

Abstract
The Drosophila grapes (grp) gene, which encodes a homolog of the Schizosaccharomyces pombe Chk1 kinase, provides a cell-cycle checkpoint that delays mitosis in response to inhibition of DNA replication (Au: OK?) [1]. Grp is also required in the undisturbed early embryonic cycles: in its absence, mitotic abnormalities appear in cycle 12 and chromosomes fail to fully separate in subsequent cycles [2,3]. In other systems, Chk1 kinase phosphorylates and suppresses the activity of Cdc25 phosphatase: the resulting failure to remove inhibitory phosphate from cyclin-dependent kinase 1 (Cdk1) prevents entry into mitosis [4,5]. Because in Drosophila embryos Cdk1 lacks inhibitory phosphate during cycles 11-13 [6], it is not clear that known actions of Grp/Chk1 suffice in these cycles. We found that the loss of grp compromised cyclin A proteolysis and delayed mitotic disjunction of sister chromosomes. These defects occurred before previously reported grp phenotypes. We conclude that Grp activates cyclin A degradation, and functions to time the disjunction of chromosomes in the early embryo. As cyclin A destruction is required for sister chromosome separation [7], a failure in Grp-promoted cyclin destruction can also explain the mitotic phenotype. The mitotic failure described previously for cycle 12 grp embryos might be a more severe form of the phenotypes that we describe in earlier embryos and we suggest that the underlying defect is reduced degradation of cyclin A.
Results and discussion
When syncytial embryos are exposed to a protein synthesis inhibitor, cycloheximide, nuclear cycles arrest in interphase and cyclin A levels decline ([6]; Figure 1, wild type, + cyc) whereas cyclin B is stable under these conditions [6]. This suggests that a steady state of synthesis and degradation maintains the interphase levels of cyclin A during the syncytial divisions. We found that grp mutants are deficient in cyclin A turnover in the presence of cycloheximide (Figure 1, grp, + cyc). This defect was seen in embryos in cycles 4-8, earlier than other reported grp phenotypes and is therefore unlikely to be secondary to these phenotypes. We infer that Grp normally destabilizes cyclin A.
Figure 1.
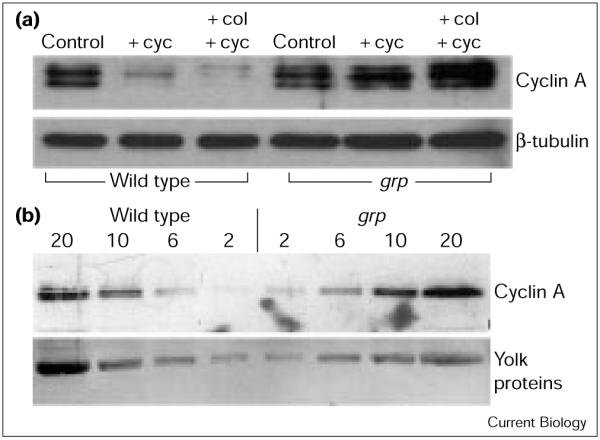
Cyclin A turnover and abundance in grp1 (grp) and wild-type embryos. (a) Defective cyclin A turnover in grp embryos. Syncytial-stage embryos (30-70 min old, cycles 4-8) were either untreated (control), or treated with cycloheximide (+ cyc), or colchicine and cycloheximide (+ col, + cyc), homogenized and extracts separated on denaturing gels and western blotted for expression of cyclin A and β-tubulin. The latter provided a loading control. (b) Higher levels of cyclin A in grp embryos. Extracts from wild-type and grp embryos (volumes loaded in each lane indicated in μl) (Au: OK?) were separated on denaturing gels and western blotted for cyclin A expression. Ponseau staining of yolk proteins indicated equal loading in corresponding lanes between wild-type and grp samples. In each case, the cyclin A signal was higher in the grp sample than in an equal volume of the wild-type sample (Au: OK?). We estimate that grp embryos have about 1.5-3-fold higher cyclin A levels than wild-type embryos. For example, the signal in the 2 μl lane for the grp sample is higher than in the corresponding lane for the wild type, but comparable to the signal in the 6 μl lane for the wild type. The signal in the 6 μl lane for grp is higher than in the corresponding lane for the wild type, but is comparable to the signal in the 10 μl lane for the wild type.
