

Abstract
Background
Retroviral vectors are regularly used to transduce stem cells and their derivatives for experimental and therapeutic purposes. Because these vectors integrate semi-randomly into the cellular genome, analysis of integranated retroviral DNA/host cell DNA junctions (IHJ) facilitates clonality studies of engrafted cells, allowing their differentiation, survival and fate to be tracked. In the case of any adverse events, IHJ analysis can allow the identification of potentially oncogenic integration sites. At present, most measures to assess IHJ are complex, insensitive and may be subject to IHJ selection bias inherent to the technology used.
Methods
We have developed and validated a simple but effective technique for generating libraries of IHJ, which we term flanking-sequence exponential anchored–polymerase chain reaction (FLEA-PCR). Flanking-sequence random anchoring is used as an alternative to restriction enzyme digestion and cassette ligation to allow consistent detection of IHJ and decrease bias.
Results
Individual clones from plasmid libraries can be sequenced and assembled using custom-written software, and FLEA-PCR smears can be analyzed by capillary electrophoresis after digestion with restriction enzymes.
Discussion
This approach can readily analyze complex mixtures of IHJ, allowing localization of these sequences to their genomic sites. This approach should simplify analysis of retroviral integration.
Keywords: gene therapy, integration-site analysis, LAM-PCR, polymerase chain reaction, retrovirus
Introduction
One of the more desirable features of retroviral vectors is that they are capable of integrating efficiently into the genome of cells. Thus a retroviral vector and its expression cassette can be introduced permanently into a target cell and any of its progeny [1-5]. As integration is semi-random [6-9], each integration site generates a unique integrant–host–junction (IHJ) that can be used as a marker to study clonal evolution of engrafted cells or analyze potentially oncogenic insertional mutagenesis [10-12].
To date most analyzes of IHJ have been performed by asymmetric polymerase chain reaction (PCR) techniques. Approaches include priming on highly repetitive genomic sequences, arbitrary priming or, most often, restriction endonuclease digestion and cassette ligation. Ligation-mediated–PCR (LM-PCR) [13,14] and, more recently, Linear-amplification-mediated PCR–(LAM-PCR) [14] are the most commonly used cassette ligation techniques. Restriction endonuclease/cassette ligation techniques may fail to amplify certain IHJ if the selected restriction site is not present near a particular IHJ. The likelihood of a proximal restriction site can be improved by using multiple restriction enzymes and cassettes, albeit at the cost of increased complexity of the procedure. Moreover, cell populations with multiple integrants (complex samples) produce templates of different lengths, and the shorter fragments may amplify more efficiently, further biasing the final PCR product. Finally, these techniques require numerous manipulations, including two exponential PCR, and complex samples must be diluted and subjected to repeated analysis.
The ideal technique should be simple yet specific and capable of amplifying all IHJ as equally as possible. It should also be capable of allowing the analysis of complex samples in a single experiment. Definitive analysis requires sequencing of IHJ fragments; hence a technique amenable to high-throughput methods would be highly convenient. We have developed and validated a simple method to generate subcloned IHJ plasmid libraries based on anchoring linear PCR products with primers that are degenerate at their extreme 3′ end. This technique of flanking-sequence exponential anchored–PCR (FLEA-PCR) amplification results in a smear of randomly anchored IHJ. As integrants yield varying fragments of random length, there is no PCR bias caused by differing amplicon lengths. Also, no bias is accrued associated with the frequency of any particular restriction site. Highly complex samples can be studied from a single experiment. Finally, the technique is highly compatible with automation and high-throughput sequencing. This approach should facilitate IHJ analysis for assessing both retroviral safety and the biologic fate of transduced cells.
Methods
Cell lines
HeLa cells, PG13 cells and 293T cells were purchased from ATCC, Manassas, VA, USA. FLYRD18 cells were a generous gift of M. Collins, Department of Immunology, University College London [15]. Peripheral blood T cells were obtained from healthy donors and isolated by Ficoll centrifugation after obtaining informed consent compliant with the Baylor College of Medicine IRB (Houston, TX, USA). Epstein–Barr virus (EBV)-specific cytotoxic T lymphocytes (CTL) were generated as described previously [16]. Briefly, peripheral blood T cells were stimulated weekly with autologous EBV-immortalized B cells and interleukin-2 (IL-2) for at least three stimulations before transduction.
Retroviral vector production and transduction
The vectors used were based on SFG retroviral vector, which has wild-type Mouse Moloney Leukemia Virus (MoMLV) long terminal repeat (LTR). Transient supernatant was generated by triple-transfection of 293T cells with vector plasmid, MoMLV gagpol plasmid (PeqPamenv) and RD114 envelope glycoprotein expression plasmid RDF [17]. Adherent cells were transduced by culturing with retroviral supernatant and 10 μg/mL polybrene (Specialty Media, Phillipsburg, NY, USA). Non-adherent cells (Jurkat T-cells and EBV-CTL) were transduced by repeated incubation of retronectin-coated plates (Takara Bio, Shiga, Japan) with supernatant and finally adding the cells (EBV-CTL with 100 IU/mL IL-2) in retroviral supernatant to the retronectin-coated plates.
Flow cytometry and cell sorting
Flow cytometric analysis was performed with a FACSCalibur System (Becton Dickinson, San Jose, CA, USA). Cell sorting was performed using a MoFlo Sorter (Cytomation, Carpinteria, CA, USA).
Primers
LTR primers were designed to SFG retroviral vector LTR. This vector contains plain MoMLV LTR. The anchor sequence is arbitrary. We picked a sequence that had a Tm of 62°C with only 21 bp length that did not form homodimers or hetrodimers with LTR primers. As sub-cloning of fragments and working with PCR products puts the procedure at risk of contamination, we changed the anchor primers often. The sequences of anchor primers used are listed in Figure 2c. All primers were synthesized by Integrated DNA Technologies (Coralsville, IA, USA). Linear primers were triethyleneglycol (TEG) biotinylated as the extra TEG spacer resulted in improved attachment to streptavidin paramagnetic beads. Primer MP797 was 5′ 6-FAM conjugated.
Figure 2.
