosphorylation preferentially effects the interaction of Bcl-2 with BH3 only proteins in the microsomal (ER-enriched) fraction.45 More recently, it has been shown that only the ER-enriched pool of Bcl-2 is subjected to regulation of Beclin binding by JNK1-mediated phosphorylation.44
Based on these results, we postulate that BH3-only proteins may differentially induce autophagy and apoptosis, by acting on (at least) two distinct subcellular compartments. BH3-only proteins (or BH3 mimetics) trigger autophagy by liberating Beclin 1 from its inhibition by Bcl-2/Bcl-XL, presumably at the level of the ER. BH3-only proteins (or BH3 mimetics) trigger apoptosis, by (directly or indirectly) stimulating the mitochondrion-permeabilizing activity of pro-apoptotic multi-domain proteins from the Bcl-2 family such as Bax and Bak.25 In apparent accord with this interpretation, Bax and Bak thus far have not been implicated in the regulation of autophagy. However, it should be noted that some interaction does occur between Beclin 1 and Bcl-2/Bcl-XL at the mitochondria. Further studies are required to determine whether such an interaction contributes to apoptosis and/or autophagy regulation.
Concluding Remarks
The results discussed in this review indicate that proteins from the Bcl-2 family not only participate in the regulation of cell death, but also are important inhibitors or inducers of autophagy in nematode, murine and human cells. At first glance, it may appear counterintuitive that pro-apoptotic BH3-only proteins participate in the induction of autophagy, which in many settings has a cytoprotective effect.5,6 There are two partially overlapping explanations to reconcile this apparent contradiction. First, the system of autophagy/apoptosis control may be dictated by distinct thresholds (in addition to the spatial regulation evoked above). Activation of BH3-only proteins at a low, subapoptotic level (that fails to engage the activation of pro-apopotic multidomain proteins such as Bax and Bak) would induce autophagy. Only when the buffering capacity of anti-apoptotic Bcl-2 proteins is exhausted would the activation of BH3-only proteins succeed in stimulating Bax or Bak and thereby induce MMP and subsequent cell death. Second, one might speculate that the pro-apoptotic proteins of the Bcl-2 family have been designed to trigger catabolic reactions in response to stress, for the mobilization of energy reserves and/or the elimination of damaged macromolecules from the organism. Such catabolic reactions would have been “invented” by evolution first at the unicellular level—and hence affect portions of the cell rather than the entire cell—and later would have been used in metazoans for the elimination of entire cells by apoptosis.52
Another major implication of the overlapping regulation of autophagy and apoptosis relates to the role of pro-apoptotic Bcl-2 proteins as tumor suppressors and that of anti-apoptotic proteins as oncogenes. The oncogenic potential of Bcl-2 family members has been attributed to disabled apoptosis,24 which is one of the hallmarks of cancer.53 However, autophagy—which turns out to be regulated by Bcl-2 proteins as well - has recently emerged as a cellular pathway that is essential for the maintenance of genomic stability and tumor suppression.37,38,54-58 Thus, overexpression of Bcl-2/Bcl-XL (or loss of BH3-only proteins) may not only participate in oncogenesis by inhibiting apoptosis, which results in improved survival of tumor cells in adverse conditions of endogenous (metabolic) or exogenous (chemotherapy-associated) stress.59 Bcl-2/Bcl-XL overexpression may also participate in oncogenesis by inhibiting autophagy, which results in genomic instability and tumor progression.
Acknowledgements
The work from the authors’ laboratories was supported by grants to B.L. from the National Institutes of Health, American Cancer Society, and Ellison Medical Foundation and by grants to G.K. from Ligue Nationale contre le Cancer (équipe labellisée), Agence Nationale de Recherche, Institut National contre le Cancer, Cancéropôle Ile-de France, European Union (Active p53, ApoSys, ChemoRes, DeathTrain, RIGHT, TransDeath) and Fondation pour la Recherche Médicale. B.L. is an HHMI investigator.