In wild-type embryos, cyclin A is unstable not only in interphase but also during mitotic arrest caused by micro-tubule destabilization. Thus, in embryos treated with colchicine, blocking protein synthesis with cycloheximide leads to a decline in cyclin A levels ([6]; Figure 1, wild type, + col, + cyc), whereas cyclin B is stable under these conditions [6]. We found that grp embryos are compromised for cyclin A proteolysis at such a colchicine-induced mitotic arrest (Figure 1, grp, + col, + cyc). As for interphase destruction of cyclin A, this defect was seen during early syncytial cycles, before the onset of other reported grp phenotypes. We conclude that Grp promotes cyclin A degradation in colchicine-arrested early embryos.
If normal levels of cyclin A are maintained by a steady state of synthesis and destruction, we would expect the levels of cyclin A to be high in grp embryos as a result of increased stability. Western blotting showed that grp embryos had slightly higher levels of cyclin A than wild-type embryos (1.5-3-fold; Figure 1b). Although this relatively small increase suggests that significant degradation of cyclin A still occurs in grp embryos, this degradation is not apparent at the arrests induced by cycloheximide or by colchicine.
As Grp is required for destruction of cyclin A in an arrested mitosis, we determined whether mitosis was disrupted in grp embryos. The mitosis-specific phosphorylation of histone H3 (PH3) apparently acts as an in vivo reporter for cyclin/Cdk activity and its disappearance at the end of mitosis requires cyclin destruction [8]. During syncytial cycles of wild-type embryos, PH3 staining is continuous along the length of the chromosome arms from metaphase until late anaphase, when loss of the epitope near kinetochores leads to graded staining ([8]; Figure 2b-d). In embryos from grp1 homozygous females or from grp1/Df females, the gradient of PH3 was seen on chromosomes in early anaphase (Figure 2a,g,h). Thus, PH3 loss is advanced with respect to chromosome segregation in grp embryos. This defect was seen at the earliest cycles scored (cycle 4 in grp1/grp1 and cycle 3 in grp1/Df), well before the onset of previously reported defects in grp embryos (cycle 11).
Figure 2.

Histone H3 dephosphorylation begins at earlier stages of chromosome segregation in grp mutants. Syncytial-stage Sevelen (wild type, wt) or grp embryos were fixed and stained for DNA (red) and with an antibody to PH3 (green). (a-e) Timing of mitotic loss of PH3 staining in a grp embryo compared with the progression of PH3 loss in wild-type mitoses; (b-e) are reproduced from [8] (Au: Is this the correct reference?). Loss of PH3 staining from the leading ends of chromosome arms in the wild type occurs as the chromosomes approach the spindle poles (d) whereas loss of PH3 in grp embryos is detected when chromosomes disjoin (a) (Au: OK?). Mitotic figures are from the M phase of cycle 10 (M10) (Au: OK? Otherwise, what does the M indicate?) and 11 (wild type) or M10 (grp). (f,g) PH3 loss in wild-type embryos compared with grp embryos during M9. (h) M12 in a grp embryo. The chromosomes of all mitotic figures show a gradient of PH3 though anaphase chromosome separation is less than that at which such PH3 gradients occur in a wild-type mitosis (see panel d). Severely mis-coordinated mitotic figures are indicated by arrowheads. (i) A portion of a later-stage (about cycle 12) grp embryo in interphase, stained for DNA: polyploid nuclei are discernible (arrowheads). These polyploid nuclei might arise if the mis-coordination of mitosis reaches a point at which the chromosomes decondense when anaphase separation of DNA masses is not yet adequate to define separate nuclei. (j) Mitotic figures (M10 or 11) in an embryo from a grp1/Df mother. As seen for the embryos from grp1/grp1 mothers (all other grp panels), the chromosomes in (j) show early loss of PH3 (arrowheads indicate particularly severe examples). Df is deficiency chromosome Df(2L)H20 (Bloomington Stock Center) that carries breakpoints at 36A08-09;36E-01-02 and therefore lacks the grp gene. The bar represents 10 μm.
The loss of PH3 during early anaphase in grp embryos could be due to the premature loss of PH3 (Figure 3b), a scenario opposite to that expected for a mutation that stabilizes cyclin A during mitosis. Alternatively, timely loss of PH3 staining but delayed chromosome separation would produce the same mis-coordination (Figure 3c). Analysis of grp embryos supports the latter hypothesis: we found an increase in the ratio of embryos with unsegregated chromosomes (prophase/metaphase) to those with segregated chromosomes (anaphase/telophase; Table 1). We conclude that Grp is required for timely chromosome segregation in syncytial mitoses.