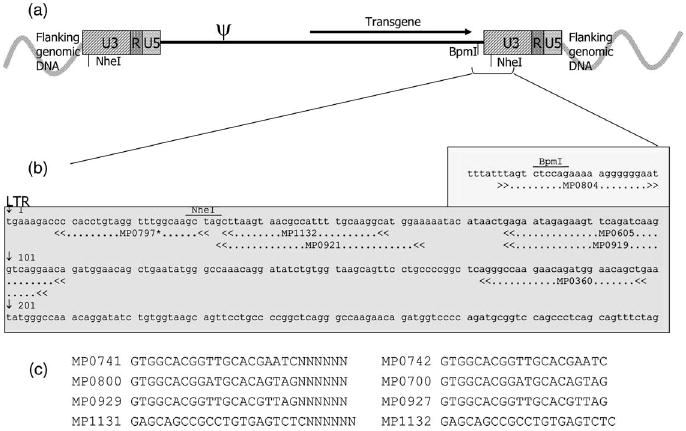
Location of FLEA-PCR primers. The plain MoMLV LTR of SFG is shown. (a) The vector integrant is bordered by 5′ and 3′ flanking genomic DNA. After integration, 5′ and 3′ LTR have identical sequences. Primers anneal close to the 5′ extremity of both LTR. (b) Close-up view showing sequences of the junction of the 3′ LTR. Note that MoMLV LTR contains a NheI restriction site very close to its 5′ end and that MoMLV internal sequences contain a BpmI site just before the 3′ LTR junction. (c) Anchoring primers and corresponding exponential PCR primers sequences. *The actual sequence of primer MP0797 is: ACCTACCTTGCCAAACCTACAGGTGGG to obliterate CviJI sites at positions 30 and 34 of the MoMLV LTR. Note: LTR primers are synthesized complementary to their annotation on the LTR sequences. They are illustrated as shown so their location on the LTR can be quickly visualized.
FLEA-PCR and LAM-PCR procedures
Genomic DNA was extracted from cells (Isoquick Kit; Orcha Research Inc., Bothwell, WA, USA). Linear PCR was performed with 47 μL Invitrogen HiFidelity supermix (contains dNTP, PCR buffer and a mixture of Taq polymerase and pfx Polymerase; Invitrogen Corp., Carlsbad, CA, USA), 100 nm TEG-bio linear primer (MP0360 or MP0605 or MP1132) and 1 μg genomic DNA. PCR was performed in a thermocycler (MJ Research, Waltham, MA, USA). Thermocycle parameters for linear PCR were: 95°C for 5 min (activate Taq) then 50 cycles of 95°C for 1 min, 60°C for 40 s and 72°C for 55 s. Next, small linear PCR fragments and unused primers were removed with a Microcon YM-100 column (Millipore, Billerica, MA, USA). Retentate was bound to streptavidin ferromagnetic beads on a shaker at room temperature overnight (Dynal, Oslo, Norway). Beads were washed with washing buffer (Kilobase binder kit; Dynal), then water, then 0.1 N NaOH and finally with water again. The shaker block was preheated to 85°C. Washed beads were resuspended in 20 μL 1 × DNA polymerase buffer (either Klenow or T7 DNA polymerase buffer, both from New England Biolabs, Beverly, MA, USA), 500 rMol dNTP, 5 μm anchoring primer and 5 μm internal blocking primer (for use with T7 DNA polymerase). Primers were annealed to the linear PCR product in this mixture on a shaking block by allowing it to cool slowly to 37°C. Once cooled, 10 IU of either Klenow or T7 DNA polymerase was added and the mixture incubated on the shaker for 1 h at 37°C. Next, the polymerase mixture was removed and the beads washed once in water. The beads were resuspended in 47 μL Invitrogen HiFidelity supermix with 500 μm anchor primer and 500 μm LTR primer. PCR was performed with the following parameters: 95°C for 5 min then 30–36 cycles of 95°C for 1 min, 60°C for 40 s and 72°C for 55 s. To obtain the most even library as little PCR was performed as possible; with high marking levels as few as 30 cycles were enough to visualize a smear. With lower marking levels up to 36 cycles were performed. If the marking was below 1%, a further nested PCR was performed using 1 μL of the first exponential PCR product as template, with Invitrogen HiFidelity supermix using thermocycle parameters as above. LAM-PCR was performed as described previously [14] using 5′ biotinylated linear primer MP0605 and Tsp509I as the restriction endonuclease. The ligation cassette was generated by annealing oligonucleotides 5′-GTACCTATGCAGCCCATAGTTGTTCCACTCCACCCATGGGC-3′ and 5′-AATTGCCCATGGGTGGAGTGGAACAACTATGGGCTGCATAGGTAC-3′. Oligonucleotides 5′-GTACC TATGCAGCCCATAGTTG-3′ and 5′-TTCCACTCCACCCATGGGC-3′ were paired with LTR primers MP1132 and MP079 for exponential PCR.
Sequencing and capillary electropheresis
Sequencing was performed using a ABI Prism 3100 genetic analyzer instrument with a big-dye terminator sequencing kit according to the manufacturer’s instructions (both from Applied Biosystems, Foster City, CA, USA). For capillary electrophoresis, buffer and unused primers were removed from the PCR product using a Microcon YM100 column. This purified PCR product was digested with 5 U CviJI (ChimerX, Milwaukee, WI, USA) by incubating in the supplied buffer and 10% DMSO at 37°C for 1 h in a total volume of 30 μL; 1 μL of this digest and 1 μL ROX 350 GeneScan size standard (Applied Biosystems) were added to 10 μL formamide (Applied Biosystems), incubated at 95°C for 10 min and then run on an ABI Prism 3100 genetic analyzer with a 36-cm capillary using POP4 polymer (Applied Biosystems).
High-resolution gels
Digested or undigested PCR product, 12 μL, was run on Elchrom EL600 precast gels (Elchrom Scientific, Cham, Switzerland) on a SEA 2000 electrophoresis apparatus (Elchrom Scientific) in 0.75 × TAE buffer, kept at a constant temperature of 47°C and constant voltage of 120 V. Gels were stained with SYBR Gold (Molecular Probes, Eugene, OR, USA) and visualized with a UV transilluminator.
Computation
The local alignment algorithm used was that of Pearson and Lipman [18,19]. Alignment with the human genome was made using the ensemble genome browser www.ensembl.org (Sanger Institute and European Bioinformatics Institute, Hinxton, Cambridge, UK. Site last accessed 1 June 2008) with NCBI 34 human genome assembly. Melting temperature (Tm) is the temperature at which an oligonucleotide duplex is 50% in single-stranded form and 50% in double-stranded form. The Tm was measured directly for any oligonucleotide or estimated using the following algorithm using known oligonucleotide/template nearest neighbor thermodynamic parameters [20]:
where ΔH° is enthalpy, ΔS° is entropy, R is the ideal gas constant (1.987 cal/K/mole) and Ct is the molar concentration of the oligonucleotide.