References
- 1.Kroemer G, El-Deiry WS, Golstein P, Peter ME, Vaux D, Vandenabeele P, Zhivotovsky B, Blagosklonny MV, Malorni W, Knight RA, Piacentini M, Nagata S, Melino G. Nomenclature Committee of Cell Death. Classification of cell death: recommendations of the Nomenclature Committee on Cell Death. Cell Death Differ. 2005;12:1463–7. doi: 10.1038/sj.cdd.4401724. [DOI] [PubMed] [Google Scholar]
- 2.Galluzzi L, Maiuri MC, Vitale I, Zischka H, Castedo M, Zotvogel L, Kroemer G. Cell death modalities: classification and pathophysiological implications. Cell Death Differ. 2007;14:1237–43. doi: 10.1038/sj.cdd.4402148. [DOI] [PubMed] [Google Scholar]
- 3.Klionsky DJ. Autophagy: from phenomenology to molecular understanding in less than a decade. Nat Rev Mol Cell Biol. 2007;8:931–7. doi: 10.1038/nrm2245. [DOI] [PubMed] [Google Scholar]
- 4.Lum JJ, DeBerardinis RJ, Thompson CB. Autophagy in metazoans: cell survival in the land of plenty. Nat Rev Mol Cell Biol. 2005;6:439–48. doi: 10.1038/nrm1660. [DOI] [PubMed] [Google Scholar]
- 5.Maiuri C, Zalckvar E, Kimchi A, Kroemer G. Self-eating and self-killing: crosstalk between autophagy and apoptosis. Nat Rev Mol Cell Biol. 2007;8:741–52. doi: 10.1038/nrm2239. [DOI] [PubMed] [Google Scholar]
- 6.Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell. 2008 doi: 10.1016/j.cell.2007.12.018. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Levine B, Yuan J. Autophagy in cell death: an innocent convict? J Clin Invest. 2005;115:2679–88. doi: 10.1172/JCI26390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rodriguez Enriquez S, Kim I, Currin RT, Lemasters JJ. Tracker dyes to probe mitochondrial autophagy (mitophagy) in rat hepatocytes. Autophagy. 2006;2:39–46. doi: 10.4161/auto.2229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Boya P, Gonzalez-Polo RA, Casares N, Perfettini JL, Dressen P, Larochett N, Metivier D, Meley D, Souquere S, Yoshimori T, Pierron G, Codogno P, Kroemer G. Inhibition of macroautophagy triggers apoptosis. Mol Cell Biol. 2005;25:1025–40. doi: 10.1128/MCB.25.3.1025-1040.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lum JJ, Bauer DE, Kong M, Harris MH, Li C, Lindsten T, Thompson CB. Growth factor regulation of autophagy and cell survival in the absence of apoptosis. Cell. 2005;120:237–48. doi: 10.1016/j.cell.2004.11.046. [DOI] [PubMed] [Google Scholar]
- 11.Danial NN, Korsmeyer SJ. Cell death: critical control points. Cell. 2004;116:205–19. doi: 10.1016/s0092-8674(04)00046-7. [DOI] [PubMed] [Google Scholar]
- 12.Levine B, Klionsky DJ. Development by self-digestion: molecular mechanisms and biological functions of autophagy. Dev Cell. 2004;6:463–77. doi: 10.1016/s1534-5807(04)00099-1. [DOI] [PubMed] [Google Scholar]
- 13.Mizushima N, Klionsky D. Protein turnover via autophagy: implications for metabolism. Annu Rev Nutr. 2007;27:19–39. doi: 10.1146/annurev.nutr.27.061406.093749. [DOI] [PubMed] [Google Scholar]
- 14.Buttner S, Eisenberg T, Herker E, Carmona-Gutierrez D, Kroemer G, Madeo F. Why yeast cells can undergo apoptosis: death in times of peace, love and war. J Cell Biol. 2006;175:521–5. doi: 10.1083/jcb.200608098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pattingre S, Tassa A, Qu X, Garuti R, Liang XH, Mizushima N, Packer M, Schneider MD, Levine B. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell. 2005;122:927–39. doi: 10.1016/j.cell.2005.07.002. [DOI] [PubMed] [Google Scholar]
