

Abstract
Tumor cell demise is an important event in the elimination of abnormal malignant cells and provides an important mechanism of natural tumor suppression. Abnormalities incapacitating these finely tuned processes provide a strong advantage for cancer clones to succeed in evading both the physiological control systems and therapeutic intervention. Expanding our knowledge of the molecular “cross-talks” that regulate tumor cell demise is crucial in guiding the successful design of future anti-cancer therapeutics. Although currently available data indicate that elimination of malignant cells often depends on classical apoptotic pathways (mitochondrial and/or death receptor pathways), the evidence is mounting that alternative apoptotic and non-apoptotic pathways may effectively contribute to tumor cell death. The assumption that every organelle is capable of sensing, amplificating and executing cell death is also a relatively novel and unexplored concept. As recently shown, the secretory pathway can be actively involved in sensing stress stimuli and possibly even initiating and propagating cell death signaling. Experimental evidence indicates that ER and Golgi apparatus can activate both pro-survival (recovery) mechanisms as well as cell suicide programs if the stress-signaling threshold is exceeded. It is thus conceivable that the fragile balance of protein trafficking between various subcellular compartments provides an exceptional therapeutic opportunity. Interestingly, a growing number of reports recognize novel therapeutic targets, including proteins in control of endoplasmic reticulum (ER) and Golgi homeostasis. Further studies are, however, needed to elucidate precise signaling pathways emanating from ER-Golgi compartment. Development of more potent and selective small-molecule drugs that activate ER-Golgi mediated cell demise is also needed. As the interest in the role of ER-Golgi network during cancer cell death has been gaining momentum, we attempt here to critically appraise current status of development of investigational anti-cancer agents that target ER and/or Golgi.
Keywords: ER, ER stress, Golgi, caspases, apoptosis, cancer, therapy
Introduction
Induction of cell death in neoplasia is a critical event defining tumor growth rate, its regression, and response to treatment [1,2,3]. Abnormalities incapacitating cell death machinery provide tumor cells with a strong survival advantage, and thereby affect the intrinsic response to oncogene activation, immune surveillance, tumor microenvironment (e.g. hypoxia and nutrient shortage), as well as to chemotherapy [1,4]. Evasion of programmed cell death (PCD) is, therefore, often considered as one of the “hallmarks of cancer” [1]. Considerable progress is still being made in our awareness of the variety of cell death modes. Multiple studies have indicated that classical caspase-dependent apoptosis is a common outcome of many anti-cancer therapies [5,6,7]. Recent reports have also characterized several alternative cell death pathways, including caspase independent PCD, cell death dependent on autophagy genes, necrosis-like PCD or mitotic catastrophe [8,9,10]. These studies have enormous impact on our understanding of cancer development and therapy [11,12,13,14]. Pharmacological inhibitor studies as well as genetic knock-out and knock-down approaches have revealed that mechanisms of cell disintegration vary depending on the cell type, cellular microenvironment (both in terms of neighboring cells as well as of intercellular matrix), and also the character and intensity of stress inducers [8,11,10,15].
In the context of cancer therapy, novel pathways, involving endoplasmic reticulum (ER), Golgi apparatus, lysosomes, autophagy and programmed necrosis all are being recognized as potential drug targets for possible therapeutic interventions [2,15,16,17,18].
Rediscovering forgotten pathways
Studies on the canonical pathways of apoptosis have provided insights into molecular mechanisms promoting cell survival or predisposing to death. The seminal discoveries of the role of Bcl-2 family members, cytochrome c/Apaf-1 and caspases in execution of cell death greatly enhanced our understanding of cell death processes, and at the same time revealed some unexpected conundrums [2,8,9]. Above all, the universality of the mitochondrial pathway of apoptosis in response to stress signals, including anti-cancer modalities, has been questioned. Undoubtedly, the fascination in the mitochondrion over-shadowed the potential importance of other organelles that can also act as stress sentinels [2,11,12,14]. Therefore, several critical questions have to be raised. Is mitochondrion self-sufficient in initiating and propagating all intrinsic death signals or does it only represent a cog in a more complex intracellular continuum? Moreover, as biological systems generally show plasticity and redundancy, do alternative back up mechanisms exist to allow cell suicide when mitochondrial pathway is abrogated? [2,8,14]
For decades our knowledge on the homeostatic functions of endoplasmic reticulum (ER), Golgi apparatus, endosomal/lysosomal compartment, cytoskeleton and plasma membrane have been intensively evolving [19]. We have gained profound insights into the dynamic interaction between all these intracellular compartments as well as structural and functional complexity in the interactive inter-organelle crosstalk that is required for cell survival [19]. Still, the assumption that each organelle is capable of sensing, amplificating and executing cell death is a relatively novel and unexplored concept [20,21,22,23]. As recently shown, the secretory pathway can be actively involved in sensing stress stimuli and possibly even initiating and propagating cell death signaling (Fig. 1) [20,22]. Moreover, experimental evidence indicates that ER and Golgi apparatus can activate both pro-survival (recovery) mechanisms as well as cell suicide programs if the stress-signaling threshold is exceeded [20,22,23]. It is conceivable that the fragile balance of protein trafficking between various subcellular compartments provides an exceptionally sensitive damage sensor [19,20,22]. Of interest is the interaction between the mitochondrion with its members of the Bcl-2 family that participates in integrating various incoming death messages, and ER. Thus, Bax overexpression has also been recently found to induce Ca2+ mobilization from ER stores, and convincing data indicate that Bcl-2 co-localizes with ER and protects cells from thapsigargin-mediated apoptosis [24,25,26]. Moreover, p53-dependent overexpression of Bax and down-regulation of Bcl-2 is known to directly influence ER (Fig. 1), lending further support to the complexity of nucleus-ER-mitochondria axis and its role in initiation and propagation of cell death processes.
Figure 1.
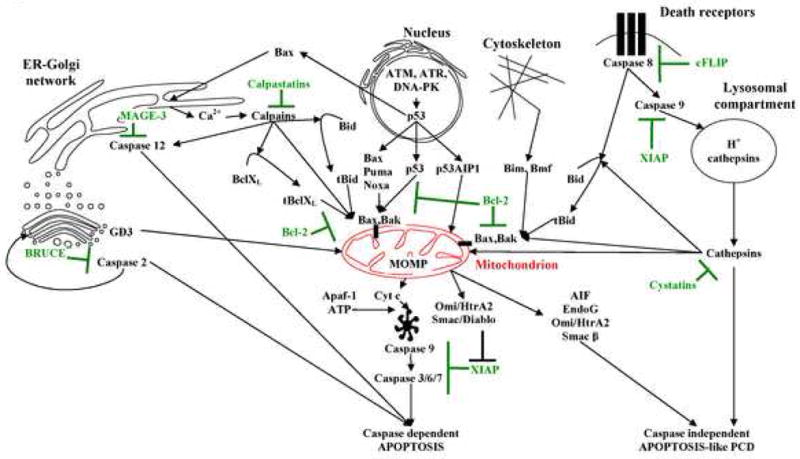
Inter-organellar network responsible for sensing, amplification and executing diverse cell death programs. Mitochondrion is shown in red. Cell death promoting pathways are shown in black whereas cell death suppressor mechanisms are depicted in green (compiled based on based on: [9, 17, 19, 20 and others]; for description refer to text).