Figure 3.
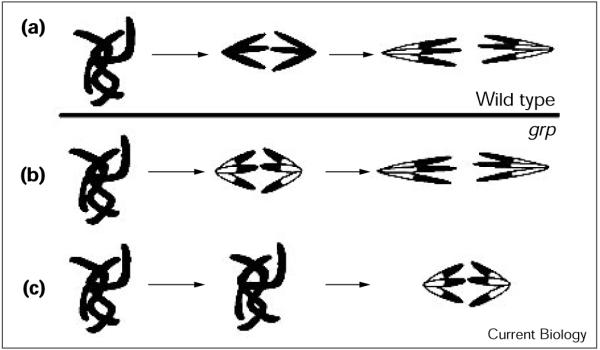
Chromosome segregation in (a) the wild type and (b,c) the two possible scenarios in grp embryos. (a) In wild-type mitoses, loss of PH3 (black) from chromosomes begins in late anaphase to reveal the DNA stain (white). (b,c) In grp embryos, either (b) PH3 loss is premature or (c) chromosome segregation is delayed but PH3 loss begins at about the normal time, resulting in histone H3 dephosphorylation at earlier stages of chromosome segregation.
Table 1. Mitotic indices in wild-type and grp embryos.
| Percentage of embryos in M phase (SD/n) | Percentage of embryos in M phase before chromosome segregation (SD) | Percentage of embryos in M phase after chromosome segregation (SD) | n | |
|---|---|---|---|---|
| Wild type | 58 (6/304) | 60 (3) | 40 (3) | 178 |
| grp1/grp1 | 64 (5/167) | 77 (3) | 23 (3) | 110 |
| grp1/Df | 60 (7/111) | 73 (5) | 27 (5) | 71 |
Syncytial embryos were fixed and stained for DNA, to determine the mitotic stage according to Edgar et al. [6]. Division cycle was determined as in Figure 2 legend. Mitotic indices are from cycles 1-11. Chromosome segregation data were from cycles 1-9 but the samples contained embryos mainly in cycles 6-9; before chromosome segregation includes prophase, prometaphase and metaphase; after segregation includes anaphase and telophase (Au: OK?). As described previously, chromosomes do not condense properly onto the metaphase plate in grp embryos [3], even before cycle 9 (data not shown); consequently, it is harder to distinguish late prophase from metaphase in grp embryos, and hence our division of mitotic stages into before and after chromosome segregation (Au: OK?). The data were from 3 and 5 different experiments, respectively, for grp and wild type.
The mitotic phenotype in grp embryos can be understood as follows. Ordinarily, the mitotic cyclins are degraded in a sequence during exit from mitosis: cyclin A is degraded before the metaphase-anaphase transition, cyclin B is degraded at the beginning of anaphase and cyclin B3 towards the end of anaphase [7]. The disappearance of PH3 can be prevented by stabilization of any of these mitotic cyclins [8]; thus, it appears that loss of PH3 marks the completion of this sequence. We suggest that grp embryos are specifically defective in the early initiation of cyclin A degradation but they do degrade cyclin A, perhaps in conjunction with the B cyclins. Eventual destruction of cyclin A would explain the ability of grp-deficient nuclei to exit mitosis and lose PH3 staining, events that can be inhibited by stable cyclin A [7,8]. Destruction of cyclin A in conjunction with the B cyclins would explain why the length of mitosis is not increased in grp embryos [1,9]. Because the expression of a stable form of cyclin A prevents chromosome disjunction [7,8], we suggest that the failure to degrade cyclin A early during mitosis in grp embryos delays chromosome disjunction. The delay in chromosome separation abbreviates anaphase and, when the abbreviation is severe, decondensation of chromosomes and entry into the next interphase occurs before the separating chromosomes reach the spindle poles (Figure 2h-j).
