

Abstract
(Z)-Trisubstituted allylic alcohols are widespread structural motifs in natural products and biologically active compounds but are difficult to directly prepare. Introduced herein is a general one-pot multicomponent coupling method for the synthesis of (Z)-α,α,β-trisubstituted allylic alcohols. (Z)-Trisubstituted vinylzinc reagents are formed in situ by initial hydroboration of 1-bromo-1-alkynes. Addition of dialkylzinc reagents induces a 1,2-metallate rearrangement that is followed by a boron-to-zinc transmetallation. The resulting vinylzinc reagents add to a variety of prochiral aldehydes to produce racemic (Z)-trisubstituted allylic alcohols. When enantioenriched aldehyde substrates are employed (Z)-trisubstituted allylic alcohols are isolated with high dr (>20:1 in many cases). For example, vinylation of enantioenriched benzyl protected α- and β-hydroxy propanal derivatives furnished the expected anti-Felkin addition products via chelation control. Surprisingly, silyl protected α-hydroxy aldehydes also afford anti-Felkin addition products. A protocol for the catalytic asymmetric addition of (Z)-trisubstituted vinylzinc reagents to prochiral aldehydes with a (−)-MIB-based catalyst has also been developed. Several additives were investigated as inhibitors of the Lewis acidic alkylzinc halide byproducts, which promote the background reaction to form the racemate. α-Ethyl and α-cyclohexyl (Z)-trisubstituted allylic alcohols can now be synthesized with excellent levels of enantioselectivity in the presence of diamine inhibitors.
1. Introduction
Chiral allylic alcohols are prevalent in natural products and are a mainstay in organic synthesis as intermediates and starting materials. As a result, their synthesis has received considerable attention.1–10 Progress has been made in the enantioselective formation of (E)-11–18 and (Z)-disubstituted19 as well as (E)-trisubstituted and tetrasubstituted20 allylic alcohols.11–13,15,21–25 General methods for the direct, efficient, and stereoselective synthesis of (Z)-trisubstituted allylic alcohols, however, remain sparse.26
One of the most common strategies to prepare (Z)-trisubstituted allylic alcohols begins with the Still-Gennari modification of the Horner-Wadsworth-Emmons (HWE) olefination.27 This popular method generates (Z)-trisubstituted α, β-unsaturated esters with good to excellent control over the double-bond geometry (Scheme 1, A). The requisite bis(trifluoroethyl) phosphonoester is prepared in 3-steps and is used with as many as 5 equiv of mildly toxic and expensive 18-crown-6 to maximize diastereoselectivity. The resulting unsaturated esters are rarely the desired intermediates, and additional synthetic manipulations are required before construction of the carbon skeleton can be resumed. Although this olefination approach is very reliable, it is not synthetically efficient and is limited to a linear two-carbon homologation.
Scheme 1.

Common Methods to Generate (Z)-Trisubstituted Allylic Alcohols and their Precursors
Another common method to generate (Z)-trisubstituted allylic alcohols involves addition of (Z)-vinylorganometallic reagents to aldehydes. The Nozaki-Hiyama-Kishi (NHK) reaction would appear to be one such example.7 Although it has been elegantly used in complex molecule syntheses, it suffers from several drawbacks. These include the toxicity of chromium, which is often used in catalytic amounts.28 Although conservation of the double bond geometry of the vinyl iodides or triflates is usually very good, (Z)-trisubstituted vinyl iodides and triflates readily isomerize to the (E)-isomer under NHK conditions (Scheme 1, B).28–32 Metallation of (Z)-vinyliodides with n-BuLi or i-PrMgBr followed by addition to chiral aldehydes is also attractive, but usually proceeds with reduced diastereoselectivity due to the reactive nature of these organometallic reagents (Scheme 1, C).33,34 Alternatively, softer nucleophiles based on Zn and Al, various additives, and Lewis acids are often employed to increase the selectivity.33–36 These methods are dependent on availability of (Z)-vinyl iodides with high stereochemical purity.
A clever method for the generation of (Z)-trisubstituted allylic alcohols was introduced by Denmark and coworkers. It involves copper catalyzed silylation of 2-butyn-1-ol with a disilane derivative, platinum catalyzed intramolecular syn-hydrosilylation, and palladium catalyzed cross-coupling with aryl iodides (Scheme 1, D).37 This 3-step process gives good yields with excellent control of the double bond geometry. To date, 2-butyn-1-ol is the only successful substrate, leaving significant potential for future development.
Silyl protected (Z)-trisubstituted allylic alcohols can be directly prepared with excellent enantiomeric excess and control of the double-bond geometry via Ng and Jamison’s novel multicomponent coupling sequence (Scheme 2). In this process, enantioenriched alkyl-substituted allenes, aromatic aldehydes, and silanes are coupled by an achiral Ni(NHC)-based catalyst with near perfect transfer of asymmetry.38 Application of this reaction to synthesis will depend on the availability of the requisite enantioenriched allene. This reaction represents the only direct method for the asymmetric synthesis of (Z)-trisubstituted allylic alcohols that simultaneously generates the (Z)-double bond and a stereocenter.
Scheme 2.

Ng and Jamison’s Synthesis of Enantioenriched (Z)-Trisubstituted Allylic Alcohols
We have been interested in the development and applications of asymmetric aldehyde vinylations for the synthesis of enantioenriched α- and β-amino acids,21,39 allylic alcohols,19,40,41 epoxy alcohols,42–45 and cyclopropyl alcohols.46,47 These studies have involved in situ generation (E)- and (Z)-disubstituted vinylzinc reagents. To expand the scope of easily accessible vinylzinc organometallics, we set out to develop in situ methods to generate (Z)-trisubstituted vinylzinc reagents.48 With the aim of introducing practical methods, we imposed the following constraints: 1) to use readily accessible starting materials, 2) to generate functional group-tolerant organometallic intermediates, and 3) to employ reactions that form exclusively (Z)-double bonds. Additionally, we sought to advance methods that would allow coupling of large fragments, as would be necessary in complex molecule synthesis.
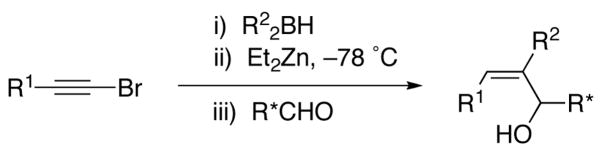
Herein we disclose the details of our synthesis of (Z)- α,α,β-trisubstituted allylic alcohols [hereafter referred to as (Z)-trisubstituted allylic alcohols]. Generation of (Z)-trisubstituted vinylzinc intermediates has been achieved via a novel 1,2-metallate rearrangement/transmetallation sequence (Scheme 3).49 Addition of the newly formed vinylzinc intermediate to prochiral aldehydes gives racemic allylic alcohols. Diastereoselective additions to enantioenriched α-hydroxy aldehyde derivatives are highly diastereoselective, giving anti-Felkin addition products even with silyl-protected substrates that normally undergo Felkin addition. The first catalytic asymmetric addition of (Z)-trisubstituted vinylzinc reagents to prochiral aldehydes enables the synthesis of enantioenriched (Z)-trisubstituted allylic alcohols. A portion of this work has been communicated.48,50
Scheme 3.

Generation and Trapping of (Z)-Trisubstituted Vinylzinc Intermediates for the Synthesis of (Z)-Trisubstituted Allylic Alcohols
2. Results and Discussion
2.1 Generation and Trapping of (Z)-Trisubstituted Vinylzinc Reagents
Our approach to the generation of (Z)-trisubstituted vinylzinc reagents is based on the work of Zweifel51 who found that hydroboration of 1-halo-1-alkynes with dicyclohexylborane was highly regioselective (Scheme 4, A).52 The resulting 1-halo-1-alkenylborane was treated with sodium methoxide, which triggered a 1,2-alkyl shift from boron to the vinylic center (also termed a 1,2-metallate rearrangement).53,54 Other nucleophiles such as Grignard reagents (B),55,56 alkyllithiums (B),55,56 and hydrides (C)57–59 also initiated similar 1,2-metallate rearrangements. The resulting (E)-vinylboranes are fairly unreactive and thus were either oxidized to ketones or protodeborated under acidic conditions to afford trans-alkenes (Scheme 4, A).
Scheme 4.