Results
Strategy of FLEA-PCR
The FLEA-PCR strategy is outlined in Figure 1. Genomic DNA is subjected to linear PCR with a biotinylated primer that anneals close to the 5′ end of the U3 region of the LTR. As a result of this PCR step, in which only one primer is used, single-stranded DNA fragments of random length are generated. The 5′ portion of these fragments contains a sequence complementary to the LTR, while the rest contains either flanking genomic sequences, termed external if the 5′ LTR is primed, or internal retroviral sequences, termed internal if the 3′ LTR is primed. These single-stranded fragments are separated from genomic DNA using streptavidin-coated paramagnetic beads. Next, primers containing a known 5′ sequence with six degenerate bases at the extreme 3′ end are annealed to the single-stranded linear PCR products. The complimentary strand is then generated using either DNA polymerase I Klenow fragment or T7 DNA polymerase. The resulting complimentary DNA strand now has known sequences at both the 5′ and 3′ end and hence can be used as template for exponential PCR. The positions of the primers used, with respect to the sequence of the LTR, in the PCR steps are shown in Figure 2.
Figure 1.
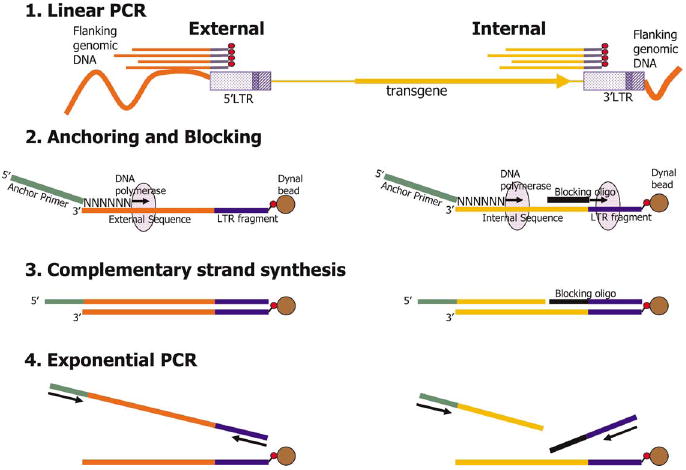
Schematic of the FLEA-PCR procedure. Linear PCR with biotinylated primer generates single-stranded biotinylated DNA fragments starting from the LTR 5′ end. If the 5′ LTR is primed, ‘external’ fragments containing flanking genomic DNA are generated. Alternatively, if the 3′ LTR is primed, ‘internal’ fragments containing retroviral internal sequences are generated. These biotinylated PCR products are separated from genomic DNA by attachment to streptavidin paramagnetic beads. Anchoring: an anchor primer mixture with a known 5′ sequence and six degenerate bases at the extreme 3′ end anneals only to the 3′ end of the linear PCR product. Complementary strand synthesis: a complementary strand is synthesized from the anchoring primer to the LTR sequences. Blocking oligonucleotide annealed to internal sequences will stall T7 DNA polymerase; hence internal complementary strands will lack LTR sequences. Exponential PCR: this newly synthesized complementary strand now has known sequences at either end and can be used as a template for exponential PCR.
FLEA-PCR generates smears from genomic DNA of transduced cells
We established the type of PCR product obtained by FLEA-PCR using genomic DNA from transduced and non-transduced cells as a template. As was expected, genomic DNA from a transduced cell produces a smear ranging from 120 nucleotides to just over 500 nucleotides. No smear is generated using genomic DNA from non-transduced cells as a template. The 120-nucleotide lower range probably represents the cut-off of the microcon column used to exclude unused primer and short linear products. Smears from simple samples (cell lines with single or few integrants) appeared similar to those from complex samples (cell lines with many different integrants or a polyclonal line; data not shown). However, once digested with a restriction endonuclease recognizing four bases, they resolved into patterns of bands related to the complexity of the sample. Figure 3 shows a smear from FLEA-PCR of a retroviral producer with multiple integrants of vector SFG.14g2a-ζ [21], digested with HaeI. Presumably each retroviral integrant should resolve to a band. We predicted, therefore, that this smear contained randomly anchored randomly amplified IHJ sequences.
Figure 3.
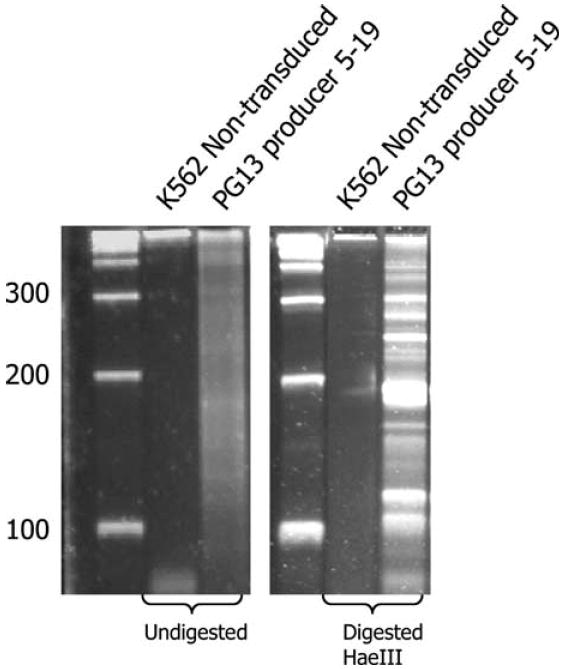
One microgram of genomic DNA from either non-transduced K562 cells or a PG13 producer line MP0451, clone 5–19, was subjected to FLEA-PCR. The PG13 producer had been derived by multiple rounds of transduction with Eco pseudotyped transient supernatant. A smear ranging from 100 bp to 400 bp was generated only from the PG13 producer line. This smear was digested with HaeIII (a four-cutter recognizing CCGG). Digested and undigested smears were run on high-resolution Elchrom gels at 50°C and 120 V for 40 min. The smear resolves into multiple bands upon digestion.