Interestingly, recent discoveries have enhanced our understanding of the transcription-independent role of p53 in apoptosis [27,28]. In analogy to BH3-only peptides p53 can reportedly translocate to mitochondria and directly interact with pro-and anti-apoptotic Bcl-2 family members [28,29,30]. In this context, we cannot exclude that transcription-independent function of p53 can in similar manner regulate cell death induced by ER stress. The evidence is also mounting that signals emanating from the ER-Golgi network may bypass mitochondrion and efficiently execute programmed cell death (Fig. 1) [20,31]. Broader understanding of these non-canonical pathways, and the transcription-independent role of p53 is expected to reveal novel molecular targets for anti-cancer drug development.
Waves of destruction: ER, calcium and beyond
The involvement of ER in stress sensing encompasses two mainstream mechanisms, namely unfolded protein response (UPR), and calcium signaling (Fig 1 and 2). UPR can be further subdivided into a transcription-dependent and transcription-independent mechanisms; the former is also involved in initial cell recovery pathways [19,32,33].
Figure 2.

Cascades of cell demise pathways in response to the ER stress (compiled based on based on: [19, 21, 33] for description refer to text).
Transcription-dependent UPR
The accumulation of unfolded proteins in response to stress stimuli (e.g. inhibition of N-linked glycosylation by tunicamycin; disruption of ER-Golgi vesicular transport by brefeldin A or deficient formation of disulfide bonds by dithiothreitol) leads to their competitive binding to ER-luminal chaperon BiP/GRP78 [32,34]. It displaces BiP/GRP78 form the ER-membrane residual serine-threonine kinase Ire1-α, allowing its dimerization and autophosphorylation on a cytosolic tail (Fig. 2) [34]. Following activation, the cytosolic fragment of Ire1-α undergoes cleavage by preselin-1 (PS-1) and translocates to nucleus where it promotes transcription of endoplasmic-resident chaperones (BiP, calreticulin) and transcription factor CHOP/GADD 153 (Fig. 2) [34,35,36]. In the context of cellular demise CHOP/GADD 153 has been shown to down-regulate Bcl-2 expression and thus initiate mitochondrial apoptotic program (Fig. 2) [37]. Moreover, tunicamycin-induced ER-stress has recently been found to up-regulate expression of TRAIL receptor DR5 through CHOP/GADD 153 transcription factor in human prostate and carcinoma cells [38]. Intriguingly, the p53-mediated trans-activation of Puma and Noxa in response to thapsigargin and tunicamycin has been demonstrated in the recent work of Li and colleagues (Fig. 2) [39]. Although ER UPR-p53 link still remains elusive it delivers novel insights on the complexity of pro-death signaling launched in response to secretory pathway stressors [19].
Transcription-independent UPR
Following activation, Ire1-α reportedly attracts adaptor protein TRAF-2 (TNFR-associated protein 2) [40]. Formation of Ire1-α/TRAF-2 complexes at the cytosolic face of ER is postulated to activate c-Jun N-terminal kinase (JNK) pathway and/or participate in recruitment, clustering and auto-activation of pro-caspase 12 molecules (Fig. 2) [40,41]. JNK reportedly inhibits Bcl-2 function leading to impaired sequestration of pro-apoptotic Bax, BH3-only proteins and regulation of ER Ca2+ stores [42]. There are also hints that JNK activity can directly modulate function of BH3-only protein Bim, hence strongly influencing mitochondrial pro-apoptotic signaling (Fig 2) [21,23,43].
The discovery of the caspase 12 as an apical enzyme involved in response to ER damage delivered novel, speculative links to the classical apoptotic machinery [44]. Although functionally present only in rodents, caspase 12, is localized specifically to the cytosolic side of ER [45,46]. Its activation in mouse embryonic fibroblasts has been reported to exclusively follow ER-targeted stress inducers such as brefeldin A, tunicamycin and thapsigargin [21]. The ER stress-activated caspase 12 was also shown to mediate cisplatin-induced apoptosis of mouse renal tubular epithelial cells [47]. On the other hand, inhibition of caspase 12 activity by overexpression of its endogenous inhibitor MAGE-3 has been reported to diminish ER-stress mediated apoptotic response in murine cells (Fig. 1) [20,48]. There are, however, still conflicting reports on the requirement of caspase 12 in the ER-stress response [21,45,46]. Apart from Ire1-α/TRAF-2-mediated auto-proteolysis, several alternative models of activation have lately been proposed for caspase 12, including: Ca2+-dependent cleavage by m-calpain; CARD domain-mediated clustering on an unidentified Apaf-1-like adaptor molecule; GRP78 mediated formation of caspase 7/caspase 12 dimers (Fig 2) [21]. Since human lack functional caspase 12 the quest is still ongoing to decipher its exact human orthologue [21,49]. In this context caspase 4 has been recently demonstrated to co-localize with and participate in ER-induced apoptosis [50]. Expanding the complexity of apoptotic pathways an isoform of pro-caspase 8, pro-caspase 8L, has also been found to be activated at the ER via interaction with Bap31 protein (Fig 2) [51].
Calcium flux in apoptosis
Stress signals culminating at the endoplasmic reticulum can also trigger rapid Ca2+ release from luminal ER stores. This process is efficiently executed by the inositol- 1,4,5-triphosphate receptors (IP3Rs) or ryanodine receptors (RyRs) serving as Ca2+ channels (Fig. 2) [24,33]. Both pro- and anti-apoptotic members of the Bcl-2 family have also been postulated as players in regulation of the ER Ca2+ homeostasis [33,52,53]. Moreover since mitochondria often are in close proximity with tubular ER network the inter-organelle information transfer is likely involved in triggering pro-apoptotic signaling [54,55]. In this regard, recent work of Boehning and colleagues illustrates novel insight into the ER-mitochondrion crosstalk at the very early stages of apoptosis induction (Fig 1). Clearly the initial cytochrome c release may initiate the activation of IP3Rs, Ca2+ discharge from ER and subsequent feedback on mitochondria through the PTP dependent MMP (Fig. 2) [33,56].
Apart from eliciting mitochondrial MMP, the rapid Ca2+ mobilization from ER lumen can also evoke activation of cytosolic Ca2+-dependent proteases, calpains. The latter are being reported to play a role in both caspase 12 processing and sole propagation of cellular dismantling (Fig. 1 and 2) [45,46,57].
Golgi - the forgotten organelle
It has been known for some time that Golgi apparatus is being dissembled during apoptosis in a manner similar to that observed during mitosis [58]. Polo-like kinase-3 (Plk3) is localized in Golgi apparatus and is likely the protein kinase that mediates fragmentation of this organelle during mitosis and apoptosis [59]. Interestingly, Plk3, an immediate DNA damage response gene product, induced by the environmental and genotoxic streass, also plays an important role in the DNA damage checkpoints activation. Over-expression of the Polo box domain of Plk3 causes cell cycle arrest and cytokinesis defects, eventually leading to apoptosis manifesting as mitotic catastrophe [60]. There are other examples of Plk3 participation in induction of apoptosis [61,62,63]. On the other hand the transcriptional silencing of Plk2 is a frequent event in B-cell malignancies and its ectopic expression leads to apoptosis of these cells [64]. The data of Syed et al (2006), however, also indicate on certain degree of redundancy between Plk1, Plk2 and Plk3 within the Plk kinases family [64]. Considering the above evidence collectively, it is possible thus that the Golgi-residing Plk3 is involved both, in signaling and execution of apoptosis, the latter by disassembling this organelle.