1,2-Metallate Rearrangements with 1-Halo-1-alkenyl Boranes
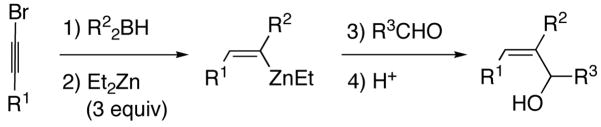
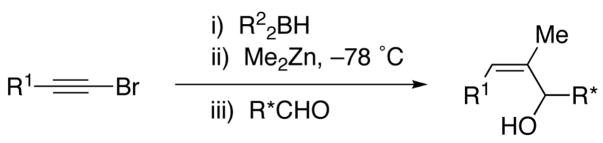
Also key to our approach were observations by Srebnik60 and Oppolzer,11 who pioneered alkenyl boron-to-zinc transmetallations to generate alkenylzinc reagents. These alkenylzinc species are significantly more reactive than their alkenylboron counterparts and readily add to aldehydes.11 By combining the seminal observations of Zweifel, Srebnik, and Oppolzer, we envisaged hydroboration of 1-halo-1-alkynes with R22BH to generate Zweifel’s intermediate 1-halo-1-alkenylboranes (Scheme 5). Rather than addition of alkoxides or highly reactive organolithium or Grignard reagents, we decided to examine the action of functional group-tolerant dialkylzinc reagents on the 1-halo-1-alkenylboranes to promote the 1,2-metallate rearrangement and subsequent boron to zinc transmetallation to generate the desired (Z)-trisubstituted vinylzinc intermediates. Trapping these intermediates with aldehydes would then furnish (Z)-trisubstituted allylic alcohols in a one-pot multicomponent coupling reaction.
Scheme 5.

One-Pot Multicomponent Coupling Reaction for the Stereospecific Synthesis of (Z)-Trisubstituted Allylic Alcohols
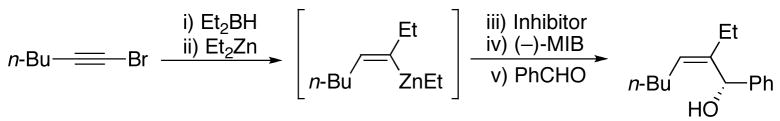
To generate the desired vinylzinc species, hydroboration of 1-bromo-1-hexyne was performed with diethylborane at rt for 30 min. The 1-bromo-1-alkenylborane was formed as a single regioisomer as confirmed by 1H NMR spectroscopy (Scheme 5, A, R2=Et). When the hydroboration was monitored by 11B{1H} NMR spectroscopy, the signal for Et2BH (25 ppm) was replaced by a new resonance at 59 ppm that was attributed to the 1-bromo-1-alkenyl borane (A, Scheme 5). Addition of one equiv of diethylzinc to the solution at −78° C and warming to room temperature produced a new resonance in the 11B{1H} NMR spectrum at 86 ppm that is consistent with BEt3 (R2=Et), the product of the 1,2-metallate rearrangement and transmetallation. Surprisingly, intermediate B was not observed under these conditions. Substitution of vinyl for methyl in Me3B results in an upfield shift of around 10 ppm [for example: Me3B, 86 ppm; Me2B(CH=CH2), 74 ppm; MeB(CH=CH2)2, 64 ppm; B(CH=CH2)3, 56 ppm].61 On the basis of this trend, it was anticipated that B should have a 11B{1H} NMR shift of ~74 ppm, similar to the known cis-Et2B[CH(Me)=CHMe] (75 ppm).61 When another equiv of diethylzinc was added to the reaction mixture, no change was observed in the 11B{1H} NMR spectrum. Next, the reaction mixture was subjected to reduced pressure at 0 °C to removed the volatile materials. Fresh toluene was added to the residual oil and the 11B{1H} NMR spectrum acquired. No detectable boron-containing products were observed, consistent with loss of the volatile triethylborane in vaccu and formation of the desired (Z)-trisubstituted vinylzinc species (D, Scheme 5).
To generate (Z)-trisubstituted allylic alcohols, 1-bromo-1-hexyne was combined with diethylborane as above. The reaction vessel was then cooled to −78 °C and 3 equiv diethylzinc were added to promote the 1,2-metallate rearrangement and the transmetallation to produce D in Scheme 5. Two equiv formaldehyde were then added to trap the (Z)-vinylzinc intermediate and the reaction was allowed to warm to room temperature. Quenching with dilute acid and purification on silica gel afforded the allylic alcohol product in 70% yield (Table 1, entry 1). Similar results were obtained with the benzyloxy-substituted 1-bromo-1-alkyne (entry 2). Unfortunately, (Z)-trisubstituted vinylzinc additions to aldehydes other than formaldehyde resulted in yields below 40%, with byproducts derived from ethyl addition to the aldehydes isolated. This byproduct was unexpected, because vinyl- and arylzinc reagents are known to add to aldehydes at least two orders of magnitude faster than alkylzinc reagents.11,40,41,44,62–64 Perhaps the steric bulk about the trisubstituted vinylzinc reagent is responsible for the reduced rate of carbonyl addition. We hypothesized that removal of the excess diethylzinc from the reaction mixture would increase the aldehyde vinylation and decrease the ethyl addition. Thus, after addition of the diethylzinc for the 1,2-metallate rearrangement and the transmetallation, the volatile contents of the reaction mixture (excess diethylzinc, triethylborane byproduct, and toluene) were removed at low pressure. Addition of fresh toluene and the aldehyde at 0 °C followed by stirring for 10–16 h, workup, and purification on silica gel afforded the desired (Z)-trisubstituted allylic alcohol with improved yields (Table 1). As seen in entries 3–8, 1-bromo-1-alkynes were successfully coupled with a series of aryl and aliphatic aldehydes providing the corresponding (Z)-trisubstituted allylic alcohols in 61–84% yield.
Table 1.
One-Pot Synthesis of (Z)-Trisubstituted Allylic Alcohols
 | |||||
|---|---|---|---|---|---|
| entry | R1 | R2 | R3CHO | yield (%) | product |
| 1 | n-Bu | Et | CH2O | 70a | 1 |
| 2 | CH2OBn | Et | CH2O | 61a | 2 |
| 3 | n-Bu | Et | Me2CHCH2CHO | 61 | 3 |
| 4 | (CH2)2OTBDPS | Et | Me2CHCH2CHO | 71 | 4 |
| 5 | (CH2)2OTBDPS | Et | p-Cl-C6H4-CHO | 84 | 5 |
| 6 | CH2OBn | Et | p-Cl-C6H4-CHO | 65 | 6 |
| 7 | (CH2)2OTBS | Et | p-Me-C6H4-CHO | 82 | 7 |
| 8 | (CH2)2OTBS | Et | p-Cl-C6H4-CHO | 84 | 8 |
| 9 | n-Bu | Cy | p-Me-C6H4-CHO | 60 | 9 |
| 10 | n-Bu | Cy | o-MeO-C6H4-CHO | 63 | 10 |
Two equiv para formaldehyde added without removal of the volatile materials.
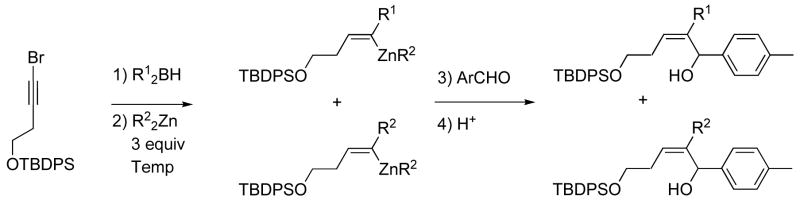
Substitution of dicyclohexylborane for diethylborane was next examined to generate the cyclohexyl substituted product (Scheme 5, R2=Cy). Interestingly, hydroboration of the bromo alkyne with dicyclohexylborane followed by addition of diethylzinc at 0 °C and p-tolualdehyde led to the expected cyclohexyl substituted product with up to 20% ethyl migration product (Table 2, entry 1, R1=Cy, R2=Et). This result prompted a brief study of various hydroborating and transmetallating reagents to examine their impact on the product mixture and to identify conditions to more strongly favor migration of the B-alkyl over the Zn-alkyl. In contrast to addition of the diethylzinc at 0 °C, when the diethylzinc addition was performed at −78 °C, Cy:Et migration increased to a synthetically useful 14:1 (Table 2, entry 2). When these conditions were used with the substrates in Table 1 the cyclohexyl-substituted allylic alcohols with p-tolualdehyde and o-anisaldehyde were obtained in approximately 60% yield (entries 9 and 10).
Table 2.
Examination of the Impact of Dialkylborane and Dialkylzinc Reagents on the 1,2-Metallate Rearrangement.
 | ||||||
|---|---|---|---|---|---|---|
| entry | R1 | additive | R2 | T (°C) | time | R1 : R2 ratio |
| 1 | Cy | Et | 0 | 16 h | Cy:Et = 4:1 | |
| 2 | Cy | Et | −78 | 30 min | Cy:Et = 14:1 | |
| 3 | Et | i-Pr | −78 | 30 min | Et:i-Pr = 5:1 | |
| 4 | Et | n-Bu | −78 | 30 min | Et:n-Bu = 4:1 | |
| 5 | Et | Me | −78 | 30 min | Et:Me = 2.5:1 | |
| 6 | Cy | Me | −78 | 30 min | Cy:Me = 3:1 | |
| 7 | Cy | t-BuLi | Et | −78 | 30 min | R2 = only H |
Next, the origin of the primary and secondary alkyl groups were reversed by using Et2BH and (i-Pr)2Zn with the dialkylzinc added at −78 °C (Table 2, entry 3). In this case, the ethyl migration product predominated (Et:i-Pr=5:1). Two primary alkyl group donors were used to compare groups with similar migratory aptitudes. In these experiment hydroboration was performed with Et2BH and transmetallation with (n-Bu)2Zn (Table 2, entry 4) or Me2Zn (entry 5) at −78 °C. With (n-Bu)2Zn, the product contained a 5:1 mixture of Et:n-Bu substituted allylic alcohols. When Me2Zn was employed, the ratio of Et:Me was closer to statistical (2.5:1, entry 5). Use of Cy2BH and ZnMe2 resulted in a 3:1 ratio of cyclohexyl to methyl migration (entry 6). Taken together, these results lead us to believe that a discrete trialkyl vinyl boronate intermediate is involved in these processes. In this intermediate, migration of the alkyl group originally attached to boron (R2, Scheme 5) is favored over the alkyl originating from the dialkylzinc. It is noteworthy that when a hydride is added to the empty site on boron (from t-BuLi, for example),58 only hydride migration to the vinylic center is observed and no alkyl migration is detected (Scheme 6 and Table 2, entry 7).19
Scheme 6.

