

Abstract
Although it is well established that exocytosis of neurotransmitters and hormones is highly regulated by numerous secretory proteins, such as SNARE proteins, there is an increasing appreciation of the importance of the chemophysical properties and organization of membrane lipids to various aspects of the exocytotic program. Based on amperometric recordings by carbon fiber microelectrodes, we show that deprivation of membrane cholesterol by methyl-β-cyclodextrin not only inhibited the extent of membrane depolarization-induced exocytosis, it also adversely affected the kinetics and quantal size of vesicle fusion in neuroendocrine PC12 cells. In addition, total internal fluorescence microscopy studies revealed that cholesterol depletion impaired vesicle docking and trafficking, which are believed to correlate with the dynamics of exocytosis. Furthermore, we found that free cholesterol is able to directly trigger vesicle fusion, albeit with less potency and slower kinetics as compared to membrane depolarization stimulation. These results underscore the versatile roles of cholesterol in facilitating exocytosis.
Introduction
Intensive research in the past four decades has revealed that the regulated secretion, or exocytosis, of neurotransmitters and hormones is achieved through the temporally and spatially coordinated actions of numerous secretory proteins (1). Most notably, complexing between SNARE proteins, involving VAMP on the vesicle and plasma membrane-associated syntaxin and SNAP-25, is believed to be essential for enabling membrane fusion (2). Although this classic protein-centric view is well established, there is an increasing appreciation of the importance of the chemophysical properties and organization of membrane lipids to various aspects of the exocytotic program (3–6). In particular, growing evidence suggests that cholesterol, an abundant and essential constituent of both plasma and vesicular membranes, is a critical regulator of exocytosis through collaboration with protein factors (7–14).
It has been demonstrated in several cell types that cholesterol acts as a prefusion organizer by stabilizing lipid membrane microdomains or rafts, where secretory proteins, particularly SNAREs, assemble to define vesicle docking and fusion sites (9,15–17). Cholesterol may promote membrane anchoring of the secretory proteins to fusion sites via cholesterol-recognition elements on proteins (18). Cholesterol deprivation disrupts cholesterol-enriched functional microdomains and inhibits exocytosis in these cells. Cholesterol is also an important factor in determining membrane properties such as fluidity, rigidity, and curvature (13,19,20). Modulation of cholesterol content in the plasma membrane changes the energy barrier for coalescence between two lipid bilayers and consequently alters the vesicle fusion kinetics (21). Furthermore, cholesterol may exert its influence on exocytosis indirectly by modulating the actin cytoskeleton (22,23).
Based on amperometric recordings by carbon fiber microelectrodes, we show that deprivation of membrane cholesterol by methyl-β-cyclodextrin (MβCD) not only inhibited the extent of membrane depolarization-induced exocytosis, it also adversely affected the kinetics and quantal size of vesicle fusion in neuroendocrine PC12 cells. In addition, total internal fluorescence microscopy (TIRFM) studies revealed that cholesterol depletion impaired vesicle docking and trafficking, which are believed to correlate with the dynamics of exocytosis (24). Furthermore, we found that free cholesterol is able to directly trigger vesicle fusion, albeit with less potency and slower kinetics as compared to membrane depolarization stimulation. These results underscore the versatile roles played by cholesterol in facilitating exocytosis.
Materials and Methods
The materials and methods used in this study are described in the Supporting Material.
Results
The frequency of exocytotic events is reduced by cholesterol depletion
MβCD sequesters cholesterol in its hydrophobic pocket (25) and has been widely used to remove cholesterol from the plasma membrane. To examine the effects of cholesterol depletion on exocytosis, we compared high K+-triggered exocytosis from the same cell before and after MβCD treatment based on amperometric measurements. Amperometry, in contrast to electrophysiological membrane capacitance measurements (26,27), directly detects exocytosis with single-vesicle resolution without interference by endocytosis. It is especially useful to reveal, with millisecond resolution, the details of the vesicle fusion kinetics catalyzed by fusion proteins (28).