Apoptotic cleavage of structural proteins localized to the Golgi has been found necessary to dismantle complex Golgi structure into separate stacks of cisternal membranes. In this context the overexpression of non-cleavable forms of structural golgin-160, p115 and GRASP65 often delay Golgi separation during caspase dependent cell death [20,65,66,67]. Yet a concept that Golgi apparatus may actually be actively involved in initiation and/or execution of cellular demise delivers a completely new insight on the secretory pathway as a watchdog of cellular fate (Fig 3) [20,22,31].
Figure 3.

Sensing and execution of cell demise pathways at Golgi apparatus. Known pathways are shown as solid lines and hypothetical signaling are depicted as dotted lines. Anti-apoptotic mechanisms are depicted in green (compiled based on [22, 31, 65, 66 and others]; for description refer to text).
The seminal discovery of caspase 2 and its co-localization to the Golgi apparatus and the nucleus prompted initial suggestions that Golgi apparatus may not only be “a passive player” in apoptosis (Fig. 3) [65]. Although caspase 2 has also been found to participate in DNA damage response in conjunction with PIDDosome complex, it is reasonable that its function may be bidirectional and depend heavily on the cellular compartmentalization [22,24,68]. Pertinent to this notion it is now generally believed that a pool of caspase 2 localized at the cytosolic side of the Golgi apparatus participates in initiation of apoptotic cascade in response to secretory pathway stresses. In support of this concept specific caspase 2-dependent cleavage of golgin 160 at Asp59 site has been shown as a very early apoptotic event that preceded caspase 3-dependent cleavage of golgin160 and poly-ADP-ribose polymerase (PARP-1) (Fig 3) [31,65]. The recent report by Machamer’s group provided further evidence that caspase-resistant mutant golgin 160 can abrogate apoptosis induced specifically by ER stress and ligation of death receptors [69]. Moreover, BRUCE (baculoviral-IAP-repeat-containing ubiquitin-conjugating enzyme) an expected negative regulator of caspase 2 activity, has been localized to the Golgi superstructure (Fig. 1) [20,70].
Besides caspase 2, the role of glycolipids and ceramides as messengers in death signaling pathways has also been raised during recent years. In the Golgi, the activity of the GD3 synthase reportedly converts ceramide to the gangliosyde GD3. The latter has been shown to translocate to the mitochondrion and solely trigger mitochondrial membrane permeability (MMP) (Fig. 3) [71,72]. Interestingly the suppression of GD3 synthase activity or its retention in the ER lumen markedly inhibits apoptosis [73]. Moreover, there are further hints that mitochondrial GD3 targets are under rigid control of anti-apoptotic Bcl-2 family members [71]. Similarly to the GD3, a semilysobisphosphatidic acid (SLBPPA) has been found to shuttle from the Golgi to mitochondria in response to death receptor stimulation, supporting the significance of inter-organelle lipid signaling pathways (Fig 3) [74].
It is conceivable that a pool of death receptors (DRs) that populate Golgi apparatus in the normal physiological conditions demonstrate yet another link between Golgi and initiation of cellular demise [19,20]. Some reports indicate that DRs may be quickly mobilized and shuttled to the cell surface during stress response after p53 or GD3 signaling [20,75]. Moreover it is tempting to speculate that during apoptotic Golgi disassembly a surge release of DRs occurs which further amplifies the death cascade (Fig 3) [19,20,22].
Not surprisingly Golgi apparatus lures as the upstream controller of many destruction cascades. The active protein transfer between ER-Golgi requires precise control mechanisms that as such are supposedly very sensitive to pathological alterations in cellular homeostasis [22]. Moreover the stability of steady-state Golgi configuration is achieved not only by a subtle balance between anterograde vis-à-vis retrograde membrane trafficking but also by the specialized structural proteins like golgins and GRAPs involved in vesicle tethering and docking interactions between separate Golgi stacks [22]. Cleavage fragments of golgins have been demonstrated to contain nuclear targeting motifs, implicating further their role in the inter-organelle signal transduction (Fig 3) [22,65,66]. Furthermore, as postulated recently, Golgi is in fact enclosed in a highly specialized exoskeleton vigorously implicated in membrane anchoring, regulation of substrate diffusion and even enzyme positioning (Fig 3) [20,22]. It is plausible to speculate that unique Golgi exoskeleton may take part in integrating a plethora of death stimuli engaging from other parts of the cytoskeleton. Although still speculative, the unique structure of exoskeleton may have evolved to sense any distressing changes in the Golgi cisternal structure. In this regard any pathological physical abnormalities like: cisternal swelling, unstacking, alterations in membrane curvature or even thickness in Golgi membranes could hypothetically initiate recruitment of specific adaptor molecules that initiate cell recovery or cell demise signals [20,22].
Finally, Golgi apparatus is conceivably equipped with effective UPR sensing mechanisms compulsory to remove any misfolded or mutated proteins that escaped aforementioned ER-UPR system [22,76]. Therefore any overload of misfolded proteins reaching Golgi may set off initial cell recovery mechanisms and when they prove to be ineffective also cell demise pathways. Keeping in mind the crucial physiological position of this organelle it is tempting to speculate that, similarly to mitochondrion and endoplasmic reticulum, Golgi is poised to sense and integrate diversified death signals [19,20,22]. Although the precise signaling pathways emanating from Golgi apparatus are still elusive it is unlikely that we will need to wait long for these questions to be answered.
Targeting secretory pathway – “Wunderwaffe” for cancer therapy?
Thanks to seminal discoveries it now becomes apparent that extensive inter-organelle cross-talks that regulate orchestrated cell dismantling are present in every cell. Even if mitochondrion reportedly stands at the nexus of controlling cell fate, redundant and failsafe mechanisms commencing from other organelles do exist and may in many cases efficiently override the classical signaling pathways (Fig 1). Thus, the present hope prevails that exploration of both the primary and the redundant back up pathways of cell destruction might prospectively provide effective means to override cancer chemo-resistance [16,18,77,78].
As the interest in the role of ER and Golgi during induction/execution of apoptosis has been gaining momentum, they simultaneously attracted interest in the development of novel anti-cancer therapies [16,19,79]. In this context, two recent studies indicated that malignant B lymphocytes feature not only proapoptotic Bcl-2 upregulation but also more elaborate endoplasmic reticulum (ER) network when compared to their normal counterparts [78,80]. Therefore, it has been postulated that ER-Golgi system may be imperative for endurance of malignant B-clones, and such reliance may reveal unique anti-cancer targets (Fig. 4A) [78]. Pertinent to the therapy of B-CLL, the successful targeting of the ER-Golgi pathway with Brefeldin A (BFA) in fludarabine refractory CLL cells has recently provided a novel approach on how to eradicate cancerous B-cells independently of their p53 status and pathological overexpression of Bcl-2, Bcl-XL, Mcl-1 and XIAP proteins [78]. Brefeldin A, a fungal 16-membered macrolide isolated from Penicillium brefeldianum, exerts ER and Golgi stress via inhibition of ADP-ribosylation factor (ARF). Subsequent decline in coatamer proteins assembly leads to the disruption of ER-Golgi vesicular transport, Golgi collapse due to the imbalance in retrograde transport and induction of apoptotic cell demise (Fig 4B, C) [78,81,82]. Apart from B-CLL cells, BFA reportedly triggered apoptosis in multiple myeloma (U266, NCI-H929), Jurkat, HeLa, leukaemia (HL60, K562, BJAB), colon (HT-29), prostate and adenoid cystic sarcoma cells [78,83,84,85,86]. Furthermore, we have recently demonstrated monotherapeutic potential of BFA against follicular lymphoma cells bearing t(14;18) translocation (Fig 4B) [87]. Brefeldin A has been reported to exert its anti-tumor activity in melanoma athymic mouse xenografts and enhance action of staurosporine and 7-hydroxystaurosporine in human promyelocytic leukemia cells [83,88]. A water-soluble prodrug form of BFA, Breflate (NSC656202), is also under development as an investigational anticancer agent and has shown promising pharmacokinetics in mice and beagle dogs [89,90].