As Grp promotes cyclin A degradation, we might detect genetic interactions between grp and cyclin A. We first tested whether grp mutants are sensitive to levels of cyclin A. Homozygous grp flies are viable but female sterile. When we introduced a single copy of a heat-inducible cyclin A transgene, however, we failed to recover homozygous grp1 progeny (Table 2). Thus, even without induction, the presence of a heat-inducible cyclin A transgene (which by itself is viable as a heterozygote or homozygote) caused grp1 to behave as a recessive lethal. The observed synthetic lethality suggests that the Grp deficiency sensitizes the fly to low levels of cyclin A expression from the transgene. In contrast to this strong interaction with increased cyclin A level, a reduction in the dose of cyclin A failed to suppress the grp1 allele (data not shown and [9]). It is perhaps not surprising that reduction of cyclin A by half did not suppress a null allele of grp. Sibon et al. [9] found that reduction in the dose of cyclin A did suppress the lethality of mei41 mutations. The mei41 gene is a homolog of the gene ATM, which is mutated in the genetic disorder ataxia-telangiectasia (Au: OK?); mei41 is thought to act upstream of grp in the checkpoint pathway, and mutations in mei41 (Au: OK? Otherwise, please clarify which gene you are referring to) result in a phenotype like grp, but less severe. Importantly, the suppression of themei41 embryonic lethality by cyclin reduction occurred without restoring interphase length. This result shows that the mitotic defect is not an inevitable consequence of premature entry into mitosis as previously thought [1]. We suggest that the mitotic defect is an anaphase failure as a result of defective metaphase destruction of cyclin A (Au: OK?).
Table 2. Synthetic lethality between grp1 mutants and flies expressing a heat-inducible cyclin A transgene (hs-cyclin A) (Au: OK?).
| Progeny |
||
|---|---|---|
| Cross | grp1/grp1 (straight winged) | grp1/CyO (curley winged) |
| grp1/CyO X | 50 | 112 |
| X | ||
| grp1/CyO | 46 | 111 |
| grp1/CyO; hs-cycA/TM6 X | 0 | 69 |
| X | ||
| grp1/CyO; hs-cycA/TM6 | 0 | 100 |
The number of grp1 homozygotes and heterozygotes recovered from crosses of grp1 flies with or without the hs-cyclin A transgene. The results of two independent crosses are shown for each experiment. The progeny from grp1; hs-cyclin A/TM6 crosses included hs-cyclin A homozygotes and hs-cyclin A/TM6 heterozygotes. Data shown are from 25°C although similar results were obtained at room temperature or 18°C.
If Grp promotes the metaphase-anaphase transition, why is it dispensable at most stages of development? Before cell cycle 12, grp embryos exhibit a defective mitosis with delayed sister chromosome separation and, yet, mitosis is successful. From this we make two inferences: first, mitosis can tolerate a limited disruption in the timing of events and, second, as anaphase occurs in the absence of Grp, there must be a backup Grp-independent mechanism that promotes sister separation slightly later. The mitotic mis-coordination in grp embryos gets progressively more severe, and cyclin A levels progressively increase during the syncytial cycles [6]. This correlation, together with the ability of reduced cyclin A dose to suppress mei41 lethality, leads us to suggest that the consequence of a defect in the grp/mei41 pathway increases in severity as cyclin A increases, until anaphase fails at mitosis 12 and 13 (see, for example, Figure 2a).
The current model for Chk1 function involves the phosphorylation and inhibition of Cdc25, in part by binding of 14-3-3 protein to the phosphorylated Cdc25 and sequestration in the cytoplasm where it is ineffective in counteracting the nuclear kinases Wee1 and Mik1 [10,11]. Thus, inhibitory phosphorylation of Cdk1 prevents its activation and the cell arrests in G2. Although this action of Chk1 appears general, it is possible that Chk1 activity has other consequences. Indeed, there is no substantial accumulation of inhibitory phosphate on Cdk1 [6], and the Cdc25Stg protein is constitutively present and nuclear during interphase of syncytial cycles 11-13 when a grp-dependent mechanism regulates the entry into mitosis ([6]; T.T.S., unpublished data). The results presented here suggest that Grp may function to destabilize cyclin A. When a Grp-dependent cell-cycle checkpoint is induced by blocking S phase with aphidicolin in cleavage-stage Drosophila embryos, Cdc25Stg is destabilized [9]. Thus, whether it is direct or indirect, Grp promotes the destruction of two cell-cycle proteins, Cdc25Stg and cyclin A. We suggest that promotion of metaphase-anaphase transition represents a second function of the grp/mei41 pathway, distinct from the checkpoint arrest of entry into mitosis. Nevertheless, a common mechanism might be involved because blocking entry into mitosis and promoting exit from mitosis both involve inhibition of cyclin/Cdk1 activity.