As shown in Fig. 1 A (top), superfusion of the cell with high K+ solution for 2 min by means of an application pipette, which depolarizes the cell membrane, gave rise to many amperometric current spikes as detected by the carbon fiber microelectrode. Each current spike corresponds to the release of catecholamine molecules from a single secretory vesicle upon its fusion with the plasma membrane. In comparison, the same cell responded to high K+ stimulation with a much-reduced number of amperometric signals after incubation with 5 mM MβCD for 30 min (Fig. 1 A, middle). The MβCD treatment removed 32.3% ± 2.8% (three cell cultures; see Fig. S1 of the Supporting Material) of membrane cholesterol, which is comparable to previous findings (10). The average time courses of the exocytotic responses (14 cells) to the first and second stimulations were normalized to the average total number of spikes resulting from the first high K+ stimulation before 5 mM MβCD treatment (Fig. 1 A, bottom). Exocytosis elicited by high K+ stimulation after cholesterol deprivation was reduced to 39.9% ± 16.0% of the normal response. This is consistent with the results of a previous study, in which a statistical comparison was performed between groups of control and MβCD-treated cells (16). Cholesterol depletion also affected the kinetics of exocytosis. The accumulative number of amperometric spikes increased exponentially with a significantly slower time constant (>140 s) in MβCD-treated cells compared to controls (∼40 s; Fig. 1 A, bottom).
Figure 1.
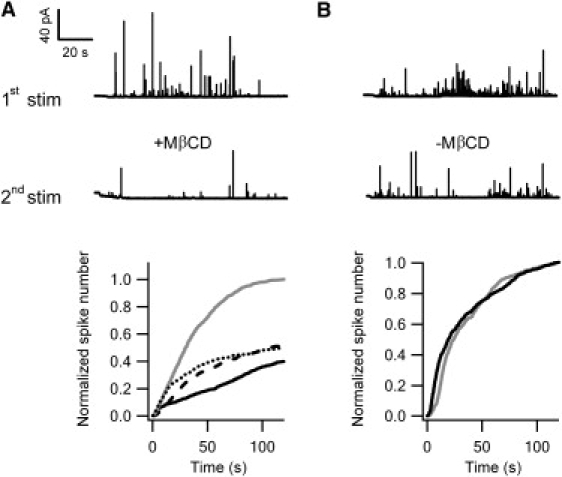
Cholesterol depletion inhibits exocytosis. The amperometric responses from two PC12 cells to high K+ stimulation are shown in the top row in A and B. The amperometric responses to the second high K+ stimulation from the same cells, with (A) or without (B) treatment with 5 mM MβCD for 30 min, are displayed in the middle row. The average time courses of the accumulative number of elicited amperometric spikes to the first (gray) and second (solid) stimulations are shown in pairs for the two protocols and are normalized to the average total spike number resulting from the first stimulation (bottom row). These traces are averaged from (A) 14 cells in which the average spike numbers in response to the first and second stimulations after 30 min incubation of 5 mM MβCD are 49.9 ± 7.5 and 19.9 ± 4.4, respectively (p < 0.001); (B) 11 cells in which the average spike numbers in response to the first and second stimulations after 30 min and without application of MβCD are 45.3 ± 3.8 and 45.5 ± 11.8, respectively. The dashed curve in A is the response (26.0 ± 5.1 events) from the cells (n = 17) whose membrane cholesterol was first depleted by 5 mM MβCD and then reloaded by the application of 5 mM MβCD-cholesterol complex; it is normalized to the average response to high K+ stimulation without any treatments. Replenishment of cholesterol provided only insignificant exocytotic recovery (p > 0.05, dashed versus solid curve). The dotted curve in A is the average response from 15 cells cultured in LPDS medium (28.1 ± 7.9 events) normalized by the average response from 15 cells cultured in FBS medium (56.1 ± 6.5 events; p < 0.001 between the two cell cultures).