Figure 4.

Targeting secretory pathway in cancer therapy: A) therapeutic window for ER-Golgi disrupting drugs; B) eradication of follicular lymphoma cells by Brefeldin A, note progressing dilation of ER leading to ER-stress and caspase-dependent apoptosis; C) autophagosome formation (green vesicles) and collapse of Golgi apparatus (red staining) in osteosarcoma U2OS cells following exposure to increasing doses of Brefeldin A. [78, 87, 99]
Since a large number of reports have demonstrated that elevated Bcl-2/Bcl-XL/Mcl-1 expression is associated with a poor clinical response, a combinatorial strategies aimed at diverse apoptotic elements may offer substantial therapeutic promise [91,92,93]. Although brefeldin A has been previously suggested as an investigational anticancer agent its potential interactions with other apoptosis-inducing compounds remain surprisingly unexplored [78,83,88,94]. Merely one study by Shao and colleagues (1996) reported synergistic induction of apoptosis upon combinatorial treatment of human promyelocytic leukemia cells with BFA and staurosporine [83]. In our recent work, we investigated the potential therapeutic utility of combined treatment between BFA vs. a small molecule Bcl-2 inhibitor (HA14-1) and BFA vs. a model trigger of death-receptor pathway (CD95 cross-linking mAb) in follicular lymphoma (FL) cells [87]. Strikingly, upon co-treatment of follicular lymphoma cells with HA14-1 and BFA we observed an enhanced cell killing, whereas the co-stimulation anti-CD95 mAb increased the efficacy of brefeldin A to a much lesser degree. Together, these data clearly point out that combinatorial targeting of diverse cell death pathways may improve anticancer action of ER/Golgi stressors [87].
Recent studies by Tinhoffer et al (2002) and Anether et al (2003) have also revealed that Tetrocarcin A, an antibiotic isolated from actinomyces, eradicated T-acute lymphoblastic leukemia (T-CLL) and B-CLL cells through ER mediated stress, independently of Bcl-2 status [95,96]. Similarly to BFA, Tetrocarcin A efficiently initiated cell death in fludarabine refractory B-CLL primary cells [96]. Of particular interest tunicamycin, another ER stress inducing antibiotic that blocks N-linked glycosylation, reportedly sensitizes human prostate cancer cells to TNF-related apoptosis-inducing ligand (TRAIL) [97]. Furthermore, pertinent to the role of SERCA pump in ER mediated apoptosis, modified thapsigargin (PSA-activated thapsigargin) has recently been postulated as an investigational drug for prostate cancer [98]. An unfolded protein response (UPR) forms an important cytoprotective mechanism, a proteasome inhibitor bortezomib augmented tunicamycin- and thapsigargin-triggered apoptosis in pancreatic cancer cells [99].
Summary and future outlook
Conventional chemotherapy is largely ineffective for the treatment of metastatic and advanced tumors. Despite recent progress in investigational therapies, survival rates are still disappointingly low and novel adjuvant and systemic therapies are urgently needed. There is a substantial expectation that endoplasmic reticulum (ER) and Golgi apparatus might provide convenient therapeutic targets for the treatment of refractory malignancies. In the broader context of ER-Golgi network it is worth noting that ubiquitin-proteasome complex has recently emerged as a major target for drug development in cancer therapy [100,101,102]. The most prominent drug, proteasome inhibitor bortezomib (Velcade) has already shown clinical activity in multiple cancers including myeloma and mantle cell lymphomas. Recent work by Hill and colleagues provided also fresh evidence that two relatively new drugs, fenretinide and bortezomib (Velcade), each acting via different cellular mechanisms, induce ER stress leading to apoptosis in melanoma cells [103]. Interestingly, novel class of ERAD inhibitors has also recently emerged [104]. Wang and colleagues reported Eeyarestatin I (EerI), a chemical inhibitor that blocks endoplasmic reticulum (ER)-associated protein degradation (ERAD), has potent antitumor activities similar to bortezomib and can synergize with bortezomib to induce cancer cell apoptosis [104]. These noteworthy results identify a novel class of anticancer agents that can directly engage ER-Golgi network to induce tumor cell death.
In closing, the secretory pathway slowly attracts interests as a prospective anti-cancer target. Further studies are necessary to elucidate precise signaling pathways emanating from ER-Golgi compartment. Development of more potent and selective small-molecule drugs that activate ER-Golgi mediated cell demise is also needed. Moreover, as ER-stress signaling is crucial for the homeostasis of differentiated tissues it is imperative determine the potential side effects of ER/Golgi targeted drugs [105]. Furthermore, an important question arises with regards to acquisition of resistance to ER targeted drugs. At the moment there is no data that can suggest acquisition of apoptosis resistance to e.g. ERAD inhibitors. Further characterization of ER stress pathways and robust in vivo models are, however, needed to determine the physiological roles of UPR and ER stress during tumour development and its ultimate therapeutic potential [105]. Recent results strongly support, however, the premise that malignant cells, engaged in intense secretory function, may be vulnerable to the interruption of ER-Golgi homeostasis. It is to be seen whether these experimental approaches find their way into clinical practice.