Comparison of Alkyl and Hydride Migration.
Insight into the ease with which 1,2-metallate rearrangement and transmetallation occur was gained by hydroboration with Et2BH and transmetallation at −78 °C with Me2Zn followed by quenching the reaction mixture with methanol after 30 min at that temperature. The ethyl and methyl substituted (Z)-olefins were obtained in a 2.7:1 ratio (Scheme 7). This result indicates that both the 1,2-metallate rearrangement and the transmetallation take place at a surprisingly low temperature. It is noteworthy that the protonolysis of vinyl boranes is typically performed with acetic acid at 0 °C.65,66
Scheme 7.

Protonalysis of Intermediates in the Generation of Trisubstituted Vinylzinc Species.
An attractive feature of this multicomponent coupling is the stereospecific nature of the 1,2-metallate rearrangement. As a result, none of the (Z)-trisubstituted allylic alcohols prepared in Table 1 were contaminated by detectable quantities of the undesired (E)-isomers.
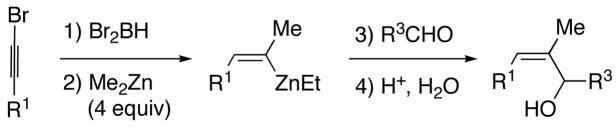
2.2. Synthesis of α-Methyl (Z)-Trisubstituted Allylic Alcohols
Naturally occurring (Z)-trisubstituted allylic alcohols often possess an α-methyl substituent. Following the procedure outlined in the previous section would require hydroboration with dimethylborane, which is a pyrophoric gas at ambient temperature.67 With the goal of developing practical and user-friendly methods, we sought to avoid using dimethylborane. To access intermediate A (Scheme 5, R2=Me) by a safer route we envisioned substituting dibromoborane for dimethylborane. Thus, hydroboration of 1-bromo-1-alkynes with dibromoborane was complete after 1 h at 70 °C or 12 h at rt (Scheme 8, X=Br). When the reaction was monitored by 11B{1H} NMR spectroscopy the Br2BH resonance (−8 ppm) diminished and was replaced by the signal for the vinyl dibromoborane (intermediate E, −1 ppm). Treatment of this intermediate with two equiv of dimethylzinc at −78 °C gave rise to 1-bromo vinyl dimethylborane F, resonating at 63 ppm in the 11B{1H} NMR spectrum. Addition of a third equiv of dimethylzinc induced the 1,2-metallate rearrangement and transmetallation, resulting in a shift in the 11B{1H} NMR spectrum to 86 ppm, consistent with formation of BMe3. We propose that the vinylzinc halide H (X=Br) is formed from MeZnBr and G, which is not observed by 11B{1H} NMR spectroscopy under the conditions of these room temperature experiments. As observed above, addition of the fourth equiv dimethylzinc did not result in a change in the 11B{1H} NMR spectrum. Also formed in this process are 3 equiv of MeZnBr byproduct (Scheme 8).
Scheme 8.

Proposed Mechanism for the Formation of α-Methyl (Z)-Trisubstituted Allylic Alcohols (X=Br or Cl)
In the synthesis of allylic alcohols, hydroboration of 1-bromo-1-hexyne with dibromoborane was performed as outlined above. Subsequently, 4 equiv of Me2Zn were added at −78 °C and the solution stirred for 30 min. The reaction vessel was then evacuated to remove the volatile materials (including excess Me2Zn). Toluene and the aldehyde substrate were then added at 0 °C. After 10–16 h the aldehyde had been consumed, as judged by TLC. The reaction mixture was next quenched with dilute acid and the product purified on silica gel. The desired (Z)-allylic alcohols were isolated in good to excellent yields (Table 3).
Table 3.
Multicomponent Synthesis of α-Methyl (Z)-Trisubstituted Allylic Alcohols
 | ||||
|---|---|---|---|---|
| entry | R1 | R3CHO | yield (%) | product |
| 1 | n-Bu | CH2O | 75a | 11 |
| 2 | (CH2)2OTBS | CH2O | 77a | 12 |
| 3 | n-Bu | Me2CHCH2CHO | 92 | 13 |
| 4 | n-Bu | p-MeC6H4CHO | 71 | 14 |
| 5 | n-Bu | p-ClC6H4CHO | 70 | 15 |
Two equiv para formaldehyde added without removal of the volatile materials.
In an effort to illustrate the potential utility of our method in natural product synthesis, (Z)-trisubstituted allylic alcohol 18, which is an intermediate in the total synthesis of (−)-hennoxazole A68 and a fragment common to the natural products (+)-migrastatin69 and (+)-discodermolide, was synthesized.70,71 A modified Corey-Fuchs procedure72,73 was employed to convert aldehyde 16 into enantioenriched 1-bromo-1-alkyne 17 in two steps. Subjecting bromoalkyne 17 to hydroboration, 1,2-metallate rearrangement/transmetallation, and addition to formaldehyde yielded the (Z)-trisubstituted allylic alcohol 18 in 73% yield without loss of ee (Scheme 9).
Scheme 9.

Synthesis of an Intermediate in the Total Synthesis of (−)-Hennoxazole A
Having developed an in situ method for the generation of (Z)-trisubstituted vinylzinc reagents and their addition to prochiral aldehydes, we desired to examine the vinylation of chiral aldehydes to control the stereochemistry at the carbinol center.
2.3 Vinylation of Enantioenriched Aldehydes Employing Diethyl- and Dicyclohexylborane
A fundamental strategy in organic synthesis makes use of one or more preexisting stereocenters in the substrate to control formation of new stereocenters. Classic examples are found in the synthesis of polyhydroxylated intermediates and natural products via carbonyl additions with chiral aldehydes.35 Excellent models for stereoselectivity have been introduced and are generally accepted. For example, use of protected α- and β-hydroxy aldehyde derivatives bearing bulky silicon-based protecting groups generally result in Felkin addition.35 In contrast, smaller protecting groups such as benzyl favor chelation control and give rise to the Cram chelation product or anti-Felkin addition products. We chose to explore addition of our (Z)-trisubstituted vinylzinc reagents to these important substrate classes.
Employing diethylborane in the generation of (Z)-trisubstituted vinylzinc reagents followed by addition to silyl protected α-hydroxy aldehydes proceeded with high diastereoselectivities (dr >15:1). For example, (S)-2-(tert-butyldimethylsilyloxy)-propanal underwent addition with excellent diastereoselectivity (>20:1 dr) and high yields (82–95%, Table 4, entries 1–3). Likewise, beginning with dicyclohexylborane (R2=Cy) resulted in α-cyclohexyl (Z)-trisubstituted allylic alcohols with >20:1 dr, albeit in slightly diminished yields (54–75%, entries 4–6). The bulkier TIPS protected aldehyde gave similar results to the TBS-protected analogue (entries 1 vs. 7). Substrates expected to undergo chelation control were next examined. Using either diethyl- or dicyclohexyl borane, addition to (S)-2-(benzyloxy)propanal or the PMB analogue, (S)-2-(4-methoxybenzyloxy)propanal, occurred with >15:1 dr and yields ranging from 52–75% (entries 8–12).
Table 4.