In summary, Grp is required for normal cyclin A turnover in the early Drosophila embryo. We have also found that grp mutant embryos are delayed (Au: OK?) in the timing of the metaphase-anaphase transition. Stable versions of cyclin A block chromosome separation at the metaphase plate, at least in cellularized Drosophila embryos, suggesting that proteolysis of cyclin A is required for this process [7,8]. Thus, the proposal that Grp activates cyclin A proteolysis can explain the mitotic phenotype as a consequence of at least temporary persistence of cyclin A.
Supplementary Material
Acknowledgements
We thank Shelly Jones and Bill Sullivan for critical reading of the manuscript and Bloomington Stock Center for fly stocks. NIH grant GM37193 to P.H.O., a Herbert Boyer fellowship to T.T.S., and a Centennial fellowship from MRC, Canada to S.D.C. supported this work.
References
- 1.Sibon OC, Stevenson VA, Theurkauf WE. DNA-replication checkpoint control at the Drosophila midblastula transition. Nature. 1997;388:93–97. doi: 10.1038/40439. [DOI] [PubMed] [Google Scholar]
- 2.Fogarty P, Kalpin RF, Sullivan W. The Drosophila maternal-effect mutation grapes causes a metaphase arrest at nuclear cycle 13. Development. 1994;120:2131–2142. doi: 10.1242/dev.120.8.2131. [DOI] [PubMed] [Google Scholar]
- 3.Fogarty P, Campbell SD, Abu-Shumays R, Phalle BS, Yu KR, Uy GL, et al. The Drosophila grapes gene is related to checkpoint gene chk1/rad27 and is required for late syncytial division fidelity. Curr Biol. 1997;7:418–426. doi: 10.1016/s0960-9822(06)00189-8. [DOI] [PubMed] [Google Scholar]
- 4.Peng CY, Graves PR, Thoma RS, Wu Z, Shaw AS, Piwnica-Worms H. Mitotic and G2 checkpoint control: regulation of 14-3-3 protein binding by phosphorylation of Cdc25C on serine-216. Science. 1997;277:1501–1505. doi: 10.1126/science.277.5331.1501. [DOI] [PubMed] [Google Scholar]
- 5.Sanchez Y, Wong C, Thoma RS, Richman R, Wu Z, Piwnica-Worms H, et al. Conservation of the Chk1 checkpoint pathway in mammals: linkage of DNA damage to Cdk regulation through Cdc25. Science. 1997;277:1497–1501. doi: 10.1126/science.277.5331.1497. [DOI] [PubMed] [Google Scholar]
- 6.Edgar BA, Sprenger F, Duronio RJ, Leopold P, O’Farrell PH. Distinct molecular mechanism regulate cell cycle timing at successive stages of Drosophila embryogenesis. Genes Dev. 1994;8:440–452. doi: 10.1101/gad.8.4.440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sigrist S, Jacobs H, Stratmann R, Lehner CF. Exit from mitosis is regulated by Drosophila fizzy and the sequential destruction of cyclins A, B and B3. EMBO J. 1995;14:4827–4838. doi: 10.1002/j.1460-2075.1995.tb00164.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Su TT, Sprenger F, DiGregorio PJ, Campbell SD, O’Farrell PH. Exit from mitosis in Drosophila syncytial embryos requires proteolysis and cyclin degradation, and is associated with localized dephosphorylation. Genes Dev. 1998;12:1495–1503. doi: 10.1101/gad.12.10.1495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sibon OC, Laurencon A, Hawley R, Theurkauf WE. The Drosophila ATM homologue Mei-41 has an essential checkpoint function at the midblastula transition. Curr Biol. 1999;9:302–312. doi: 10.1016/s0960-9822(99)80138-9. [DOI] [PubMed] [Google Scholar]
- 10.Lopez-Girona A, Furnari B, Mondesert O, Russell P. Nuclear localization of Cdc25 is regulated by DNA damage and a 14-3-3 protein. Nature. 1999;397:172–175. doi: 10.1038/16488. [DOI] [PubMed] [Google Scholar]
- 11.Russell P. Checkpoints on the road to mitosis. Trends Biochem Sci. 1998;23:399–402. doi: 10.1016/s0968-0004(98)01291-2. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.