- 16.Maiuri MC, Criollo A, Tasdemir E, Vicencio JM, Tajeddine N, Hickman JA, Geneste O, Kroemer G. BH3-only proteins and BH3 mimetics induce autophagy by competitively disrupting the interaction between Beclin 1 and Bcl-2/Bcl-X(L) Autophagy. 2007;3:374–6. doi: 10.4161/auto.4237. [DOI] [PubMed] [Google Scholar]
- 17.Erlich S, Mizrachy L, Segev O, Lindenboim L, Zmira O, Adi-Harel S, Hirsch JA, Stein R, Pinkas-Kramarski R. Differential interactions between Beclin 1 and Bcl-2 family members. Autophagy. 2007;3:561–8. doi: 10.4161/auto.4713. [DOI] [PubMed] [Google Scholar]
- 18.Maiuri MC, Le Toumelin G, Criollo A, Rain JC, Gautier F, Juin P, Tasdemir E, Pierron G, Troulinaki K, Tavernarakis N, Hickman JA, Geneste O, Kroemer G. Functional and physical interaction between Bcl-X(L) and a BH3-like domain in Beclin-1. EMBO J. 2007;26:2527–39. doi: 10.1038/sj.emboj.7601689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liang C, E X, Jung JU. Downregulation of autophagy by herpesvirus Bcl-2 homologs. Autophagy. 2007 doi: 10.4161/auto.5210. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 20.Daido S, Kanzawa T, Yamamoto A, Takeuchi H, Kondo Y, Kondo S. Pivotal role of the cell death factor BNIP3 in ceramide-induced autophagic cell death in malignant glioma cells. Cancer Res. 2004;64:4286–93. doi: 10.1158/0008-5472.CAN-03-3084. [DOI] [PubMed] [Google Scholar]
- 21.Hamacher Brady A, Brady NR, Logue SE, Sayen MR, Jinno M, Kirshenbaum LA, Gottlieb RA, Gustafsson AB. Response to myocardial ischemia/reperfusion injury involves Bnip3 and autophagy. Cell Death Differ. 2007;14:146–57. doi: 10.1038/sj.cdd.4401936. [DOI] [PubMed] [Google Scholar]
- 22.Abedin MJ, Wang D, McDonnell MA, Lehmann U, Kelekar A. Autophagy delays apoptotic death in breast cancer cells following DNA damage. Cell Death Differ. 2007;14:500–10. doi: 10.1038/sj.cdd.4402039. [DOI] [PubMed] [Google Scholar]
- 23.Rashmi R, Pillai SG, Vijayalingam S, Ryerse J, Chinnadurai G. BH3-only protein BIK induces caspase-independent cell death with autophagic features in Bcl-2 null cells. Oncogene. 2007 doi: 10.1038/sj.onc.1210783. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Adams JM, Cory S. The Bcl-2 apoptotic switch in cancer development and therapy. Oncogene. 2007;26:1324–37. doi: 10.1038/sj.onc.1210220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kroemer G, Galluzzi L, Brenner C. Mitochondrial membrane permeabilization in cell death. Physiol Rev. 2007;87:99–163. doi: 10.1152/physrev.00013.2006. [DOI] [PubMed] [Google Scholar]
- 26.Galonek HL, Hardwick JM. Upgrading the BCL-2 network. Nat Cell Biol. 2006;8:1317–9. doi: 10.1038/ncb1206-1317. [DOI] [PubMed] [Google Scholar]
- 27.Certo M, Del Gaizo Moore V, Nishino M, Wei G, Korsmeyer S, Armstrong SA, Letai M. Mitochondria primed by death signals determine cellular addiction to antiapoptotic BCL-2 family members. Cancer Cell. 2006;9:351–65. doi: 10.1016/j.ccr.2006.03.027. [DOI] [PubMed] [Google Scholar]
- 28.Wei MC, Zong WX, Cheng EH, Lindsten T, Panoutsakopoulou V, Ross AJ, Roth KA, MacGregor GR, Thompson CB, Korsmeyer SJ. Proapoptotic BAX and BAK: a requisite gateway to mitochondrial dysfunction and death. Science. 2001;292:624–6. doi: 10.1126/science.1059108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pagliari LJ, Kuwawna T, Bonzon C, Newmeyer DD, Tu S, Beere HM, Green DR. The multidomain proapoptotic molecules Bax and Bak are directly activated by heat. Proc Natl Acad Sci USA. 2005;102:17975–80. doi: 10.1073/pnas.0506712102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liang XH, Kleeman LK, Jiang HH, Gordon G, Goldman JE, Berry G, Herman B, Levine B. Protection against fatal Sindbis virus encephalitis by Beclin, a novel Bcl-2-interacting protein. J Virol. 1998;72:8586–96. doi: 10.1128/jvi.72.11.8586-8596.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liang XH, Jackson S, Seaman M, Brown K, Kempkes B, Hibshoosh H, Levine B. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature. 1999;402:672–6. doi: 10.1038/45257. [DOI] [PubMed] [Google Scholar]