Acknowledgments
Supported by NCI CA RO1 28 704 (ZD), L’Oreal-UNESCO “For Women In Science” 2007 Award (JS).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100(1):57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 2.Broker LE, Kruyt FA, Giaccone G. Cell death independent of caspases: a review. Clin Cancer Res. 2005;11(9):3155–62. doi: 10.1158/1078-0432.CCR-04-2223. [DOI] [PubMed] [Google Scholar]
- 3.Crighton D, Ryan KM. Splicing DNA-damage responses to tumour cell death. Biochim Biophys Acta. 2004;1705(1):3–15. doi: 10.1016/j.bbcan.2004.09.001. [DOI] [PubMed] [Google Scholar]
- 4.Meng XW, Lee SH, Kaufmann SH. Apoptosis in the treatment of cancer: a promise kept? Curr Opin Cell Biol. 2006;18(6):668–76. doi: 10.1016/j.ceb.2006.10.008. [DOI] [PubMed] [Google Scholar]
- 5.Lowe SW, Ruley HE, Jacks T, Housman DE. p53-dependent apoptosis modulates the cytotoxicity of anticancer agents. Cell. 1993;74(6):957–67. doi: 10.1016/0092-8674(93)90719-7. [DOI] [PubMed] [Google Scholar]
- 6.Kaufmann SH, Gores GJ. Apoptosis in cancer: cause and cure. Bioessays. 2000;22(11):1007–17. doi: 10.1002/1521-1878(200011)22:11<1007::AID-BIES7>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 7.Ghobrial IM, Witzig TE, Adjei AA. Targeting apoptosis pathways in cancer therapy. CA Cancer J Clin. 2005;55(3):178–94. doi: 10.3322/canjclin.55.3.178. [DOI] [PubMed] [Google Scholar]
- 8.Leist M, Jaattela M. Four deaths and a funeral: from caspases to alternative mechanisms. Nat Rev Mol Cell Biol. 2001;2(8):589–98. doi: 10.1038/35085008. [DOI] [PubMed] [Google Scholar]
- 9.Jaattela M. Programmed cell death: many ways for cells to die decently. Ann Med. 2002;34(6):480–8. doi: 10.1080/078538902321012423. [DOI] [PubMed] [Google Scholar]
- 10.Lockshin RA, Zakeri Z. Programmed cell death and apoptosis: origins of the theory. Nat Rev Mol Cell Biol. 2001;2(7):545–50. doi: 10.1038/35080097. [DOI] [PubMed] [Google Scholar]
- 11.Okada H, Mak TW. Pathways of apoptotic and non-apoptotic death in tumour cells. Nat Rev Cancer. 2004;4(8):592–603. doi: 10.1038/nrc1412. [DOI] [PubMed] [Google Scholar]
- 12.Edinger AL, Thompson CB. Death by design: apoptosis, necrosis and autophagy. Curr Opin Cell Biol. 2004;16(6):663–9. doi: 10.1016/j.ceb.2004.09.011. [DOI] [PubMed] [Google Scholar]
- 13.Abraham MC, Shaham S. Death without caspases, caspases without death. Trends Cell Biol. 2004;14(4):184–93. doi: 10.1016/j.tcb.2004.03.002. [DOI] [PubMed] [Google Scholar]
- 14.Hail N, Jr, Carter BZ, Konopleva M, Andreeff M. Apoptosis effector mechanisms: a requiem performed in different keys. Apoptosis. 2006;11(6):889–904. doi: 10.1007/s10495-006-6712-8. [DOI] [PubMed] [Google Scholar]
- 15.Levine B, Yuan J. Autophagy in cell death: an innocent convict? J Clin Invest. 2005;115(10):2679–88. doi: 10.1172/JCI26390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kroemer G, Jaattela M. Lysosomes and autophagy in cell death control. Nat Rev Cancer. 2005;5(11):886–97. doi: 10.1038/nrc1738. [DOI] [PubMed] [Google Scholar]
- 17.Kim R. Recent advances in understanding the cell death pathways activated by anticancer therapy. Cancer. 2005;103(8):1551–60. doi: 10.1002/cncr.20947. [DOI] [PubMed] [Google Scholar]
- 18.Kim R, Emi M, Tanabe K. Role of mitochondria as the gardens of cell death. Cancer Chemother Pharmacol. 2006;57(5):545–53. doi: 10.1007/s00280-005-0111-7. [DOI] [PubMed] [Google Scholar]
- 19.Ferri KF, Kroemer G. Organelle-specific initiation of cell death pathways. Nat Cell Biol. 2001;3(11):E255–63. doi: 10.1038/ncb1101-e255. [DOI] [PubMed] [Google Scholar]
- 20.Maag RS, Hicks SW, Machamer CE. Death from within: apoptosis and the secretory pathway. Curr Opin Cell Biol. 2003;15(4):456–61. doi: 10.1016/s0955-0674(03)00075-9. [DOI] [PubMed] [Google Scholar]
- 21.Momoi T. Caspases involved in ER stress-mediated cell death. J Chem Neuroanat. 2004;28(1–2):101–5. doi: 10.1016/j.jchemneu.2004.05.008. [DOI] [PubMed] [Google Scholar]
- 22.Hicks SW, Machamer CE. Golgi structure in stress sensing and apoptosis. Biochim Biophys Acta. 2005;1744(3):406–14. doi: 10.1016/j.bbamcr.2005.03.002. [DOI] [PubMed] [Google Scholar]
- 23.Szegezdi E, Logue SE, Gorman AM, Samali A. Mediators of endoplasmic reticulum stress-induced apoptosis. EMBO Rep. 2006;7(9):880–5. doi: 10.1038/sj.embor.7400779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Orrenius S, Zhivotovsky B, Nicotera P. Regulation of cell death: the calcium-apoptosis link. Nat Rev Mol Cell Biol. 2003;4(7):552–65. doi: 10.1038/nrm1150. [DOI] [PubMed] [Google Scholar]
- 25.Scorrano L, Korsmeyer SJ. Mechanisms of cytochrome c release by proapoptotic BCL-2 family members. Biochem Biophys Res Commun. 2003;304(3):437–44. doi: 10.1016/s0006-291x(03)00615-6. [DOI] [PubMed] [Google Scholar]
- 26.Scorrano L, Oakes SA, Opferman JT, Cheng EH, Sorcinelli MD, Pozzan T, Korsmeyer SJ. BAX and BAK regulation of endoplasmic reticulum Ca2+: a control point for apoptosis. Science. 2003;300(5616):135–9. doi: 10.1126/science.1081208. [DOI] [PubMed] [Google Scholar]
- 27.Chipuk JE, Maurer U, Green DR, Schuler M. Pharmacologic activation of p53 elicits Bax-dependent apoptosis in the absence of transcription. Cancer Cell. 2003;4(5):371–81. doi: 10.1016/s1535-6108(03)00272-1. [DOI] [PubMed] [Google Scholar]
- 28.Chipuk JE, Green DR. p53’s believe it or not: lessons on transcription-independent death. J Clin Immunol. 2003;23(5):355–61. doi: 10.1023/a:1025365432325. [DOI] [PubMed] [Google Scholar]
- 29.Chipuk JE, Bouchier-Hayes L, Green DR. Mitochondrial outer membrane permeabilization during apoptosis: the innocent bystander scenario. Cell Death Differ. 2006;13(8):1396–402. doi: 10.1038/sj.cdd.4401963. [DOI] [PubMed] [Google Scholar]
- 30.Skommer J, Wlodkowic D, Deptala A. Larger than life: Mitochondria and the Bcl-2 family. Leuk Res Leuk Res. 2007 Mar;31(3):277–86. doi: 10.1016/j.leukres.2006.06.027. [DOI] [PubMed] [Google Scholar]
- 31.Machamer CE. Golgi disassembly in apoptosis: cause or effect? Trends Cell Biol. 2003;13(6):279–81. doi: 10.1016/s0962-8924(03)00101-6. [DOI] [PubMed] [Google Scholar]
- 32.Rutkowski DT, Kaufman RJ. A trip to the ER: coping with stress. Trends Cell Biol. 2004;14(1):20–8. doi: 10.1016/j.tcb.2003.11.001. [DOI] [PubMed] [Google Scholar]