Since the second stimulation was applied 30 min after the first, however, it can be argued that the observed decrease in the frequency of exocytotic events may be simply due to deterioration of cells. This possibility was ruled out by control experiments (11 cells), which showed that the paired high K+ stimulations, 30 min apart and without MβCD application, elicited similar exocytotic responses (Fig. 1 B). When MβCD is preloaded with cholesterol to saturation, it can be used as a cholesterol donor to replenish cholesterol in the cell membrane (12). Using this strategy, cholesterol molecules carried by a 5 mM MβCD-cholesterol complex were reloaded back into the cells after initial MβCD depletion. After reloading was completed, the level of membrane cholesterol was elevated to 167.6% ± 25.4% of the normal level without MβCD depletion (three cell cultures; Fig. S1), similar to previously reported values (29). Despite the full recovery of the membrane cholesterol level, however, the rescue of MβCD-induced inhibition by cholesterol reloading was insignificant (dashed curve in Fig. 1 A). This raises a concern about the specificity of the MβCD treatment.
As an alternative strategy, cellular cholesterol was metabolically deprived (30). Specifically, cells were cultured in a medium supplemented with lipoprotein-deficient serum (LPDS) in place of fetal bovine serum (FBS) 4–6 days before the experiments to block cholesterol synthesis and deplete cholesterol stores (31). In the LPDS-cultured cells, exocytotic responses triggered by high K+ were significantly reduced to ∼50% of those in the FBS-cultured cells (Fig. 1 A, bottom), similar to the consequence caused by MβCD. This experiment corroborates the notion that cholesterol is crucial in supporting exocytosis.
The rate and quantal size of vesicular release, and prespike foot are reduced by cholesterol depletion
It is also evident from Fig. 1 that the amplitudes of the amperometric events became generally smaller after depletion of membrane cholesterol. This suggests that cholesterol is involved in the process of quantal vesicle fusion. To investigate this phenomenon, individual amperometric signals in the same experiments presented in Fig. 1 were extracted and analyzed. The normalized averaged amperometric spikes from the first and second stimulations are displayed in pairs for the two different experimental protocols in Fig. 2 A. The average amplitude of the amperometric spikes after cholesterol depletion was decreased to 62.6% ± 4.6%. In line with this, the rate of release, or the rate of fusion pore expansion, which is quantified by the linear slope as the amperometric spike rises from 35% to 90% of the peak amplitude, is reduced to 56.0% ± 6.5% (Fig. 2 C). The quantal size of vesicle release (Q), which is the total charge (integration) of the amperometric current spike, was also reduced to 61.9% ± 3.9% by cholesterol depletion (Fig. 2 D). Again, the control experiments confirmed that these observations were not due to deterioration of cells. The average amplitude, rise slope, and quantal size resulting from the second high K+ stimulation in the control cells were even slightly enhanced, possibly because of activity-dependent potentiation. Replenishment of cholesterol in the cholesterol-depleted cells completely rescued the reductions in amperometric amplitude, fusion rate, and quantal size (Fig. 2, B–D), suggesting that our observations were due to specific effects of MβCD on cholesterol depletion. In addition, when cholesterol was depleted by LPDS culturing, the rise slope and quantal size also decreased to 29.4% ± 2.1% and 71.7% ± 3.1%, respectively (Fig. 2, B–D).
Figure 2.
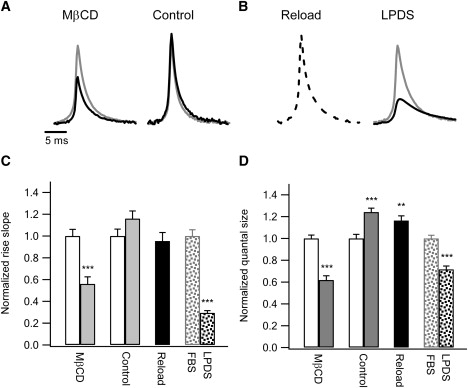