Diastereoselective Synthesis of Allylic Alcohols with α-Ethyl and α-Cyclohexyl Substituents
 | |||||||
|---|---|---|---|---|---|---|---|
| entry | R1 | R2 | RCHO | yield (%) | dra | product | major product |
| 1 | n-Bu | Et | 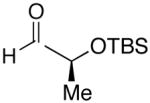 |
82 | >20:1 | 19 | 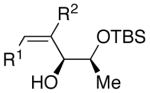 |
| 2 | (CH2)3Cl | Et | 92 | >20:1 | 20 | ||
| 3 | (CH2)2OTBDPS | Et | 95 | >20:1 | 21 | ||
| 4 | n-Bu | Cy | 75 | >20:1 | 22 | ||
| 5 | (CH2)3Cl | Cy | 54 | >20:1 | 23 | ||
| 6 | (CH2)2OTBDPS | Cy | 60 | >20:1 | 24 | ||
| 7 | n-Bu | Et | 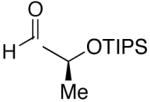 |
74 | >20:1 | 25 | 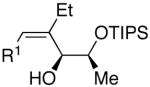 |
| 8 | n-Bu | Et | 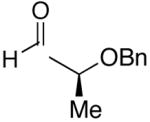 |
75 | 18:1 | 26 | 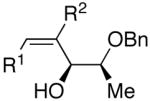 |
| 9 | (CH2)2OTBDPS | Et | 75 | 16:1 | 27 | ||
| 10 | n-Bu | Cy | 68 | 15:1 | 28 | ||
| 11 | (CH2)2OTBDPS | Cy | 52 | 16:1 | 29 | ||
| 12 | n-Bu | Et | 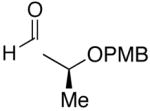 |
71 | 20:1 | 30 | 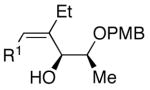 |
| 13 | 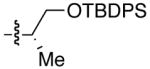 |
Et | 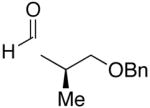 |
74 | 12:1 | 31 | 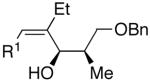 |
| 14 | 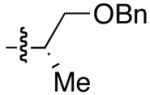 |
Et | 87 | 13–18:1 | 32 | ||
| 15 | 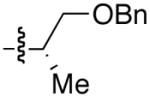 |
Et | 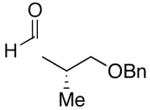 |
65 | 7–12:1 | 33 | 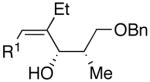 |
| 16 | n-Bu | Et | 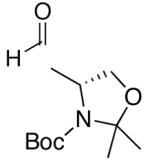 |
92 | >20:1 | 34 | 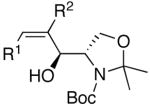 |
| 17 | n-Bu | Cy | 29 | >20:1 | 35 | ||
| 18 | n-Bu | Et | 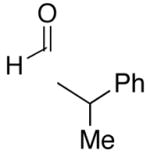 |
91 | 1:8 | 36 | 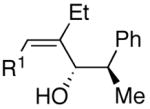 |
| 19 | (CH2)2OTBDPS | Et | 95 | 1:8.5 | 37 | ||
Diastereomeric ratio (anti-Felkin : Felkin) based on 1H NMR of crude product. Relative stereochemistry was determined by X-ray crystallography, Mosher ester analysis, or Heathcock’s analysis. See Supporting Inormation for details.
Use of enantioenriched protected α-methyl β-hydroxy propanals was examined with an enantioenriched 1-bromo-1-alkyne (entries 13–15) to determine both the impact of the aldehyde β-alkoxy group and the alkyne α-stereocenter on the diastereoselectivity of the addition. Generation of the vinylzinc intermediate from the TBDPS protected alkyne was followed by treatment with the β-benzyloxy (R)-aldehyde. The addition afforded the product in 74% isolated yield with high diastereoselectivity (12:1, entry 13). Under the same conditions, use of the benzyloxy-protected alkyne resulted in higher diastereoselectivity (13–18:1) and yield (87%, entry 14). Use of the enantiomeric aldehyde caused a decrease in the diastereoselectivity (7–12:1 dr) and yield (67%, entry 15). This latter combination is the mismatched pair.
A common starting material for the preparation of enantio- and diastereoenriched amino alcohol derivatives is Garner’s aldehyde.74 This aldehyde proved to be an excellent substrate when reactions were initiated with diethylborane (92% yield, >20:1 dr, entry 16), giving a highly functionalized allylic alcohol. In contrast, the analogous reaction employing dicyclohexylborane proved more challenging, yielding less than 30% allylic alcohol product (entry 17). The major component isolated from the reaction mixture was the olefin (E)-(1-cyclohexyl)-1-hexene, which presumably arises from protonation of the unreacted vinylzinc intermediate. Isolation of the (E)-olefin suggests that the vinylzinc intermediate may be too bulky to efficiently add to the sterically hindered Garner aldehyde. Additions to racemic 2-phenyl propanal exhibited excellent yields and good selectivity (>8:1, entries 18 and 19). As seen Table 4, addition of (Z)-vinylzinc reagents to protected α-hydroxy and amino hydroxy aldehydes proceeded with excellent stereochemical control. Determination of the stereochemical outcome in the vinylation of chiral aldehydes was the next task at hand.
As outlined above, bulky silyl protecting groups normally favor Felkin addition whereas smaller benzyloxy groups lead to chelate formation and anti-Felkin or chelation controlled addition.35 Analysis of the stereochemical outcome of additions to protected α-hydroxy aldehydes in Table 4 was performed by the modified Mosher method (entries 1–12).75,76 As expected, benzyloxy and PMB protected aldehydes underwent chelation-controlled addition to afford anti-Felkin products. To our surprise, Mosher analysis of the TBS-protected addition products (entries 1–6) indicated that they to underwent anti-Felkin addition. Although TBS protected α-hydroxy aldehydes nearly always undergo Felkin addition, a few examples of anti-Felkin additions have been reported.77–82 These unexpected results inspired us to examine the bulkier TIPS protected derivative. We were again surprised to find that the product was derived from anti-Felkin addition. To confirm these unexpected results, allylic alcohols 19, 25 and 30 were deprotected to generate 1,2-diols (Scheme 10). All three diols had matching 1H and 13C{1H} NMR spectra and optical rotations. The diols were then independently converted to the acetonides and carbonates, which again revealed identical spectra, consistent with the assignments made by the modified Mosher method. X-ray structure determination of 32 confirmed that addition to Garner’s aldehyde gave the anti-Felkin product (Figure 1). On the basis of Lodge and Heathcock’s analysis,83,84 it was determined that additions to 2-phenylpropanal gave the expected Felkin addition product.
Scheme 10.

Stereochemical Correlation Between Compounds 19, 25, and 30
Figure 1.

ORTEP drawing 35 (entry 17, Table 4) with 30% probability thermal ellipsoids
2.4 Diastereoselective Synthesis of α-Methyl (Z)-Trisubstituted Allylic Alcohols
As with the prochiral aldehydes in Section 2.2, diastereoselective additions of α-methyl (Z)-trisubstituted vinylzinc reagents to chiral aldehydes were initiated primarily with dihaloboranes, X2BH (X=Cl, Br). Dimethylborane was used for comparison purposes.
The results of the diastereoselective synthesis of α-methyl (Z)-trisubstituted allylic alcohols are illustrated in Table 5. Although most reactions were conducted with Cl2BH, Br2BH gave nearly identical yields and diastereoselectivities. Additions to (S)-2-(tert-butyldimethylsilyloxy)-propanal proceeded in 67–78% yield with excellent diastereoselectivities (>20:1). Modified Mosher analysis again indicated that anti-Felkin addition predominated (entries 1–5). To compare additions beginning with X2BH, Me2BH, and R2BH (R=Et, Cy), Me2BH was prepared85 by conproportionation of a 2:1 mixture of Me3B86 and BH3·SMe2 in diethyl ether. After hydroboration of 1-bromo-1-hexyne with Me2BH, the procedure employed in Table 3 with dimethylzinc was applied to (S)-2-(tert-butyldimethylsilyloxy)-propanal. As can be seen from entries 4 and 5 (Table 5), the results with Me2BH were similar to those with Cl2BH (entries 1 and 3), and yields were almost as high as those with diethylborane (Table 4, entries 1 and 3). Additions to (S)-2-(benzyloxy)propanal initiated with Cl2BH resulted in 51–57% yields and high facial selectivity (≥17:1 dr, entries 6 and 7). When the β-benzyloxy derivative (R)-3-(benzyloxy)-2-methylpropanal was used, the addition product was isolated in 59–70% with high diastereoselectivity favoring anti-Felkin addition (>9:1, entries 8–10). The slight decrease in diastereoselectivity relative to the α-protected analogues is likely due to the increased ring size of the chelated substrate. Garner’s aldehyde gave lower yield (47%, entry 11) but excellent diastereoselectivity (>20:1) and was assigned as the anti-Felkin addition product in analogy to 32 (Table 4, entry 16). The diastereoselectivity in the addition to 2-phenylpropanal decreased from 8.5:1 with the ethyl-substituted vinylzinc (Table 4, entry 19) to 4:1 with the methyl-substituted analogue (Table 5, entry 12). This decrease is attributed to the smaller size of the vinyl zinc reagent in the latter case. It is possible that the lower yields in Table 5 relative to those in Table 4 are due to the reactivity and the Lewis acidity of the haloboranes, which are known to cleave ethers.
Table 5.