FLEA-PCR allows reliable characterization of IHJ from simple samples
Next we sought to demonstrate that these PCR-generated smears did indeed contain informative IHJ sequences. HeLa cells were transduced with retroviral vector SFG.eGFP (a plain MoMLV-based vector) at a low multiplicity of infection (MOI) (7% transduction) so that the transduced cells would be most likely have a single integrant. Twenty eGFP-positive cells were single-cell sorted and expanded. The products of FLEA-PCR from genomic DNA from each of these HeLa clones resulted in a smear. To allow characterization, the smears were subcloned into pTOPO to generate plasmid libraries. Twelve plasmids from each library were then directly sequenced. As the LTR primer MP1132 anneals to +33 from the 5′ end of the LTR (Figure 2), the first 33 bp of LTR amplified by PCR were unprimed. All characterized plasmids contained this sequence. This LTR sequence was preceded either by an unknown flanking sequence precisely at the junction tgaaa or, in approximately half the clones (52%), the internal retroviral sequence that precedes the 3′ LTR. Alignment of different IHJ sequences from the same library demonstrated identical flanking sequences of varying length (Figure 4b). Hence, as predicted, FLEA-PCR generated a library of randomly anchored IHJ. The lengths of flanking DNA obtained ranged from 0 to 469 with mean of 134 bp. All external sequences could be matched to genomic sequences with an alignment of >99% (Figure 4a). In order to establish definitively that the site of integrations was correct, we designed primers based on predicted sequences that annealed approximately 200 bp downstream of the 3′ LTR from integrants in six randomly selected HeLa cell clones. Using an LTR forward primer and these 3′ flanking primers, we obtained bands of the predicted length (Figure 4c) in all six clones. Direct sequencing of these bands revealed a 3′ LTR genomic DNA junction that was predicted from the site of integration established previously and the 4 bp duplication characteristic of MoMLV retroviral integration (Figure 4d). From simple samples at least, this technique produced fragments of flanking genomic DNA containing an LTR junction with high precision and no PCR artifact.
Figure 4.

Results from a study of HeLa cell clones. The FLEA-PCR-amplified smear from genomic DNA from 20 HeLa clones was subcloned to create a plasmid library of IHJ. Twelve plasmids from each library were sequenced. (a) Summary of sequencing results showing the length of the longest sequence of flanking genomic DNA from each library, the BLAT score and percentage identity with genomic sequences, 10 bp of flanking DNA and unprimed LTR sequences and the genomic locus elucidated from the human genome database. *A precise locus could not be elucidated in the two clones marked because integration occurred in a repeated region of genomic DNA. (b) The results of sequencing plasmids from the same library from two HeLa clones. Plasmids all contain the same IHJ but contain different lengths of flanking genomic sequence, presumably caused by the random length of linear PCR products. The highlighted text details the genomic locus and percentage identity of the longest clone against genomic sequences. The top sequence (in bold) is the genomic sequence, the next sequence lines down are FLEA-PCR clones, aligned against the genomic sequence. (c) Reverse primers were designed to anneal to the 3′ flanking sequence from six HeLa clones predicted by the human genome sequence database. These reverse primers and a forward primer some 200 bp in from the 3′ extremity of the LTR were used to amplify the 3′ IHJ. The DNA fragments amplified where all of the expected length. NT, non-transduced HeLa genomic DNA. (d) These fragments were then sequenced. The immediate junctional sequences are shown along with the 5′ junction. Sequences were all as expected based on human genome data. The characteristic 4-bp repeat of MoMLV integration was also present.
FLEA-PCR allows study of IHJ from samples containing as little as 0.1% transduced cells
Next, we sought to study the sensitivity of the method. We made serial dilutions of randomly selected transduced HeLa cell clones with non-transduced cells. Genomic DNA from this mixture was studied with FLEA-PCR. Using linear PCR primer MP0360, which anneals further 3′ on the LTR (Figure 2), we could perform nested exponential PCR using first primer MP0605 and secondly primer MP1132. A smear could be visualized by ethidium bromide staining in samples containing 3% transduced cells (Figure 5a). With a second step of exponential PCR, it was possible to generate smears from samples containing 0.1% transduced cells (Figure 5b). To ensure that the procedure remained accurate and that no artifact was generated, we sequenced the external IHJ generated from 0.1% transduced samples. Although they were shorter as a consequence of using a linear primer set deeper in the LTR, they were otherwise identical to sequences obtained from the original clone at 100% marking. Hence FLEA-PCR allowed study of IHJ from samples where as little as 0.1% of cells were transduced.
Figure 5.
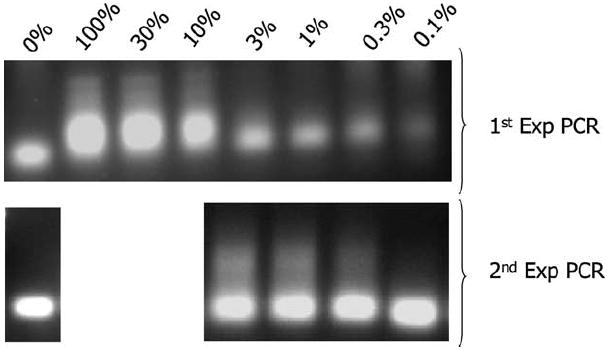
Sensitivity of FLEA-PCR. Smears derived from FLEA-PCR of transduced HeLa cell clone MP647 genomic DNA diluted in non-transduced genomic DNA. (a) Smears after 33 cycles of exponential PCR with primers MP0360 and MP1134. (b) Nested PCR amplification of this product with primers MP0605 and MP1134.
FLEA-PCR allows generation of a representative plasmid library of subcloned IHJ
Having shown the approach could reproducibly and robustly detect integration sites in single clones and moderately complex samples, we determined its value in populations of cells with large numbers of multiple integrants (highly complex samples). Cytotoxic lymphocytes (EBV-CTL) were transduced with clinical-grade vector RV04002B coding for an artificial T-cell receptor [21]. (This is a typical therapeutic product we administer as adoptive immunotherapy to patients.) Transduction efficiency was approximately 10% (Figure 6b). We transduced 5 × 106 T cells and expanded them for an additional week by stimulating with autologous LCL and IL-2. Genomic DNA from these cells was subjected to FLEA-PCR. The subsequent smear (Figure 6b) obtained was subcloned into pTOPO to generate a plasmid library. Sixty plasmids from this library were studied. Initial analysis was performed by restriction endonuclease digestion. MoMLV LTR conveniently contains a NheI site at its 5′ end, while retroviral sequences just before the 3′ LTR contain a BpmI site (Figure 2). All characterized plasmids contained a NheI site (indicating the presence of an LTR sequence; Figure 6c) and 14 out of the 48 contained a BpmI site (indicating the additional presence of an LTR internal sequence; Figure 6d). Forty plasmids without a BpmI site and eight with were sequenced. A summary of sequencing results is presented in Figure 6e. All sequences contained the 33-bp of unprimed LTR. All external sequences (i.e. those without a BpmI site) could be aligned with a human genome sequence and a precise localization of integration was possible for 82% of sequences. The remainder could be aligned to multiple different loci in the genome, suggesting that integration had occurred in a repeated genomic element. An example of alignment of an external sequence is given in Figure 6f. Plasmids containing BpmI sites were all internal retroviral sequences in this experiment, and these were randomly anchored in a similar fashion to external sequences. An example of an internal sequence aligned to the parental vector is shown in Figure 6g. Hence FLEA-PCR could generate representative plasmid libraries of subcloned IHJ from complex samples and external sequences could be selected by their lack of a BpmI site. These sequences could all be validated by detection of a +33-bp unprimed LTR sequence, and the flanking sequences could be used to track clones and determine their integration site by alignment to the human genome sequence database.