- 32.Kametaka S, Okano T, Ohsumi M, Ohsumi Y. Apg14p and Apg6/Vps30p form a protein complex essential for autophagy in the yeast, Saccharomyces cerevisiae. J Biol Chem. 1998;273:22284–91. doi: 10.1074/jbc.273.35.22284. [DOI] [PubMed] [Google Scholar]
- 33.Kihara A, Kabeya Y, Ohsumi Y, Yoshimori T. Beclin-phosphatidylinositol 3-kinase complex functions at the trans-Golgi network. EMBO Rep. 2001;2:330–5. doi: 10.1093/embo-reports/kve061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liang C, Feng P, Ku B, Dotan I, Canaani D, Oh BH, Jung JU. Autophagic and tumour suppressor activity of a novel Beclin1-binding protein UVRAG. Nat Cell Biol. 2006;8:688–99. doi: 10.1038/ncb1426. [DOI] [PubMed] [Google Scholar]
- 35.Fimia GM, Stoykova A, Romagnoli A, Giunta L, Di Bartolomeo S, Nardacci R, Corazzari M, Fuoco C, Ucar A, Schwartz P, Gruss P, Piacentini M, Chowdhury K, Cecconi F. Ambra1 is a novel regulator of autophagy and controls nervous system development. Nature. 2007;447:1121–5. doi: 10.1038/nature05925. [DOI] [PubMed] [Google Scholar]
- 36.Takahasi Y, Coppola D, Matsushita N, Cualing HD, Sun M, Sato Y, Liang C, Jung JU, Cheng JC, Mul JJ, Pledger WJ, Wang HG. Bif-1 interacts with Beclin 1 through UVRAG and regulates autophagy and tumorigenesis. Nat Cell Biol. 2007;9:1142–51. doi: 10.1038/ncb1634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yue Z, Jin S, Yang C, Levine AJ, Heintz N. Beclin 1, an autophagy gene essential for early embryonic development, is a haploinsufficient tumor suppressor. Proc Natl Acad Sci USA. 2003;100:15077–82. doi: 10.1073/pnas.2436255100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Qu X, Yu J, Bhagat G, Furuya N, Hibshoosh H, Troxel A, Rosen J, Eskelinen EL, Mizushima N, Ohsumi Y, Cattoretti G, Levine B. Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. J Clin Invest. 2003;112:1809–20. doi: 10.1172/JCI20039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Furuya N, Yu J, Byfield M, Pattingre S, Levine B. The evolutionarily conserved domain of Beclin 1 is required for Vps34 binding, autophagy and tumor suppressor function. Autophagy. 2005;1:46–52. doi: 10.4161/auto.1.1.1542. [DOI] [PubMed] [Google Scholar]
- 40.Criollo A, Maiuri MC, Tasdemir E, Vitale I, Fiebig AA, Andrews D, Molgo J, Diaz J, Lavandero S, Harper F, Pierron G, di Stefano D, Rizzuto R, Szabadkai G, Kroemer G. Regulation of autophagy by the inositol trisphosphate receptor. Cell Death Differ. 2007;14:1029–39. doi: 10.1038/sj.cdd.4402099. [DOI] [PubMed] [Google Scholar]
- 41.Oberstein A, Jeffrey P, Shi Y. Crystal structure of the BCL-XL-Beclin 1 peptide complex: Beclin 1 is a novel BH3-only protein. J Biol Chem. 2007:13123–32. doi: 10.1074/jbc.M700492200. [DOI] [PubMed] [Google Scholar]
- 42.Feng W, Huang S, Wu H, Zhang M. Molecular basis of Bcl-XL’s target recognition versatility revealed by the structure of Bcl-XL in complex with the BH3 domain of Beclin-1. J Mol Biol. 2007;372:223–35. doi: 10.1016/j.jmb.2007.06.069. [DOI] [PubMed] [Google Scholar]