Diastereoselective Synthesis of α-Methyl Substituted Allylic Alcohols
 | |||||||
|---|---|---|---|---|---|---|---|
| entry | R1 | R2 | RCHO | yield (%) | dra | product | major product |
| 1 | n-Bu | Cl | 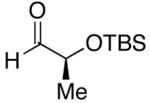 |
68 | >20:1 | 41 | 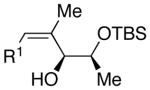 |
| 2 | (CH2)3Cl | Cl | 67 | >20:1 | 42 | ||
| 3 | (CH2)2OTBDPS | Cl | 78 | >20:1 | 43 | ||
| 4 | n-Bu | Me | 78 | >20:1 | 41 | ||
| 5 | (CH2)2OTBDPS | Me | 75 | >20:1 | 43 | ||
| 6 | n-Bu | Cl | 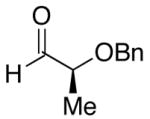 |
51 | >20:1 | 44 | 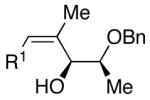 |
| 7 | (CH2)2OTBDPS | Cl | 57 | 17:1 | 45 | ||
| 8 | n-Bu | Br | 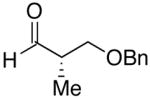 |
70 | 17:1 | 46 | 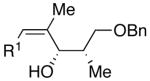 |
| 9 | CH2OTBDPS | Br | 63 | 9–13:1 | 47 | ||
| 10 | (CH2)2OTBDPS | Br | 59 | 11:1 | 48 | ||
| 11 | (CH2)2OTBDPS | Cl | 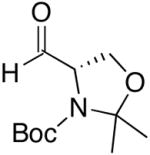 |
47 | >20:1 | 49 | 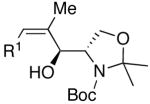 |
| 12 | (CH2)2OTBDPS | Cl | 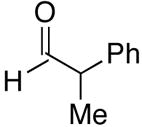 |
55 | 1:4 | 50 | 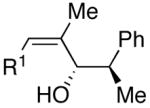 |
Diastereomeric ratio (anti-Felkin : Felkin) based on 1H NMR of crude product. Relative stereochemistry was determined by X-ray crystallography, Mosher ester analysis, or Heathcock’s analysis. See Supporting Information for details.
2.5 Catalytic Asymmetric (Z)-Trisubstituted Vinylzinc Additions
The results of prior sections demonstrate that excellent stereochemical control can be achieved in diastereoselective additions of (Z)-trisubstituted vinylzinc reagents to protected chiral α- and β-hydroxy aldehydes. The utility of enantioenriched allylic alcohols inspired investigations into a catalytic asymmetric version of this reaction using prochiral aldehydes.
To advance a catalytic asymmetric (Z)-vinylation of aldehydes we adapted the optimized procedure employed in Table 4. Thus, after hydroboration of 1-bromo-1-hexyne with diethylborane, 3 equiv diethylzinc were added at −78 °C, and after 30 min the volatile materials were removed under reduced pressure. Addition of fresh toluene, 5–20 mol% of Nugent’s enantioenriched amino alcohol (−)-MIB,87,88 and benzaldehyde resulted in formation of the desired allylic alcohol (Table 6). Unfortunately, the product was racemic (entries 1–3). Although MIB forms one of the fastest and most enantioselective catalysts for addition of alkylzinc and vinylzinc reagents to aldehydes,2 the unexpected absense of enantioselectivity in these additions indicated that the MIB-based zinc complex did not promote the addition under these conditions.
Table 6.
Impact of Inhibitors on the Catalytic Asymmetric Generation of α-Ethyl (Z)-Trisubstituted Allylic Alcohols
 | ||||
|---|---|---|---|---|
| entry | inhibitor | equiva | (−)-MIB (mol %) | ee (%) |
| 1 | none | – | 5 | 0 |
| 2 | none | – | 10 | 0 |
| 3 | none | – | 20 | 0 |
| 4 | DiMPEG | 0.1 | 5 | 0 |
| 5 | DiMPEG | 0.2 | 5 | 0 |
| 6 | DiMPEG | 0.5 | 10 | 0 |
| 7 | TMEDA | 0.1 | 5 | 2 |
| 8 | TMEDA | 0.2 | 5 | 2 |
| 9 | TMEDA | 0.6 | 5 | 16 |
| 10 | TMEDA | 1 | 5 | 96 |
| 11 | TEEDA | 1 | 5 | 97 |
| 12 | TMPDA | 1 | 5 | 92 |
| 13 | DIEDA | 1 | 5 | 73 |
| 14 | DMEDA | 1 | 5 | 72 |
With respect to equivalents of bromoalkynes used in the reaction.
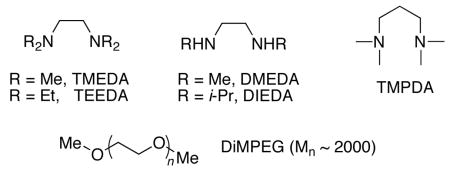
After consideration of the proposed reaction mechanism (Scheme 5) we suspected that the zinc halide byproduct might be sufficiently Lewis acidic to promote the background reaction and generate the racemate. We had encountered related problems in the asymmetric arylation of aldehydes with aryl bromides89 and in the asymmetric vinylation of aldehydes to afford (Z)-disubstituted allylic alcohols.19 Both of these processes generated LiCl byproducts and gave racemic products in the presence of the MIB-based catalyst. Rather than filtration of the reaction mixtures to remove the LiCl, which would be difficult and impractical in large-scale applications, we desired to inhibit the LiCl from promoting the additions. Inhibition of the LiCl was accomplished by addition of diamines such as N,N,N′N′-tetraethylethylene diamine (TEEDA) and the tetramethyl analogue TMEDA. In the presence of these diamines, the abovementioned arylations and vinylations exhibited enantioselectivities >90% for most substrates. More closely related to the present study, in which zinc halides are the byproduct, Bolm and coworkers observed that dimethyl poly(ethylene glycol) (DiMPEG) inhibited zinc bromide, albeit at the cost of the yield (8–31%).90
On the basis of Bolm’s results with ZnBr2,90 we first tested 0.1–0.5 equiv DiMPEG (Mn~2000) as a potential inhibitor in the vinylation of benzaldehyde, but to no avail (0% ee, entries 4–6, Table 6). Although LiCl, which we had successfully inhibited in the arylation and vinylation reactions above, and zinc halides are quite different, they both have at least two open coordination sites. In contrast, the MIB-base catalyst seems to have only a single accessible coordination site. We therefore believed that our diamine-based inhibitor strategy was worthy of exploration in the context of the enantioselective (Z)-vinylation. Thus, a series of diamines were examined beginning with TMEDA. We were encouraged by our initial results; as the amount of TMEDA was increased from 0.1 to 0.6 equiv the enantioselectivity went from 2% to 16% (entries 7–9). We were gratified to find that a further increase in the amount of diamine to 1 equiv resulted in product of 96% ee (entry 10)! Virtually identical enantioselectivity was observed with the tetraethylethylene diamine, TEEDA (97% ee, entry 11). Interestingly, 1 equiv 1,3-tetramethylpropylene diamine (TMPDA) resulted in a drop in the product enantioselectivity to 92%, likely because the decreased stability associated with the larger metallocycle formed on chelation of the diamine to zinc (entry 12). The secondary diamines N,N′-diisopropyl- and N,N′-dimethylethylene diamines were less effective at slowing the zinc halide-promoted reaction than their tetraalkyl counterparts. On the basis of these optimization experiments, and the lower cost of TMEDA relative to TEEDA, we employed 1 equiv TMEDA with a series of aldehydes and 1-bromo-1-alkynes.
The modified procedure for the catalytic asymmetric vinylation initially follows the protocol employed in Table 1 for prochiral alkyl and aryl aldehydes using diethylborane and diethylzinc. After removal of the volatile materials under reduced pressure, toluene, TMEDA, and (−)-MIB were added sequentially at 0 °C followed by the aldehyde. Reactions were monitored until the aldehyde had been consumed (TLC), quenched, and purified on silica gel. (Z)-Trisubstituted allylic alcohols were isolated in 63–90% yield with >94% ee using bromoalkynes with aliphatic substituents or derived from silyl protected propargyl and homopropargyl alcohols (Table 7, entries 1–6). Beginning the process with dicyclohexylborane provided the α-cyclohexyl-substituted allylic alcohols with high enantioselectivities (77–95%) and slightly diminished yields (50–80%, entries 7–9).
Table 7.