Figure 6.
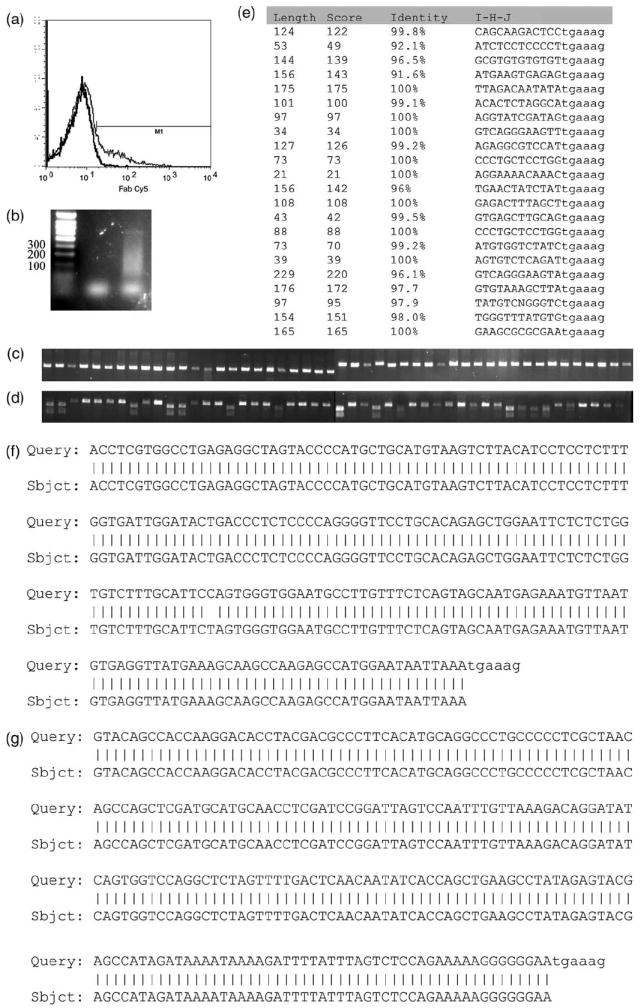
Results from study of a plasmid library of a subcloned FLEA-PCR smear derived from a polyclonal primary T-cell line. (a) Transduction of T cells stained with anti-Fab antibody showing limited transduction efficiency (12%). Histograms of transduced and non-transduced cells are overlaid (b). FLEA-PCR smear from a non-transduced K562 genomic DNA negative control (left lane) and transduced T-cell line (right lane). (c) Forty-eight plasmids from a subcloned library digested with NheI. DNA fragments were cloned into pTOPO, which does not contain a NheI site. All the plasmids generated subsequently contained a NheI, site strongly suggesting that subcloned fragments are derived from IHJ, as the extreme 5′ end of the MoMLV LTR contains a NheI site (which was unprimed in this experiment). (d) Forty-eight plasmids from a subcloned library digested with BpmI. pTOPO contains a single BpmI site. Plasmids cut twice hence contain inserts with a BpmI site most probably derived from the 3′ retroviral LTR. (e) Sequencing of plasmids containing inserts without a BpmI site. The length of flanking genomic sequence is detailed along with the BLAT score and the percentage identity with a human genomic database sequence. A fragment of the IHJ is also shown with the LTR in lower case. (f) An example of alignment between a FLEA-PCR-derived flanking sequence (Query) with a human genome database sequence (Sbjct). (g) An example of alignment between a FLEA-PCR-derived internal sequence (Query) with a retroviral map (Sbjct). In both (f) and (g) the beginning of the LTR (‘tgaaag’) is shown in lower case. (h) FLEA-PCR compared with LAM-PCR on a mix of eight HeLa clones. The lanes are: ladder, FLEA-PCR using non-transduced HeLa cell genomic DNA as template; LAM-PCR with non-transduced HeLa genomic DNA; FLEA-PCR smear of mixed clones (single round of PCR); LAM-PCR (after nested PCR). Sequencing results of FLEA-PCR are summarized to the left of the gel, with number of times an IHJ was sequenced followed by the HeLa cell clone it belongs to and the junctional sequence with the LTR in bold lower case. To the right of the gel, LAM-PCR sequences are summarized with the Tsp501I site in bold upper case, genomic DNA sequences, LTR sequences in bold lower case and finally the HeLa clone this junction belongs to.
To see how completely a complex sample could be studied by FLEA-PCR, genomic DNA from eight randomly selected HeLa clones, characterized above, were mixed and used as a template for FLEA-PCR. To ensure our technique was not inferior to existing methods, we also used this mix as a template for LAM-PCR with Tsp509I as the restriction endonuclease (Figure 6f). The FLEA-PCR smear was TOPO-TA cloned and 64 external clones (as determined by restriction digestion with BpMI) were sequenced. Nested LAM-PCR product bands were cut out of a high-resolution agarose gel and individually subcloned into pTopo and sequenced. All eight integrants were identified by FLEA-PCR. The subcloned fragment length provided from 69 to 479 bp of flanking genomic DNA, with a mean length of 182 bp. Representation of HeLa clones in the library ranged from 14 sequences for clone 639 to three sequences for clone 653, with a mean of eight sequences. Five of eight HeLa clones were identified by LAM-PCR. Notably, two of the non-identified clones (637 and 660) had integration sites with Tsp50I sites further than 500 bp from the integration sites.