- 33.Walter L, Hajnoczky G. Mitochondria and endoplasmic reticulum: the lethal interorganelle crosstalk. J Bioenerg Biomembr. 2005;37(3):191–206. doi: 10.1007/s10863-005-6600-x. [DOI] [PubMed] [Google Scholar]
- 34.Bertolotti A, Zhang Y, Hendershot LM, Harding HP, Ron D. Dynamic interaction of BiP and ER stress transducers in the unfolded-protein response. Nat Cell Biol. 2000;2(6):326–32. doi: 10.1038/35014014. [DOI] [PubMed] [Google Scholar]
- 35.Katayama T, Imaizumi K, Sato N, Miyoshi K, Kudo T, Hitomi J, Morihara T, Yoneda T, Gomi F, Mori Y, Nakano Y, Takeda J, Tsuda T, Itoyama Y, Murayama O, Takashima A, St George-Hyslop P, Takeda M, Tohyama M. Presenilin-1 mutations downregulate the signalling pathway of the unfolded-protein response. Nat Cell Biol. 1999;1(8):479–85. doi: 10.1038/70265. [DOI] [PubMed] [Google Scholar]
- 36.Patil C, Walter P. Intracellular signaling from the endoplasmic reticulum to the nucleus: the unfolded protein response in yeast and mammals. Curr Opin Cell Biol. 2001;13(3):349–55. doi: 10.1016/s0955-0674(00)00219-2. [DOI] [PubMed] [Google Scholar]
- 37.McCullough KD, Martindale JL, Klotz LO, Aw TY, Holbrook NJ. Gadd153 sensitizes cells to endoplasmic reticulum stress by down-regulating Bcl2 and perturbing the cellular redox state. Mol Cell Biol. 2001;21(4):1249–59. doi: 10.1128/MCB.21.4.1249-1259.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shiraishi T, Yoshida T, Nakata S, Horinaka M, Wakada M, Mizutani Y, Miki T, Sakai T. Tunicamycin enhances tumor necrosis factor-related apoptosis-inducing ligand-induced apoptosis in human prostate cancer cells. Cancer Res. 2005;65(14):6364–70. doi: 10.1158/0008-5472.CAN-05-0312. [DOI] [PubMed] [Google Scholar]
- 39.Li J, Lee B, Lee AS. Endoplasmic reticulum stress-induced apoptosis: multiple pathways and activation of p53-up-regulated modulator of apoptosis (PUMA) and NOXA by p53. J Biol Chem. 2006;281(11):7260–70. doi: 10.1074/jbc.M509868200. [DOI] [PubMed] [Google Scholar]
- 40.Yoneda T, Imaizumi K, Oono K, Yui D, Gomi F, Katayama T, Tohyama M. Activation of caspase-12, an endoplastic reticulum (ER) resident caspase, through tumor necrosis factor receptor-associated factor 2-dependent mechanism in response to the ER stress. J Biol Chem. 2001;276(17):13935–40. doi: 10.1074/jbc.M010677200. [DOI] [PubMed] [Google Scholar]
- 41.Urano F, Bertolotti A, Ron D. IRE1 and efferent signaling from the endoplasmic reticulum. J Cell Sci. 2000;113(Pt 21):3697–702. doi: 10.1242/jcs.113.21.3697. [DOI] [PubMed] [Google Scholar]
- 42.Bassik MC, Scorrano L, Oakes SA, Pozzan T, Korsmeyer SJ. Phosphorylation of BCL-2 regulates ER Ca2+ homeostasis and apoptosis. EMBO J. 2004;23(5):1207–16. doi: 10.1038/sj.emboj.7600104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lei K, Davis RJ. JNK phosphorylation of Bim-related members of the Bcl2 family induces Bax-dependent apoptosis. Proc Natl Acad Sci U S A. 2003;100(5):2432–7. doi: 10.1073/pnas.0438011100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.van de Craen M, Vandenabeele P, Declercq W, Van den Brande I, Van Loo G, Molemans F, Schotte P, Van Criekinge W, Beyaert R, Fiers W. Characterization of seven murine caspase family members. FEBS Lett. 1997;403(1):61–9. doi: 10.1016/s0014-5793(97)00026-4. [DOI] [PubMed] [Google Scholar]
- 45.Nakagawa T, Zhu H, Morishima N, Li E, Xu J, Yankner BA, Yuan J. Caspase-12 mediates endoplasmic-reticulum-specific apoptosis and cytotoxicity by amyloid-beta. Nature. 2000;403(6765):98–103. doi: 10.1038/47513. [DOI] [PubMed] [Google Scholar]
- 46.Nakagawa T, Yuan J. Cross-talk between two cysteine protease families. Activation of caspase-12 by calpain in apoptosis. J Cell Biol. 2000;150(4):887–94. doi: 10.1083/jcb.150.4.887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Liu H, Baliga R. Endoplasmic reticulum stress-associated caspase 12 mediates cisplatin-induced LLC-PK1 cell apoptosis. J Am Soc Nephrol. 2005;16:1985–92. doi: 10.1681/ASN.2004090768. [DOI] [PubMed] [Google Scholar]
- 48.Morishima N, Nakanishi K, Takenouchi H, Shibata T, Yasuhiko Y. An endoplasmic reticulum stress-specific caspase cascade in apoptosis. Cytochrome c-independent activation of caspase-9 by caspase-12. J Biol Chem. 2002;277(37):34287–94. doi: 10.1074/jbc.M204973200. [DOI] [PubMed] [Google Scholar]
- 49.Fischer H, Koenig U, Eckhart L, Tschachler E. Human caspase 12 has acquired deleterious mutations. Biochem Biophys Res Commun. 2002;293(2):722–6. doi: 10.1016/S0006-291X(02)00289-9. [DOI] [PubMed] [Google Scholar]
- 50.Hitomi J, Katayama T, Eguchi Y, Kudo T, Taniguchi M, Koyama Y, Manabe T, Yamagishi S, Bando Y, Imaizumi K, Tsujimoto Y, Tohyama M. Involvement of caspase-4 in endoplasmic reticulum stress-induced apoptosis and Abeta-induced cell death. J Cell Biol. 2004;165(3):347–56. doi: 10.1083/jcb.200310015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Breckenridge DG, Nguyen M, Kuppig S, Reth M, Shore GC. The procaspase-8 isoform, procaspase-8L, recruited to the BAP31 complex at the endoplasmic reticulum. Proc Natl Acad Sci U S A. 2002;99(7):4331–6. doi: 10.1073/pnas.072088099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Thomenius MJ, Distelhorst CW. Bcl-2 on the endoplasmic reticulum: protecting the mitochondria from a distance. J Cell Sci. 2003;116(Pt 22):4493–9. doi: 10.1242/jcs.00829. [DOI] [PubMed] [Google Scholar]
- 53.Chen R, Valencia I, Zhong F, McColl KS, Roderick HL, Bootman MD, Berridge MJ, Conway SJ, Holmes AB, Mignery GA, Velez P, Distelhorst CW. Bcl-2 functionally interacts with inositol 1,4,5-trisphosphate receptors to regulate calcium release from the ER in response to inositol 1,4,5-trisphosphate. J Cell Biol. 2004;166(2):193–203. doi: 10.1083/jcb.200309146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mannella CA, Buttle K, Rath BK, Marko M. Electron microscopic tomography of rat-liver mitochondria and their interaction with the endoplasmic reticulum. Biofactors. 1998;8(3–4):225–8. doi: 10.1002/biof.5520080309. [DOI] [PubMed] [Google Scholar]
- 55.Rizzuto R, Pinton P, Carrington W, Fay FS, Fogarty KE, Lifshitz LM, Tuft RA, Pozzan T. Close contacts with the endoplasmic reticulum as determinants of mitochondrial Ca2+ responses. Science. 1998;280(5370):1763–6. doi: 10.1126/science.280.5370.1763. [DOI] [PubMed] [Google Scholar]