Catalytic Asymmetric Synthesis of α-Ethyl and α-Cyclohexyl (Z)-Trisubstituted Allylic Alcohols
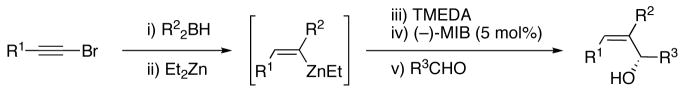 | |||||||
|---|---|---|---|---|---|---|---|
| entry | bromoalkyne (R1) | R2 | aldehyde | product | yield (%) | ee (%) | product |
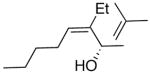
| 1 | n-Bu | Et | 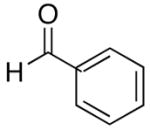 |
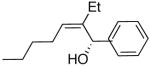 |
65 | 95 | 51 |
| 2 | n-Bu | Et | 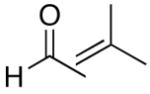 |
 |
90 | 94 | 52 |
| 3 | (CH2)2OTBDPS | Et | 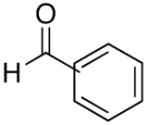 |
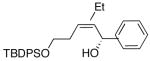 |
90 | 97 | 53 |
| 4 | (CH2)2OTBDPS | Et | 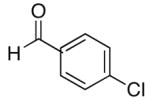 |
75 | 93 | 54 | |
| 5 | (CH2)2OTBDPS | Et | 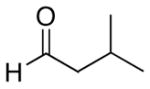 |
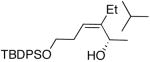 |
87 | 95 | 55 |
| 6 | CH2OTBDPS | Et | 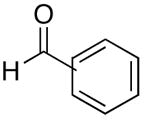 |
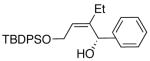 |
63 | 94 | 56 |
| 7 | n-Bu | Cy | 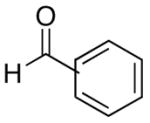 |
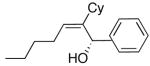 |
50 | 95 | 57 |
| 8 | (CH2)2OTBDPS | Cy | 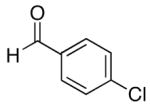 |
80 | 92 | 58 | |
| 9 | (CH2)2OTBDPS | Cy | 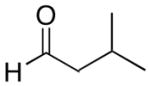 |
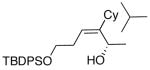 |
52 | 77 | 59 |
Attempts to adapt this procedure to the synthesis of α-methyl-substituted allylic alcohols with Br2BH and Cl2BH and varying amounts of DiMPEG or diamine inhibitor resulted in enantioselectivities below 30% and low yields (15–40%). We hypothesize that the low enantioselectivities arise from the additional 2 equiv of zinc halide byproduct generated when using X2BH, as outlined in Scheme 8. Increasing the equivalents of diamine to inhibit the additional zinc halide (>300 mol%) also appeared to inhibit the Lewis acidic MIB-based catalyst (5–20 mol%).
Use of Me2BH was plagued with problems of a different sort. Like the synthesis of α-ethyl- and α-cyclohexyl-substituted allylic alcohols, use of Me2BH generates only 1 equiv zinc halide. The use of TMEDA was ineffective, however, and levels of enantioselectivity did not surpass 24%. The issue could be that the gaseous Me2BH is generated as a solution in diethyl ether, which coordinates to the boron. Traces of Lewis basic solvent inhibit the MIB-based Lewis acid catalyst. Thus, we believe that a different strategy will be necessary for the enantioselective synthesis of α-methyl (Z)-trisubstituted allylic alcohols.
3. Summary and Outlook
Reported herein is a simple and efficient method for the stereospecific generation of (Z)-trisubstituted vinylzinc reagents. Beginning with readily accessible 1-halo-1-alkynes, hydroboration with diethyl- or dicyclohexylborane provide 1-halo-1-alkenylboranes with excellent regioselectivity. The key to success of this method is our discovery that dialkylzinc reagents can both induce the 1,2-metallate rearrangement with formation of a C-C bond and promote the boron-to-zinc transmetallation to generate the requisite (Z)-trisubstituted vinylzinc reagents. These reagents smoothly add to prochiral aldehydes to generate a variety of (Z)-trisubstituted allylic alcohols. It is noteworthy that no contamination by the thermodynamically more favorable (E)-allylic alcohols was observed by 1H NMR spectroscopy. In sharp contrast, the NHK reaction with (Z)-trisubstituted vinyl iodides and tosylates results in formation of the isomerized (E)-trisubstituted allylic alcohols.28–31
For the special case of α-methyl-substituted allylic alcohols, X2BH (X=Cl, Br) was substituted for the pyrophoric gas Me2BH in the generation of 1-bromo-1-alkenyl boranes. Here, the 3 equiv of dimethylzinc plays three roles by 1) promoting metathesis of the B-Br bonds forming B-Me bonds, 2) inducing the 1,2-metallate rearrangement with creation of a C-Me bond, and 3) orchestrating the boron-to-zinc transmetallation. The resulting vinylzinc reagent undergoes additions to prochiral aldehydes to produce racemic α-methyl (Z)-trisubstituted allylic alcohols in good yields with complete control of the double bond geometry.
The addition of (Z)-trisubstituted vinylzinc reagents to chiral protected α- or β-hydroxy aldehydes proceeded with high diastereoselectivities (>20:1 in most cases). With benzyloxy-substituted aldehydes, the expected anti-Felkin addition products formed via chelation control and were isolated in good yields. Vinylation of α-siloxy protected propanals also proceed with excellent stereochemical control (>20:1 dr). Surprisingly, the resulting products are derived from anti-Felkin addition. Although further investigations are needed to understand the mechanism of stereoinduction in this system, our suspicion is that the zinc halide byproduct is behaving as a chelating Lewis acid.
Finally, significant progress has been made in the catalytic asymmetric addition of (Z)-trisubstituted vinylzinc reagents to aldehydes to prepare enantioenriched allylic alcohols. The successful generation of α-ethyl and α-cyclohexyl (Z)-trisubstituted allylic alcohols suggests that other primary and secondary alkyl groups could be installed by use of readily accessible R2BH derivatives in the hydroboration of 1-halo-1-alkynes. Further research is necessary to develop the methyl 1,2-metallate rearrangement, transmetallation, and asymmetric addition, which has proven to be capricious.
The ability to access (Z)-trisubstituted vinylzinc reagents with near perfect control of the double bond geometry, and their propensity to add to prochiral and enantioenriched aldehydes with high enantio- and diastereoselectivity, render this method valuable in the synthesis of diverse (Z)-trisubstituted allylic alcohols that are otherwise difficult to prepare.
4. Experimental Section
General Considerations
All reactions were performed under a nitrogen atmosphere with oven-dried glassware using standard Schlenk or vacuum line techniques. Toluene was dried through an alumina column. THF and diethyl ether were distilled from Na/benzophenone. Thin-layer chromatography (TLC) and 11B NMR were used to monitor reaction progress. The 1H NMR and 13C{1H} NMR spectra were obtained on Brüker AM-500, DMX-360 and DMX 300 Fourier transform NMR spectrometers at 500, 360, and 300 MHz for 1H and 125, 90, 75 MHz for 13C{1H} NMR, respectively. Chemical shifts are reported in units of parts per million (ppm) downfield from tetramethylsilane or residual protonated solvent for 1H and 13C{1H} NMR; 11B NMR is calibrated to an external BF3·OEt2 standard. All coupling constants are reported in Hz. A vertical asymptote at 4.5 ppm appears on some 1H NMR spectra and at 100 ppm in some 13C{1H} NMR spectra, which are due to the iNMR processing software (ver. 0.7). The infrared spectra were obtained using a Perkin-Elmer Spectrum 100 series FT-IR spectrometer. All reagents were purchased from Aldrich or Acros unless otherwise described. 1-Haloalkynes were prepared according to literature procedures.82,91–95 Chiral aldehydes were prepared by literature methods;74,96–98 either Swern oxidation of the primary alcohol or DIBAL-H reduction of the methyl ester was performed to generate the crude chiral aldehydes just prior to reaction. These aldehydes were used without further purification due to their sensitivity. All commercially available aldehyde substrates were distilled prior to use. Diastereomeric ratios were determined by 1H NMR of crude reaction products. Flash chromatography was preformed on Silicycle silica gel (230–400 mesh). Analysis of enantiomeric excess was performed using a Hewlett-Packard 1100 Series HPLC and a Chiralcel OD-H chiral stationary phase column.
Cautionary Note
Dialkylzinc reagents are pyrophoric and must be handled with caution. In experiments involving removal of volatile materials that contain pyrophoric alkyl boron and/or alkyl zinc species, a cold trap was inserted between the Schlenk line and the reaction vessel and cooled with liquid nitrogen. After the volatile materials had been removed, the liquid nitrogen coolant was removed and the trap backfilled and then purged with nitrogen gas. While purging, a dilute solution of isopropanol in hexanes was then added followed by dropwise addition of water to destroy the alkyl boron and alkyl zinc species.
General Procedure for the Diastereoselective Addition to Chiral Aldehydes using Dialkylboranes
To the 1-bromo-1-alkyne (1 mmol) in a 3 times evacuated and N2 flushed 10 mL Schlenk flask was added toluene (1 mL) and dialkylborane (1 mmol) at 0 °C and warmed to rt over 30 min. Following hydroboration, dialkylzinc (3 mmol, 1–2 M in toluene) was added at −78 °C and stirred for 30 min. Warming to 0 °C, the flask was evacuated to dryness under reduced pressure. Toluene (1 mL) was then added, followed by the aldehyde (0.66 mmol). The reaction was stirred at 0°C and allowed to warm to rt. The reaction was stirred for 7–16 h until complete (by TLC). After completion, the reaction mixture was quenched by cautions addition of sat. NH4Cl (1 mL) followed 2N HCl (1 mL) and 5 mL of EtOAc. After separation, the aqueous layer was extracted with EtOAc (2 × 10 mL), and the combined organic layers were washed with NaHCO3 and brine, dried over MgSO4, and concentrated. The crude product was purified by column chromatography on silica gel.