Blocking internal band synthesis results in a highly informative plasmid library allowing high-throughput sequencing
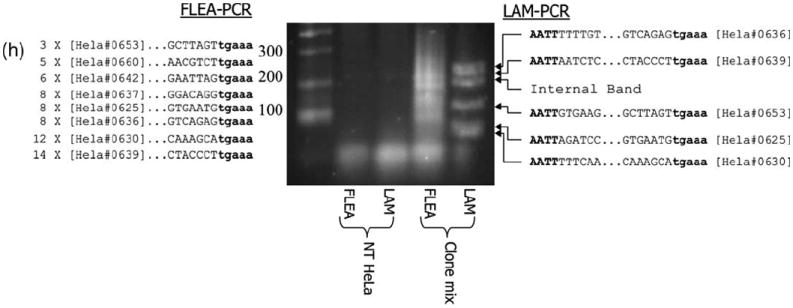
Although analysis of a single sample by FLEA-PCR can yield considerable information, practical applications will often require analysis of libraries from multiple samples obtained at different time-points. Internal bands, however, do not provide any information and, as the internal junction is represented in a 1:1 molar ratio with all IHJ, it appears at this ratio in a typical library. Hence half of sequenced plasmids are uninformative and their sequencing is wasteful. Screening by restriction digest to identify a BpMI site partially solves this problem but is difficult to automate. As the retroviral internal sequences just before the 3′ LTR are known and are similar in nearly all oncoretroviral vectors, we looked to see if it was possible to block their synthesis during the phase of complementary strand generation. An oligonucleotide annealing to internal linear PCR products was designed (MP0804). T7 DNA polymerase was substituted for Klenow as it has neither strand displacement nor 5′ to 3′ endonuclease properties. A HeLa cell clone was restudied, this time with and without a blocking oligonucleotide added to the polymerase mixture during complementary strand synthesis. Eighty clones from the subsequent plasmid library were digested with BpMI for both conditions. The numbers of internal sequences dropped from 50% to < 5% (P < 0.01; Figure 7). The resulting reduction of internal sequences allowed efficient analysis of a plasmid library without prior analysis by restriction digest.
Figure 7.
Effects of blocking oligonucleotides on FLEA-PCR-amplified libraries. A genomic DNA HeLa cell clone was subjected to FLEA-PCR using T7 DNA polymerase with and without addition of internal sequences blocking oligonucleotide. A plasmid library for each condition was generated by subcloning the subsequent smear. Plasmids from each library were digested with BpmI. Digested clones from the library generated without blocking oligonucleotides (a) and with blocking oligonucleotides (b) are shown.
Analysis of 6FAM-labeled FLEA-PCR smears digested by CviJI allows their analysis by capillary electrophoresis
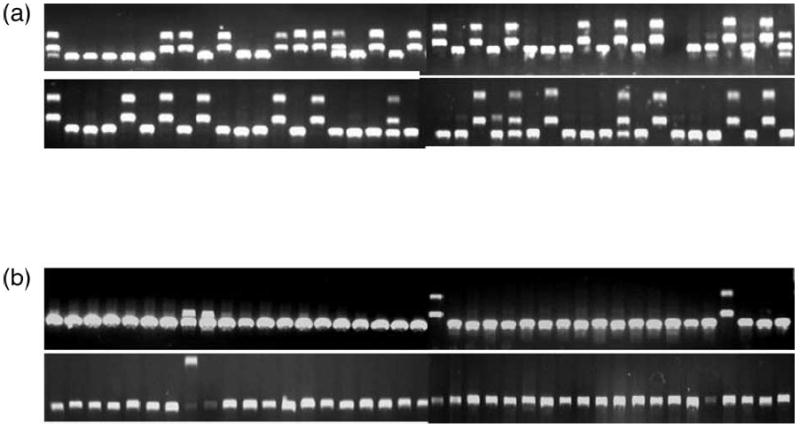
Conveniently, LAM-PCR allows clonality to be assessed by studying the sizes of each exponential PCR product. The FLEA-PCR smear provides no such direct information. However, digestion of this smear with a restriction endonuclease resolves it into distinct bands. The choice of restriction endonuclease in LAM-PCR is limited because digestion is performed at an early stage, and hence any enzyme that digests the LTR as far as the linear primer cannot be used. The FLEA-PCR smear can, however, be digested with almost any endonuclease, including the RCGY-recognizing restriction enzyme CviJI [22]. This endonuclease cuts on average every 64 nucleotides, increasing the probability of resolving every IHJ. By using primers that anneal as far to the 5′ edge of the LTR as possible (linear primer MP0921 and LTR primer MP0797; Figure 2a), the ‘bandwidth’ of the FLEA-PCR smear is extended as far as possible. The primer MP0797 5′ end was 6FAM conjugated. Although each IHJ may generate multiple fragments after digestion, if they contain several CviJI sites only the most 3′ fragment containing a piece of the LTR and primer MP0797 is 6FAM conjugated. Hence visualization of 6FAM-conjugated fragments on a high-resolution gel, or by capillary electrophoresis and optical detection of the digest, can be used to enumerate the IHJ present in the FLEA-PCR smear. We sorted Jurkat T cells transduced at a low MOI with SFG.eGFP at 2, 4, 8, 16 and 32 GFP-positive T cells/well. These populations were expanded, genomic DNA extracted and FLEA-PCR amplified with primers MP0921 and MP0797. The subsequent smears were digested with CviJI and analyzed by capillary electrophoresis. A clear proportional increase in the number of distinct spikes between 20 bp and 120 bp could be seen in these progressively more complex samples (Figure 8). Hence FLEA-PCR smears can be studied by using capillary electrophoresis after appropriate restriction enzyme digest.
Figure 8.
Capillary electrophoresis analysis of a CviJI digest of a 6-FAM-labeled smear. SFG.eGFP-transduced Jurkat T cells were FACS sorted and 2, 4, 8, 16 and 32 transduced cells placed in each well and expanded. Genomic DNA from these populations was subjected to FLEA-PCR with a 6-FAM-conjugated LTR primer. The subsequent smear was digested with CviJI and run on a capillary device (shown in blue) with a GeneScan ROX 350-bp size marked (shown in red).
Discussion
Analysis of IHJ can facilitate safety studies of retroviral gene therapy and permit clonality studies of engrafted cells, allowing their differentiation, survival and fate to be tracked [23-25]. As interest increases in the use of genetically modified cells for therapeutic purposes, the ability to follow the transduced cells and their progeny in vivo will become ever more critical, to both improve the effectiveness of these therapeutic agents and assure their safety [11]. We have developed and validated a simple but effective technique for generating libraries of IHJ. Random anchoring is used as an alternative to restriction enzyme digestion and cassette ligation to allow consistent detection of IHJ and decrease bias. Libraries can be randomly sequenced and assembled using custom-written software and FLEA-PCR smears can be analyzed by capillary electrophoresis after restriction enzyme digest.