- 56.Boehning D, Patterson RL, Sedaghat L, Glebova NO, Kurosaki T, Snyder SH. Cytochrome c binds to inositol (1,4,5) trisphosphate receptors, amplifying calcium-dependent apoptosis. Nat Cell Biol. 2003;5(12):1051–61. doi: 10.1038/ncb1063. [DOI] [PubMed] [Google Scholar]
- 57.Guicciardi ME, Gores GJ. Calpains can do it alone: implications for cancer therapy. Cancer Biol Ther. 2003;2(2):153–4. [PubMed] [Google Scholar]
- 58.Sesso A, Fujiwara DT, Jaeger M, Jaeger R, Li TC, Monteiro MM, Correa H, Ferreira MA, Schumacher RI, Belisario J, Kachar B, Chen EJ. Structural elements common to mitosis and apoptosis. Tissue Cell. 1999;31(3):357–71. doi: 10.1054/tice.1999.0042. [DOI] [PubMed] [Google Scholar]
- 59.Ruan Q, Wang Q, Xie S, Fang Y, Darzynkiewicz Z, Guan K, Jhanwar-Uniyal M, Dai W. Polo-like kinase 3 is Golgi localized and involved in regulation of Golgi fragmentation during the cell cycle. Exp Cell Res. 2004;294:51–9. doi: 10.1016/j.yexcr.2003.10.022. [DOI] [PubMed] [Google Scholar]
- 60.Jiang N, Wang X, Jhanwar-Uniyal M, Darzynkiewicz Z, Dai W. Polo box domain of Plk3 functions as a centrosome localization signal, overexpression of which causes mitotic arrest, cytokinesis defects, and apoptosis. J Biol Chem. 2006;281:10577–82. doi: 10.1074/jbc.M513156200. [DOI] [PubMed] [Google Scholar]
- 61.Wang L, Dai W, Lu L. Stress-induced c-Jun activation mediated by Polo-like kinase 3 in corneal epithelial cells. J Biol Chem. 2007;282:32121–7. doi: 10.1074/jbc.M702791200. [DOI] [PubMed] [Google Scholar]
- 62.Wang Q, Xie S, Chen J, Fukasawa K, Naik U, Traganos F, Darzynkiewicz Z, Jhanwar-Uniyal M, Dai W. Cell cycle arrest and apoptosis induced by human Polo-like kinase 3 is mediated through perturbation of microtubule integrity. Mol Cell Biol. 2002;22:3450–9. doi: 10.1128/MCB.22.10.3450-3459.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Li Z, Niu J, Uwagawa T, Peng B, Chiao PJ. Function of polo-like kinase 3 in NF-kappaB-mediated proapoptotic response. J Biol Chem. 2005;280:16843–50. doi: 10.1074/jbc.M410119200. [DOI] [PubMed] [Google Scholar]
- 64.Syed N, Smith P, Sullivan A, Spender LC, Dyer M, Karran L, O’Nions J, Allday M, Hoffman I, Crawford D, Griffin B, Farrel PJ, Crook T. Transcriptional silencing of Polo-like kinase 2 (SNK/PLK2) is a frequent event in B-cell malignancies. Blood. 2006;107:250–6. doi: 10.1182/blood-2005-03-1194. [DOI] [PubMed] [Google Scholar]
- 65.Mancini M, Machamer CE, Roy S, Nicholson DW, Thornberry NA, Casciola-Rosen LA, Rosen A. Caspase-2 is localized at the Golgi complex and cleaves golgin-160 during apoptosis. J Cell Biol. 2000;149(3):603–12. doi: 10.1083/jcb.149.3.603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Chiu R, Novikov L, Mukherjee S, Shields D. A caspase cleavage fragment of p115 induces fragmentation of the Golgi apparatus and apoptosis. J Cell Biol. 2002;159(4):637–48. doi: 10.1083/jcb.200208013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lane JD, Lucocq J, Pryde J, Barr FA, Woodman PG, Allan VJ, Lowe M. Caspase-mediated cleavage of the stacking protein GRASP65 is required for Golgi fragmentation during apoptosis. J Cell Biol. 2002;156(3):495–509. doi: 10.1083/jcb.200110007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Tinel A, Tschopp J. The PIDDosome, a protein complex implicated in activation of caspase-2 in response to genotoxic stress. Science. 2004;304(5672):843–6. doi: 10.1126/science.1095432. [DOI] [PubMed] [Google Scholar]
- 69.Maag RS, Mancini M, Rosen A, Machamer CE. Caspase-resistant Golgin-160 disrupts apoptosis induced by secretory pathway stress and ligation of death receptors. Mol Biol Cell. 2005;16(6):3019–27. doi: 10.1091/mbc.E04-11-0971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hauser HP, Bardroff M, Pyrowolakis G, Jentsch S. A giant ubiquitin-conjugating enzyme related to IAP apoptosis inhibitors. J Cell Biol. 1998;141(6):1415–22. doi: 10.1083/jcb.141.6.1415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Malisan F, Testi R. GD3 ganglioside and apoptosis. Biochim Biophys Acta. 2002;1585(2–3):179–87. doi: 10.1016/s1388-1981(02)00339-6. [DOI] [PubMed] [Google Scholar]
- 72.Rippo MR, Malisan F, Ravagnan L, Tomassini B, Condo I, Costantini P, Susin SA, Rufini A, Todaro M, Kroemer G, Testi R. GD3 ganglioside directly targets mitochondria in a bcl-2-controlled fashion. FASEB J. 2000;14(13):2047–54. doi: 10.1096/fj.99-1028com. [DOI] [PubMed] [Google Scholar]
- 73.Tomassini B, Malisan F, Franchi L, Nicolo C, Calvo GB, Saito T, Testi R. Calnexin suppresses GD3 synthase-induced apoptosis. FASEB J. 2004;18(13):1553–5. doi: 10.1096/fj.04-1675fje. [DOI] [PubMed] [Google Scholar]
- 74.Cristea IM, Degli Esposti M. Membrane lipids and cell death: an overview. Chem Phys Lipids. 2004;129(2):133–60. doi: 10.1016/j.chemphyslip.2004.02.002. [DOI] [PubMed] [Google Scholar]
- 75.Bennett M, Macdonald K, Chan SW, Luzio JP, Simari R, Weissberg P. Cell surface trafficking of Fas: a rapid mechanism of p53-mediated apoptosis. Science. 1998;282(5387):290–3. doi: 10.1126/science.282.5387.290. [DOI] [PubMed] [Google Scholar]
- 76.Arvan P, Zhao X, Ramos-Castaneda J, Chang A. Secretory pathway quality control operating in Golgi, plasmalemmal, and endosomal systems. Traffic. 2002;3(11):771–80. doi: 10.1034/j.1600-0854.2002.31102.x. [DOI] [PubMed] [Google Scholar]
- 77.Linder S, Shoshan MC. Lysosomes and endoplasmic reticulum: targets for improved, selective anticancer therapy. Drug Resist Updat. 2005;8(4):199–204. doi: 10.1016/j.drup.2005.06.004. [DOI] [PubMed] [Google Scholar]
- 78.Carew JS, Nawrocki ST, Krupnik YV, Dunner K, McConkey DJ, Keating MJ, et al. Targeting endoplasmic reticulum protein transport: a novel strategy to kill malignant B cells and overcome fludarabine resistance in CLL. Blood. 2006;107(1):222–231. doi: 10.1182/blood-2005-05-1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Jaattela M. Multiple cell death pathways as regulators of tumour initiation and progression. Oncogene. 2004;23(16):2746–56. doi: 10.1038/sj.onc.1207513. [DOI] [PubMed] [Google Scholar]
- 80.Carew JS, Nawrocki ST, Xu RH, Dunner K, McConkey DJ, Wierda WG, et al. Increased mitochondrial biogenesis in primary leukemia cells: the role of endogenous nitric oxide and impact on sensitivity to fludarabine. Leukemia. 2004;18(12):1934–40. doi: 10.1038/sj.leu.2403545. [DOI] [PubMed] [Google Scholar]