General Procedure for the Diastereoselective Addition to Chiral Aldehydes using Dihaloborane
To a 1-bromo-1-alkyne (1 mmol) in a 3 times evacuated and N2 flushed 10 mL Schlenk flask was added toluene (1 mL) and HBX2 (X=Cl or Br, 1 mmol) at 0 °C. Following hydroboration, at 70 °C for 1 h, dialkylzinc (4.5 mmol, 1–2 M in toluene) was added at −78 °C and stirred for 30 min. Warming to 0 °C, the flask was evacuated to dryness under reduced pressure. Toluene (1 mL) was added followed by the aldehyde (0.66 mmol, generally neat). The reaction was stirred at 0°C and allowed to warm to rt. The reaction was stirred for 7–16 h until complete (by TLC). After completion, the reaction mixture was quenched by cautions addition of sat. NH4Cl (2 mL) followed 2N HCl (1 mL) and 5 mL of EtOAc. After separation, the aqueous layer was extracted with EtOAc (2 × 10 mL), and the combined organic layers were washed with NaHCO3 and brine, dried over MgSO4, and concentrated. The crude product was purified by column chromatography on silica gel.
General Procedure for the Catalytic Asymmetric Addition to Prochiral Aldehydes
To 1-bromo-1-alkyne (1 mmol) in a 3 times evacuated and N2 flushed 10 mL Schlenk flask was added toluene (1 mL) and dialkylborane (1 mmol) at 0 °C. Following hydroboration, dialkylzinc (4.8 mmol, 1–2 M in toluene) was added at −78 °C and stirred for 30 min. Warming to 0 °C, the flask was evacuated to dryness under reduced pressure. Toluene (1 mL) was added followed by TMEDA (1 mmol). After stirring for 2 min, (−)-MIB (5 mol%) was added followed by addition of aldehyde (0.5 mmol, generally neat). The reaction was stirred at 0°C and allowed to warm to rt overnight. The reaction was quenched by cautious addition of sat. NH4Cl (1 mL) followed 2N HCl (1 mL) and 5 mL of EtOAc. After separation, the aqueous layer was extracted with EtOAc (2 × 10 mL), and the combined organic layers were washed with NaHCO3 and brine, dried over MgSO4, and concentrated. The crude product was purified by column chromatography on silica gel.
Supplementary Material
Acknowledgments
We thank the NSF (CHE-065210) and NIH (National Institute of General Medical Sciences, GM58101) for partial support of this work. We are also grateful to Akzo Nobel Polymer Chemicals for a gift of dialkylzinc reagents and to a referee for helpful comments on B to C migrations.
Footnotes
Supporting Information Available: Procedures, full characterization, the CIF file for compound 32, and stereochemical assignments of new compounds are available (PDF). This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Walsh PJ, Kozlowski MC. Fundamentals of Asymmetric Catalysis. University Science Books; Sausalito: 2008. [Google Scholar]
- 2.Pu L, Yu HB. Chem Rev. 2001;101:757–824. doi: 10.1021/cr000411y. [DOI] [PubMed] [Google Scholar]
- 3.Corey EJ, Helal CJ. Angew Chem Int Ed. 1998;37:1986–2012. doi: 10.1002/(SICI)1521-3773(19980817)37:15<1986::AID-ANIE1986>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- 4.Ohkuma T, Koizumi M, Doucet H, Pham T, Kozawa M, Murata K, Katayama E, Yokozawa T, Ikariya T, Noyori R. J Am Chem Soc. 1998;120:13529–13530. [Google Scholar]
- 5.Noyori R, Ohkuma T. Angew Chem Int Ed Engl. 2001;40:40–73. [PubMed] [Google Scholar]
- 6.Gao Y, Klunder JM, Hanson RM, Masamune H, Ko SY, Sharpless KB. J Am Chem Soc. 1987;109:5765–5780. [Google Scholar]
- 7.Furstner A. Chem Rev. 1999;99:991–1045. doi: 10.1021/cr9703360. [DOI] [PubMed] [Google Scholar]
- 8.Ng S, Ho C, Schleicher K, Jamison T. Pure Appl Chem. 2008;80:929–939. doi: 10.1351/pac200880050929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Moslin R, Miller-Moslin K, Jamison T. Chem Commun. 2007:4441–4449. doi: 10.1039/b707737h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Montgomery J. Angew Chem Int Ed. 2004;43:1433–7851. [Google Scholar]
- 11.Oppolzer W, Radinov RN. Helv Chim Acta. 1992;75:170–173. [Google Scholar]
- 12.Oppolzer W, Radinov RN. J Am Chem Soc. 1993;115:1593–1594. [Google Scholar]
- 13.Oppolzer W, Radinov RN, Brabander JD. Tetrahedron Lett. 1995;36:2607–2610. [Google Scholar]
- 14.Oppolzer W, Radinov RN, El-Sayed E. J Org Chem. 2001;66:4766–4770. doi: 10.1021/jo000463n. [DOI] [PubMed] [Google Scholar]
- 15.Wipf P, Xu W. Tetrahedron Lett. 1994;35:5197–5200. [Google Scholar]
- 16.Wipf P, Xu W, Takahashi H, Jahn H, Coish PDG. Pure Appl Chem. 1997;69:639–644. [Google Scholar]
- 17.Wipf P, Kendall C. Chem Eur J. 2002;8:1778–1784. doi: 10.1002/1521-3765(20020415)8:8<1778::aid-chem1778>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- 18.Sprout CM, Richmond ML, Seto CT. J Org Chem. 2005;70:7408–7417. doi: 10.1021/jo051342w. [DOI] [PubMed] [Google Scholar]
- 19.Salvi L, Jeon SJ, Fisher EL, Carroll PJ, Walsh PJ. J Am Chem Soc. 2007;129:16119–16125. doi: 10.1021/ja0762285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yang Y, Zhu SF, Zhou CY, Zhou QL. J Am Chem Soc. 2008;130:14052–14053. doi: 10.1021/ja805296k. [DOI] [PubMed] [Google Scholar]
- 21.Chen YK, Lurain AE, Walsh PJ. J Am Chem Soc. 2002;124:12225–12231. doi: 10.1021/ja027271p. [DOI] [PubMed] [Google Scholar]
- 22.Miller KM, Huang WS, Jamison TF. J Am Chem Soc. 2003;125:3442–3443. doi: 10.1021/ja034366y. [DOI] [PubMed] [Google Scholar]
- 23.Chan J, Jamison TF. J Am Chem Soc. 2003;125:11514–11515. doi: 10.1021/ja0373925. [DOI] [PubMed] [Google Scholar]
- 24.Negishi E. Dalton Trans. 2005:827–848. doi: 10.1039/b417134a. [DOI] [PubMed] [Google Scholar]
- 25.Baxter RD, Montgomery J. J Am Chem Soc. 2008;130:9662–9663. doi: 10.1021/ja803774s. [DOI] [PubMed] [Google Scholar]
- 26.Oblinger E, Montgomery J. J Am Chem Soc. 1997;119:9065–9066. [Google Scholar]
- 27.Still WC, Gennari C. Tetrahedron Lett. 1983;24:4405–4408. [Google Scholar]
- 28.Furstner A, Shi N. J Am Chem Soc. 1996;118:2533–2534. [Google Scholar]
- 29.Jin H, Uenishi JI, Christ WJ, Kishi Y. J Am Chem Soc. 1986;108:5644–5646. [Google Scholar]
- 30.Takai K, Kimura K, Kuroda T, Hiyama T, Nozaki H. Tetrahedron Lett. 1983;24:5281–5284. [Google Scholar]
- 31.Takai K, Tagashira M, Kuroda T, Oshima K, Utimoto K, Nozaki H. J Am Chem Soc. 1986;108:6048–6050. doi: 10.1021/ja00279a068. [DOI] [PubMed] [Google Scholar]
- 32.Dutheuil G, Lei X, Pannecoucke X, Quirion JC. J Org Chem. 2005;70:1911–1914. doi: 10.1021/jo048093g. [DOI] [PubMed] [Google Scholar]
- 33.Spino C, Granger MC, Boisvert L, Beaulieu C. Tetrahedron Lett. 2002;43:4183–4185. [Google Scholar]
- 34.Marshall JA, Eidam P. Org Lett. 2004;6:445–448. doi: 10.1021/ol0363568. [DOI] [PubMed] [Google Scholar]
- 35.Mengel A, Reiser O. Chem Rev. 1999;99:1191–1224. doi: 10.1021/cr980379w. [DOI] [PubMed] [Google Scholar]
- 36.Spino C, Granger MC, Tremblay MC. Org Lett. 2002;4:4735–4737. doi: 10.1021/ol027236n. [DOI] [PubMed] [Google Scholar]
- 37.Denmark SE, Pan W. Org Lett. 2003;5:1119–1122. doi: 10.1021/ol0342002. [DOI] [PubMed] [Google Scholar]
- 38.Ng SS, Jamison TF. Tetrahedron. 2005;61:11405–11417. [Google Scholar]
- 39.Lurain AE, Walsh PJ. J Am Chem Soc. 2003;125:10677–10683. doi: 10.1021/ja035213d. [DOI] [PubMed] [Google Scholar]
- 40.Li H, Walsh PJ. J Am Chem Soc. 2004;126:6538–6539. doi: 10.1021/ja049206g. [DOI] [PubMed] [Google Scholar]
- 41.Li H, Walsh PJ. J Am Chem Soc. 2005;127:8355–8361. doi: 10.1021/ja0425740. [DOI] [PubMed] [Google Scholar]
- 42.Lurain AE, Carroll PJ, Walsh PJ. J Org Chem. 2005;70:1262–1268. doi: 10.1021/jo048345d. [DOI] [PubMed] [Google Scholar]
- 43.Lurain AE, Maestri A, Kelly AR, Carroll PJ, Walsh PJ. J Am Chem Soc. 2004;126:13608–13609. doi: 10.1021/ja046750g. [DOI] [PubMed] [Google Scholar]
- 44.Hussain MH, Walsh PJ. Acc Chem Res. 2008;41:883–893. doi: 10.1021/ar800006h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rowley Kelly A, Lurain AE, Walsh PJ. J Am Chem Soc. 2005;127:14668–14674. doi: 10.1021/ja051291k. [DOI] [PubMed] [Google Scholar]
- 46.Kim HY, Lurain AE, García-García P, Carroll PJ, Walsh PJ. J Am Chem Soc. 2005;127:13138–13139. doi: 10.1021/ja0539239. [DOI] [PubMed] [Google Scholar]
- 47.Kim HY, Salvi L, Carroll PJ, Walsh PJ. J Am Chem Soc. 2009;131:954–962. doi: 10.1021/ja806989n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chen YK, Walsh PJ. J Am Chem Soc. 2004;126:3702–3703. doi: 10.1021/ja0396145. [DOI] [PubMed] [Google Scholar]
- 49.This intermediate generated by our method has recently been employed by Negishi and coworkers: Huang Z, Negishi E. J Am Chem Soc. 2007;129:14788–14792. doi: 10.1021/ja0772039.