To characterize IHJ, a variety of strategies must be used to facilitate PCR amplification when only one primer can anneal to a known sequence (the LTR). The first of these random priming PCR usually depends on priming highly repetitive sequences (e.g. ALU sequences) as well as the LTR [26,27]. By chance, if a junction is near an ALU repeat, it will be amplified. The main limitation of this technique is the amplification of repeat–repeat sequences, which greatly outnumber the repeat–LTR sequences. Also, an IHJ may not contain a nearby repeat and therefore may not be detected. Arbitrary primed PCR [28] is a variation of this technique where low initial annealing temperatures allow primers to anneal anywhere, while higher subsequent annealing temperatures then restrict the PCR to IHJ. Further manipulations are usually required to remove random fragments of genomic sequences amplified by the degenerate primers only [26,29,30]. A second basic approach, restriction digest and ligation-based techniques, has evolved from inverse PCR amplification [31], which requires circularization of digested fragments through complex cassettes designed to overcome cassette–cassette amplification (vectorette [32-34] and splinkterette [35,36] PCR), to the most utilized technique, LAM-PCR [14]. These methods rely on the presence of a particular restriction endonuclease close to the IHJ. For instance, if using Tsp509I, 104 out of 562 IHJ we have identified lack a Tsp509I site within 500 bp and may not be detected by LAM-PCR. Furthermore, every IHJ has a distinct fragment length even when serving as templates for the exponential PCR phase. A shorter fragment may ‘swamp’ longer fragments, creating additional bias. In samples with more than one IHJ, the internal band is present in molar excess and, if short, can completely dominate a reaction
In FLEA-PCR, linear PCR fragments containing IHJ are separated from genomic DNA. Instead of complementary strand synthesis, digestion and cassette ligation, these linear fragments are randomly anchored and can be used immediately as a template for exponential PCR. This greatly simplifies the procedure and also eliminates bias consequent to restriction site occurrence and different amplicon length. Although the subsequent smear does not reveal any immediate information, a representative plasmid library can easily be generated by subcloning and definitive analysis performed by sequencing. These libraries can be studied using high-throughput methods and may be amenable to analysis by massively parallel signature sequencing [37]. We have shown that such a library contains IHJ sequences. We have confirmed the validity of these IHJ sequences by amplifying the opposite (3′) IHJ by designing primers based on predicted sequences and also by showing the 4-bp duplication characteristic of MoMLV integration. If a further nested PCR step is added, FLEA-PCR is highly sensitive, allowing characterization of rare integrants in a background of non-transduced cells. Representative libraries of IHJ from polyclonal populations or from cells with multiple integrants can be generated in a single experiment. Almost no PCR artifact is generated and sequences can easily be validated by the presence of unprimed LTR sequences. Uninformative internal sequences can be blocked at the complementary strand synthesis phase. This technique should facilitate clonal evolution and lineage studies and may also be useful for the long-term safety follow-up of patients who have received retrovirally transduced cells.
Acknowledgments
This work was supported in part by a British Society for Haematology Society Fellowship to M. A. Pule, a Doris Duke Distinguished Clinical Scientist Award to H. E. Heslop, NCI PO1 CA94237, a Specialized Center of Research grant from the Leukemia and Lymphoma Society, and the GCRC at Baylor College of Medicine (RR00188).
Footnotes
Declaration of interest: The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
References
- 1.Schmidt M, Zickler P, Hoffmann G, et al. Polyclonal long-term repopulating stem cell clones in a primate model. Blood. 2002;100:2737–43. doi: 10.1182/blood-2002-02-0407. [DOI] [PubMed] [Google Scholar]
- 2.Brenner MK, Rill DR, Moen RC, et al. Gene-marking to trace origin of relapse after autologous bone-marrow transplantation. Lancet. 1993;341:85–6. doi: 10.1016/0140-6736(93)92560-g. [DOI] [PubMed] [Google Scholar]
- 3.Heslop HE, Rooney CM. Adoptive cellular immunotherapy for EBV lymphoproliferative disease. Immunol Rev. 1997;157:217–22. doi: 10.1111/j.1600-065x.1997.tb00984.x. [DOI] [PubMed] [Google Scholar]
- 4.Kohn DB, Hershfield MS, Carbonaro D, et al. T lymphocytes with a normal ADA gene accumulate after transplantation of transduced autologous umbilical cord blood CD34+ cells in ADA-deficient SCID neonates. Nat Med. 1998;4:775–80. doi: 10.1038/nm0798-775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hacein-Bey-Abina S, Le Deist F, Carlier F, et al. Sustained correction of X-linked severe combined immunodeficiency by ex vivo gene therapy. New Engl J Med. 2002;346:1185–93. doi: 10.1056/NEJMoa012616. [DOI] [PubMed] [Google Scholar]
- 6.Schroder AR, Shinn P, Chen H, et al. HIV-1 integration in the human genome favors active genes and local hotspots. Cell. 2002;110:521–9. doi: 10.1016/s0092-8674(02)00864-4. [DOI] [PubMed] [Google Scholar]
- 7.Wu X, Li Y, Crise B, Burgess SM. Transcription start regions in the human genome are favored targets for MLV integration. Science. 2003;300:1749–51. doi: 10.1126/science.1083413. [DOI] [PubMed] [Google Scholar]
- 8.Laufs S, Gentner B, Nagy KZ, et al. Retroviral vector integration occurs in preferred genomic targets of human bone marrow-repopulating cells. Blood. 2003;101:2191–8. doi: 10.1182/blood-2002-02-0627. [DOI] [PubMed] [Google Scholar]
- 9.Mitchell RS, Beitzel BF, Schroder AR, et al. Retroviral DNA integration: ASLV, HIV, and MLV show distinct target site preferences. PLoS Biol. 2004;2:E234. doi: 10.1371/journal.pbio.0020234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Donahue RE, Kessler SW, Bodine D, et al. Helper virus induced T cell lymphoma in nonhuman primates after retroviral mediated gene transfer. J Exp Med. 1992;176:1125–35. doi: 10.1084/jem.176.4.1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hacein-Bey-Abina S, von Kalle C, Schmidt M, et al. A serious adverse event after successful gene therapy for X-linked severe combined immunodeficiency. New Engl J Med. 2003;348:255–6. doi: 10.1056/NEJM200301163480314. [DOI] [PubMed] [Google Scholar]