- 81.Donaldson JG, Cassel D, Kahn RA, Klausner RD. ADP-ribosylation factor, a small GTP-binding protein, is required for binding of the coatomer protein beta-COP to Golgi membranes. Proc Natl Acad Sci U S A. 1992;89(14):6408–12. doi: 10.1073/pnas.89.14.6408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Cheung HH, Lynn Kelly N, Liston P, Korneluk RG. Involvement of caspase-2 and caspase-9 in endoplasmic reticulum stress-induced apoptosis: a role for the IAPs. Exp Cell Res. 2006;312(12):2347–57. doi: 10.1016/j.yexcr.2006.03.027. [DOI] [PubMed] [Google Scholar]
- 83.Shao RG, Shimizu T, Pommier Y. Brefeldin A is a potent inducer of apoptosis in human cancer cells independently of p53. Exp Cell Res. 1996;227(2):190–6. doi: 10.1006/excr.1996.0266. [DOI] [PubMed] [Google Scholar]
- 84.Wallen E, Sellers RG, Peehl DM. Brefeldin A induces p53-independent apoptosis in primary cultures of human prostatic cancer cells. Urol. 2000;164:836–41. doi: 10.1097/00005392-200009010-00058. [DOI] [PubMed] [Google Scholar]
- 85.Salles FT, Hespanhol AM, Jaeger RG, Marques MM. Brefeldin-A induces apoptosis in human adenoid cystic carcinoma cultured cells. Oral Oncol. 2004;40(6):585–90. doi: 10.1016/j.oraloncology.2003.12.007. [DOI] [PubMed] [Google Scholar]
- 86.Boya P, Cohen I, Zamzami N, Vieira HL, Kroemer G. Endoplasmic reticulum stress-induced cell death requires mitochondrial membrane permeabilization. Cell Death Differ. 2002;9(4):465–7. doi: 10.1038/sj.cdd.4401006. [DOI] [PubMed] [Google Scholar]
- 87.Wlodkowic D, Skommer J, Pelkonen J. Brefeldin A triggers apoptosis associated with mitochondrial breach and enhances HA14-1- and anti-Fas-mediated cell killing in follicular lymphoma cells. Leukemia Res. 2007;31(12):1687–700. doi: 10.1016/j.leukres.2007.03.008. [DOI] [PubMed] [Google Scholar]
- 88.Sausville EA, Duncan KL, Senderowicz A, Plowman J, Randazzo PA, Kahn R, Melpeis L, Grever MR. antiproliferative effect in vitro and antitumor activity in vivo of brefeldin A. Cancer J Sci Am. 1996;2(1):52–8. [PubMed] [Google Scholar]
- 89.Phillips LR, Supko JG, Malspeis L. Analysis of Brefeldin A in plasma by gas chromatography with electron capture. Anal Biochem. 1993;211:16–22. doi: 10.1006/abio.1993.1225. [DOI] [PubMed] [Google Scholar]
- 90.Phillips LR, Wolfe TL, Malspeis L, Supko JG. Analysis of brefeldin A and the prodrug breflate in plasma by gas chromatography with mass selective detection. J Pharm Biomed Anal. 1998;16(8):1301–9. doi: 10.1016/s0731-7085(97)00142-8. [DOI] [PubMed] [Google Scholar]
- 91.Brown JM, Attardi LD. The role of apoptosis in cancer development and treatment response. Nat Rev Cancer. 2005;5(3):231–7. doi: 10.1038/nrc1560. [DOI] [PubMed] [Google Scholar]
- 92.Green DR, Kroemer G. Pharmacological manipulation of cell death: clinical applications in sight? J Clin Invest. 2005;115(10):2610–7. doi: 10.1172/JCI26321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Skommer J, Wlodkowic D, Deptala A. Larger than life: Mitochondria and the Bcl-2 family. Leuk Res Leuk Res. 2007 Mar;31(3):277–86. doi: 10.1016/j.leukres.2006.06.027. [DOI] [PubMed] [Google Scholar]
- 94.Chapman JR, Tazaki H, Mallouh C, Konno S. Mechanism of Brefeldin A-Induced Growth Inhibition and Cell Death in Human Prostatic Carcinoma Cells. Mol Urol. 1999;3(1):11–16. [PubMed] [Google Scholar]
- 95.Tinhoffer I, Anether G, Senfter M, Pfaller K, Bernhard D, Hara M, Greil R. Stressful death of TALL tumor cells after treatment with the anti-tumor agent Tetrocarcin-A. FASEB J. 2002;16:1295–1297. doi: 10.1096/fj.02-0020fje. [DOI] [PubMed] [Google Scholar]
- 96.Anether G, Tinhoffer I, Greil R. Tetrocarcin-A-induced ER stress mediates apoptosis in B-CLL cells via a Bcl-2-independent pathway. Blood. 2003;101(11):4561–4567. doi: 10.1182/blood-2002-08-2501. [DOI] [PubMed] [Google Scholar]
- 97.Shiraishi T, Yoshida T, Nakata S, Horinaka M, Wakada M, Mizutani Y, Miki T, Sakai T. Tunicamycin enhances tumor necrosis factor-related apoptosis-inducing ligand-induced apoptosis in human prostate cancer cells. Cancer Res. 2005;65(14):6364–70. doi: 10.1158/0008-5472.CAN-05-0312. [DOI] [PubMed] [Google Scholar]
- 98.Denmeade SR, Isaacs JT. The SERCA pump as a therapeutic target: making a “smart bomb” for prostate cancer. Cancer Biol & Ther. 2005;4(1):14–22. doi: 10.4161/cbt.4.1.1505. [DOI] [PubMed] [Google Scholar]
- 99.Nawrocki ST, Carew JS, Pino MS, Highshaw RA, Dunner K, Huang P, Abbruzzese JL, McConkey DJ. Bortezomib sensitizes pancreatic cancer cells to endoplasmic reticulum stress-mediated apoptosis. Cancer Res. 2005;65(24):11658–11666. doi: 10.1158/0008-5472.CAN-05-2370. [DOI] [PubMed] [Google Scholar]
- 100.Hoeller D, Dikic I. Targeting the ubiquitin system in cancer therapy. Nature. 2009;26;458(7237):438–44. doi: 10.1038/nature07960. [DOI] [PubMed] [Google Scholar]
- 101.Bennett MK, Kirk CJ. Development of proteasome inhibitors in oncology and autoimmune diseases. Curr Opin Drug Discov Devel. 2008;11(5):616–25. [PubMed] [Google Scholar]
- 102.Burger AM, Seth AK. The ubiquitin-mediated protein degradation pathway in cancer: therapeutic implications. Eur J Cancer. 2004;40(15):2217–29. doi: 10.1016/j.ejca.2004.07.006. [DOI] [PubMed] [Google Scholar]
- 103.Hill DS, Martin S, Armstrong JL, Flockhart R, Tonison JJ, Simpson DG, Birch-Machin MA, Redfern CP, Lovat PE. Combining the endoplasmic reticulum stress-inducing agents bortezomib and fenretinide as a novel therapeutic strategy for metastatic melanoma. Clin Cancer Res. 2009;15(4):1192–8. doi: 10.1158/1078-0432.CCR-08-2150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Wang Q, Mora-Jensen H, Weniger MA, Perez-Galan P, Wolford C, Hai T, Ron D, Chen W, Trenkle W, Wiestner A, Ye Y. ERAD inhibitors integrate ER stress with an epigenetic mechanism to activate BH3-only protein NOXA in cancer cells. Proc Natl Acad Sci U S A. 2009;106(7):2200–5. doi: 10.1073/pnas.0807611106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Boelens J, Lust S, Offner F, Bracke ME, Vanhoecke BW. The endoplasmic reticulum: a target for new anticancer drugs. In Vivo. 2007;21(2):215–26. [PubMed] [Google Scholar]