- 50.Chen, Y. K. Ph.D., University of Pennsylvania, 2004.
- 51.Zweifel G, Arzoumanian H. J Am Chem Soc. 1967;89:5086–5088. [Google Scholar]
- 52.Nelson DJ, Blue CD, Brown HC. J Am Chem Soc. 1982;104:4913–4917. [Google Scholar]
- 53.Kocienski P, Barber C. Pure Appl Chem. 1990;62:1933–1940. [Google Scholar]
- 54.Jarowicki K, Kocienski PJ. Synlett. 2005:167–169. [Google Scholar]
- 55.Brown HC, Imai T, Bhat NG. J Org Chem. 1986;51:5277–5282. [Google Scholar]
- 56.Brown HC, Soundararajan R. Tetrahedron Lett. 1994;35:6963–6966. [Google Scholar]
- 57.Brown HC, Imai T. Organometallics. 1984;3:1392–1395. [Google Scholar]
- 58.Campbell JB, Molander GA. J Organomet Chem. 1978;156:71–79. [Google Scholar]
- 59.Negishi E, Williams RM, Lew G, Yoshida T. J Organomet Chem. 1975;92:C4–C6. [Google Scholar]
- 60.Srebnik M. Tetrahedron Lett. 1991;32:2449–2452. [Google Scholar]
- 61.Nöth H, Wrachmeyer B. In: In Nuclear Magnetic Resonance Spectroscopy of Boron Compounds. Diehl P, Fluck E, Kosfeld R, editors. Springer-Verlag; Berlin: 1978. [Google Scholar]
- 62.Bolm C, Hildebrand JP, Muniz K, Hermanns N. Angew Chem, Int Ed. 2001;40:3285–3308. [Google Scholar]
- 63.Rudolph J, Rasmussen T, Bolm C, Norrby PO. Angew Chem Int Ed. 2003;42:3002–3005. doi: 10.1002/anie.200351150. [DOI] [PubMed] [Google Scholar]
- 64.Schmidt F, Stemmler RT, Rudolph J, Bolm C. Chem Soc Rev. 2006;35:454–470. doi: 10.1039/b600091f. [DOI] [PubMed] [Google Scholar]
- 65.Soderquist JA, Rane AM, Matos K, Ramos J. Tetrahedron Leters. 1995;36:6847–6850. [Google Scholar]
- 66.Molander GA, Ellis NM. J Org Chem. 2008;73:6841–6844. doi: 10.1021/jo801191v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Dimethylborane can be synthesized from a metathesis reaction with a borane adduct and trimethylborane. Trimethylborane is a pyrophoric gas at standard temperature and pressure and is also quite expensive (>$1100/10 g).
- 68.Yokokawa F, Asano T, Shioiri T. Tetrahedron. 2001;57:6311–6327. [Google Scholar]
- 69.Nakae KYY, Sawa T, Homma Y, Hamada M, Takeuchi T, Imoto M. J Antibiot. 2000;53:1130–1136. doi: 10.7164/antibiotics.53.1130. [DOI] [PubMed] [Google Scholar]
- 70.Gunasekera SP, Gunasekera M, Longley RE, Schulte GK. J Org Chem. 1990;55:4912–4915. [Google Scholar]
- 71.Gunasekera SP, Gunasekera M, Longley RE, Schulte GK. J Org Chem. 1991;56:1346–1346. [Google Scholar]
- 72.Corey EJ, Fuchs PL. Tetrahedron Lett. 1972;13:3769–3772. [Google Scholar]
- 73.Grandjean D, Pale P, Chuche J. Tetrahedron Lett. 1994;35:3529–3530. [Google Scholar]
- 74.Garner P, Park JM. Org Synth. 1992;70:18–24. [Google Scholar]
- 75.Dale JA, Dull DL, Mosher HS. J Org Chem. 1969;34:2543–2549. [Google Scholar]
- 76.Hoye TR, Jeffrey CS, Shao F. Nat Protoc. 2007;2:2451–2458. doi: 10.1038/nprot.2007.354. [DOI] [PubMed] [Google Scholar]
- 77.Chen X, Hortelano ER, Eliel EL, Frye SV. J Am Chem Soc. 1990;112:6130–6131. [Google Scholar]
- 78.Chen X, Hortelano ER, Eliel EL, Frye SV. J Am Chem Soc. 1992;114:1778–1784. [Google Scholar]
- 79.Jeon SJ, Fisher EL, Carroll PJ, Walsh PJ. J Am Chem Soc. 2006;128:9618–9619. doi: 10.1021/ja061973n. [DOI] [PubMed] [Google Scholar]
- 80.Li H, Carroll PJ, Walsh PJ. J Am Chem Soc. 2008;130:3521–3531. doi: 10.1021/ja077664u. [DOI] [PubMed] [Google Scholar]
- 81.Reetz MT, Huellmann MJ. J Chem Soc, Chem Commun. 1986;21:1600–1602. [Google Scholar]
- 82.Henry KJ, Grieco PA, Jagoe CT. Tetrahedron Lett. 1992;33:1817–1820. [Google Scholar]
- 83.Lodge EP, Heathcock CH. J Am Chem Soc. 1987;109:3353–3361. [Google Scholar]
- 84.Maier ME, Oost T. J Organomet Chem. 1995;505:95–107. [Google Scholar]
- 85.Stampf EJ, Odom JD. J Organomet Chem. 1977;131:171–178. [Google Scholar]
- 86.Brown HC, Racherla US. J Org Chem. 1986;51:427–432. [Google Scholar]
- 87.Nugent WA. J Chem Soc, Chem Commun. 1999:1369–1370. [Google Scholar]
- 88.Chen YK, Jeon SJ, Walsh PJ, Nugent WA. Org Synth. 2006;82:87–90. [Google Scholar]
- 89.Kim JG, Walsh PJ. Angew Chem Int Ed. 2006;45:4175–4178. doi: 10.1002/anie.200600741. [DOI] [PubMed] [Google Scholar]
- 90.Rudolph J, Hermanns N, Bolm C. J Org Chem. 2004;69:3997–4000. doi: 10.1021/jo0495079. [DOI] [PubMed] [Google Scholar]
- 91.Kohnen AL, Dunetz JR, Danheiser RL. Org Synth. 2007;84:88–101. [PMC free article] [PubMed] [Google Scholar]
- 92.Webb JA, Klijn JE, Hill PA, Bennett JL, Goroff NS. J Org Chem. 2004;69:660–664. doi: 10.1021/jo035584c. [DOI] [PubMed] [Google Scholar]
- 93.Murray RE. Synth Commun. 1980;10:345–349. [Google Scholar]
- 94.Grandjean D, Pale P, Chuche J. Tetrahedron Lett. 1994;35:3529–3530. [Google Scholar]
- 95.Corey EJ, Fuchs PL. Tetrahedron Lett. 1972;13:3769–3772. [Google Scholar]
- 96.Roush WR, Palkowitz AD, Ando K. J Am Chem Soc. 1990;112:6348–6359. [Google Scholar]
- 97.Enders D, von Berg S, Jandeleit B. Org Synth. 2002;78:177–182. [Google Scholar]
- 98.Marshall JA, Yanik MM, Adams ND, Ellis KC, Chobanain HR. Org Synth. 2005;81:157–164. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.