- 12.Hacein-Bey-Abina S, von Kalle C, Schmidt M, et al. LMO2-associated clonal T cell proliferation in two patients after gene therapy for SCID-X1. Science. 2003;302:415–9. doi: 10.1126/science.1088547. [DOI] [PubMed] [Google Scholar]
- 13.Mueller PR, Wold B. In vivo footprinting of a muscle specific enhancer by ligation mediated PCR. Science. 1989;246:780–6. doi: 10.1126/science.2814500. [DOI] [PubMed] [Google Scholar]
- 14.Schmidt M, Hoffmann G, Wissler M, et al. Detection and direct genomic sequencing of multiple rare unknown flanking DNA in highly complex samples. Hum Gene Ther. 2001;12:743–9. doi: 10.1089/104303401750148649. [DOI] [PubMed] [Google Scholar]
- 15.Cosset FL, Takeuchi Y, Battini JL, et al. High-titer packaging cells producing recombinant retroviruses resistant to human serum. J Virol. 1995;69:7430–6. doi: 10.1128/jvi.69.12.7430-7436.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rooney CM, Smith CA, Ng CY, et al. Use of gene-modified virus-specific T lymphocytes to control Epstein–Barr-virus-related lymphoproliferation. Lancet. 1995;345:9–13. doi: 10.1016/s0140-6736(95)91150-2. [DOI] [PubMed] [Google Scholar]
- 17.Cosset FL, Takeuchi Y, Battini JL, et al. High-titer packaging cells producing recombinant retroviruses resistant to human serum. J Virol. 1995;69:7430–6. doi: 10.1128/jvi.69.12.7430-7436.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pearson WR, Lipman DJ. Improved tools for biological sequence comparison. Proc Natl Acad Sci USA. 1988;85:2444–8. doi: 10.1073/pnas.85.8.2444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Eddy SR. What is dynamic programming? Nat Biotechnol. 2004;22:909–10. doi: 10.1038/nbt0704-909. [DOI] [PubMed] [Google Scholar]
- 20.SantaLucia J, Jr, Allawi HT, Seneviratne PA. Improved nearest-neighbor parameters for predicting DNA duplex stability. Biochemistry. 1996;35:3555–62. doi: 10.1021/bi951907q. [DOI] [PubMed] [Google Scholar]
- 21.Rossig C, Bollard CM, Nuchtern JG, et al. Epstein–Barr virus-specific human T lymphocytes expressing antitumor chimeric T-cell receptors: potential for improved immunotherapy. Blood. 2002;99:2009–16. doi: 10.1182/blood.v99.6.2009. [DOI] [PubMed] [Google Scholar]
- 22.Swaminathan N, Mead DA, McMaster K, et al. Molecular cloning of the three base restriction endonuclease R.CviJI from eukaryotic Chlorella virus IL-3A. Nucleic Acids Res. 1996;24:2463–9. doi: 10.1093/nar/24.13.2463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kuramoto K, Follman D, Hematti P, et al. The impact of low-dose busulfan on clonal dynamics in nonhuman primates. Blood. 2004;104:1273–80. doi: 10.1182/blood-2003-08-2935. [DOI] [PubMed] [Google Scholar]
- 24.Kuramoto K, Follmann DA, Hematti P, et al. Effect of chronic cytokine therapy on clonal dynamics in nonhuman primates. Blood. 2004;103:4070–7. doi: 10.1182/blood-2003-08-2934. [DOI] [PubMed] [Google Scholar]
- 25.Shi PA, Angioletti MD, Donahue RE, et al. In vivo gene marking of rhesus macaque long-term repopulating hematopoietic cells using a VSV-G pseudotyped versus amphotropic oncoretroviral vector. J Gene Med. 2004;6:367–73. doi: 10.1002/jgm.514. [DOI] [PubMed] [Google Scholar]
- 26.Puskas LG, Fartmann B, Bottka S. Restricted PCR: amplification of an individual sequence flanked by a highly repetitive element from total human DNA. Nucleic Acids Res. 1994;22:3251–2. doi: 10.1093/nar/22.15.3251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Minami M, Poussin K, Brechot C, Paterlini P. A novel PCR technique using Alu-specific primers to identify unknown flanking sequences from the human genome. Genomics. 1995;29:403–8. doi: 10.1006/geno.1995.9004. [DOI] [PubMed] [Google Scholar]
- 28.Parker JD, Rabinovitch PS, Burmer GC. Targeted gene walking polymerase chain reaction. Nucleic Acids Res. 1991;19:3055–60. doi: 10.1093/nar/19.11.3055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sorensen AB, Duch M, Jorgensen P, Pedersen FS. Amplification and sequence analysis of DNA flanking integrated proviruses by a simple two-step polymerase chain reaction method. J Virol. 1993;67:7118–24. doi: 10.1128/jvi.67.12.7118-7124.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gentner B, Laufs S, Nagy KZ, et al. Rapid detection of retroviral vector integration sites in colony-forming human peripheral blood progenitor cells using PCR with arbitrary primers. Gene Ther. 2003;10:789–94. doi: 10.1038/sj.gt.3301935. [DOI] [PubMed] [Google Scholar]
- 31.Ochman H, Gerber AS, Hartl DL. Genetic applications of an inverse polymerase chain reaction. Genetics. 1988;120:621–3. doi: 10.1093/genetics/120.3.621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Riley J, Butler R, Ogilvie D, et al. A novel, rapid method for the isolation of terminal sequences from yeast artificial chromosome (YAC) clones. Nucleic Acids Res. 1990;18:2887–90. doi: 10.1093/nar/18.10.2887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Arnold C, Hodgson IJ. Vectorette PCR: a novel approach to genomic walking. PCR Methods Appl. 1991;1:39–42. doi: 10.1101/gr.1.1.39. [DOI] [PubMed] [Google Scholar]
- 34.Carteau S, Hoffmann C, Bushman F. Chromosome structure and human immunodeficiency virus type 1 cDNA integration: centromeric alphoid repeats are a disfavored target. J Virol. 1998;72:4005–14. doi: 10.1128/jvi.72.5.4005-4014.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Devon RS, Porteous DJ, Brookes AJ. Splinkerettes: improved vectorettes for greater efficiency in PCR walking. Nucleic Acids Res. 1995;23:1644–5. doi: 10.1093/nar/23.9.1644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ivics Z, Hackett PB, Plasterk RH, Izsvak Z. Molecular reconstruction of Sleeping Beauty, a Tc1-like transposon from fish, and its transposition in human cells. Cell. 1997;91:501–10. doi: 10.1016/s0092-8674(00)80436-5. [DOI] [PubMed] [Google Scholar]
- 37.Brenner S, Johnson M, Bridgham J, et al. Gene expression analysis by massively parallel signature sequencing (MPSS) on microbead arrays. Nat Biotechnol. 2000;18:630–4. doi: 10.1038/76469. [DOI] [PubMed] [Google Scholar]