- 43.Oltersdorf T, Elmore SW, Shoemaker AR, Armstrong RC, Auger DJ, Belli BA, Bruncko M, Deckwerth TL, Dinges J, Hajduk PJ, Joseph MK, Kitada S, Korsmeyer SJ, Kunzer AR, Letai A, Li C, Mitten MJ, Nettesheim DG, Ng S, Nimmer PM, O’Connor JM, Oleksijew A, Petros AM, Reed JC, Shen W, Tahir SK, Thompson CB, Tomaselli KJ, Wang B, Wendt MD, Zhang H, Fesik SW, Rosenberg SH. An inhibitor of Bcl-2 family proteins induces regression of solid tumors. Nature. 2005;435:677–81. doi: 10.1038/nature03579. [DOI] [PubMed] [Google Scholar]
- 44.Wei Y, Pattingre S, Bassik M, Sinha S, Levine B. JNK1-mediated phosphorylation of Bcl-2 regulates starvation-induced autophagy. Mol Cell. 2008 doi: 10.1016/j.molcel.2008.06.001. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bassik M, Scorrano L, Oakes SA, Pozzan T, Korsmeyer SJ. Phosphorylation of BCL-2 regulates ER Ca2+ homeostasis and apoptosis. EMBO J. 2004;23:1207–16. doi: 10.1038/sj.emboj.7600104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tasdemir E, Maiuri MC, Tajeddine N, Vitale I, Criollo A, Vicencio JM, Hickman JA, Geneste O, Kroemer G. Cell cycle-dependent induction of. 2007;6:2263–7. doi: 10.4161/cc.6.18.4681. [DOI] [PubMed] [Google Scholar]
- 47.Abeliovich H. Mitophagy: the life-or-death dichotomy includes yeast. Autophagy. 2007;3:275–7. doi: 10.4161/auto.3915. [DOI] [PubMed] [Google Scholar]
- 48.Bernales S, McDonald KL, Walter P. Autophagy counterbalances endoplasmic reticulum expansion during the unfolded protein response. PLoS Biol. 2006;4:423. doi: 10.1371/journal.pbio.0040423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhu W, Cowie A, Wasfy GW, Penn LZ, Leber B, Andrews DW. Bcl-2 mutants with restricted subcellular location reveal spatially distinct pathways for apoptosis in different cell types. EMBO J. 1996;15:4130–41. [PMC free article] [PubMed] [Google Scholar]
- 50.Gonzalez-Polo RA. The apoptosis/autophagy paradox: autophagic vacuolization before apoptotic death. J Cell Sci. 2005;118:3091–102. doi: 10.1242/jcs.02447. [DOI] [PubMed] [Google Scholar]
- 51.Criollo A, Vicencio JM, Tasdemir E, Maiuri MC, Lavandero S, Kroemer G. The inositol trisphosphate receptor in the control of autophagy. Autophagy. 2007;3:350–3. doi: 10.4161/auto.4077. [DOI] [PubMed] [Google Scholar]
- 52.Golstein P, Kroemer G. Redundant cell death mechanisms as relics and backups. Cell Death Differ. 2005;12:1490–6. doi: 10.1038/sj.cdd.4401607. [DOI] [PubMed] [Google Scholar]
- 53.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 54.Marino G, Salvador-Montoliu N, Fuevo A, Knecht E, Mizushima N, Lopez-Otin C. Tissue-specific autophagy Alterations and increased tumorigenesis in mice deficient in Atg4C/autophagin-3. J Biol Chem. 2007;282:18573–83. doi: 10.1074/jbc.M701194200. [DOI] [PubMed] [Google Scholar]
- 55.Karantza Wadsworth V, Patel S, Kravchuk O, Chen G, Mathew R, Jin S, White E. Autophagy mitigates metabolic stress and genome damage in mammary tumorigenesis. Genes Dev. 2007;21:1621–35. doi: 10.1101/gad.1565707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mathew R, Kongara S, Beaudoin B, Karp CM, Bray K, Degenhardt K, Chen G, Jin S, White E. Autophagy suppresses tumor progression by limiting chromosomal instability. Genes Dev. 2007;21:1367–81. doi: 10.1101/gad.1545107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Levine B. Cell biology: autophagy and cancer. Nature. 2007;446:745–47. doi: 10.1038/446745a. [DOI] [PubMed] [Google Scholar]
- 58.Mathew R, Karantza-Wadsworth V, White E. Role of autophagy in cancer. Nat Rev Cancer. 2007;7:961–7. doi: 10.1038/nrc2254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Pattingre S, Levine B. Bcl-2 inhibition of autophagy: a new route to cancer? Cancer Res. 2006;66:2885–8. doi: 10.1158/0008-5472.CAN-05-4412. [DOI] [PubMed] [Google Scholar]