Cholesterol depletion impairs vesicle fusion. Amperometric spikes obtained in the experiments presented in Fig. 1 were individually extracted and analyzed. (A) The average spikes of the amperometric responses to the first (gray) and second (solid) stimulations are shown in pairs for the two protocols. The amplitudes for the two paired spikes are normalized based on the average amperometric amplitude responding to the first stimulation, which is 17.31 ± 0.69 pA (left, 5 mM MβCD protocol) or 21.69 ± 0.96 pA (right, control). (B) The average spike from the cells replenished with cholesterol after initial depletion is shown as the dashed trace on the left. Its amplitude (20.03 ± 1.14 pA, 17 cells) is normalized to the amplitude of the gray trace shown in A (left). Average spikes from cells cultured in FBS medium (gray, 16.40 ± 0.66 pA) or LPDS medium (solid, 5.40 ± 0.21 pA) are shown in pairs on the right. (C) The statistics of the rise slope of vesicle fusion defined by the linear slope between 35% and 90% of the peak amperometric amplitude. In the 5 mM MβCD protocol (the first paired columns on the left, normalized to the response from the first stimulation), the rise slopes are 10.10 ± 0.62 pA/ms (blank) and 5.66 ± 0.66 pA/ms (gray), from the first and second stimulations, respectively. In the control experiments (the second paired columns, normalized to the response from the first stimulation), the rise slopes are 14.01 ± 0.89 pA/ms (blank) and 16.25 ± 1.00 pA/ms (gray), from the first and second stimulations, respectively. The rise slope after reloading of cholesterol (solid, normalized to the first response in the MβCD protocol) is 9.63 ± 0.80 pA/ms. Rise slopes from FBS-medium cultured cells (dotted gray, 10.54 ± 0.61 pA/ms) and LPDS-medium cultured cells (dotted, 3.10 ± 0.22 pA/ms) are normalized and displayed in pairs on the right. (D) The statistics of the quantal size defined by the total charge integration of the amperometric current spike. The data are normalized as described in C. In the 5 mM MβCD protocol (the first paired columns), the quantal sizes are 64.22 ± 2.01 fC (blank) and 39.76 ± 2.51 fC (gray), from the first and second stimulations, respectively. In the control experiments (the second paired columns), the quantal sizes are 60.34 ± 2.28 fC (blank) and 74.88 ± 2.19 fC (gray), from the first and second stimulations, respectively. The quantal size after reloading of cholesterol (solid) is 74.75 ± 2.78 fC. Quantal sizes from FBS-medium cultured cells (dotted gray, 74.14 ± 2.20 fC) and LPDS-medium cultured cells (dotted, 53.13 ± 2.28 fC) are normalized and displayed in pairs on the right. ∗∗p < 0.01, ∗∗∗p < 0.001.
A substantial energy barrier needs to be overcome to enable fusion between the vesicular and plasma membranes. When the driving force provided by fusion proteins barely overcomes such an energy barrier, a small foot preceding the amperometric spike occurs as a result of slow expansion of the fusion pore (32). As shown in Fig. S5, cholesterol depletion by MβCD or metabolic depletion reduced the percentage of footed events and the foot size.
Vesicle fusion time is increased after cholesterol depletion
In a previous study of rat chromaffin cells (33), it was suggested that small and large vesicles may differ in terms of the molecular machinery (i.e., the number and/or composition of proteins and/or lipids) required to drive fusion pore expansion. For small vesicles, the halfwidth time of the amperometric spike (t1/2), which indicates the duration of vesicle fusion, linearly scales with the cubic root of the quantal size (Q1/3), which reflects the vesicle size. For large vesicles, t1/2 levels off at a plateau, meaning that large vesicles dilate relatively more rapidly. It has been speculated that additional fusion machines are accommodated in the fusion complex of large vesicles and account for their expedited fusion (33). As shown in Fig. 3 (open circles), a similar trend was also seen in PC12 cells. Specifically, for vesicles > 3.5 fC1/3, t1/2 no longer increases linearly with Q1/3, but instead remains at ∼3.2 ms. Of interest, the capping of t1/2 was relieved by MβCD, i.e., a linear t1/2 and Q1/3 relation resulted (Fig. 3, gray circles). These data suggest that cholesterol is critical for ensuring the quick release of large vesicles. Moreover, for a given vesicle size (Q1/3), the cholesterol-depleted cells took a longer time to discharge their contents, and this effect was more prominent for large vesicles. The MβCD-induced slowdown in vesicle fusion is caused by depletion of membrane cholesterol rather than nonspecific actions of MβCD, because t1/2 and its saturation at large Q1/3 were recovered after the cholesterol-depleted cells were reloaded with cholesterol (Fig. 3, solid circles). Metabolic depletion of cholesterol by LPDS medium similarly impaired the fast fusion of large vesicles, and increased the fusion time to an even greater extent as compared to MβCD.
Figure 3.
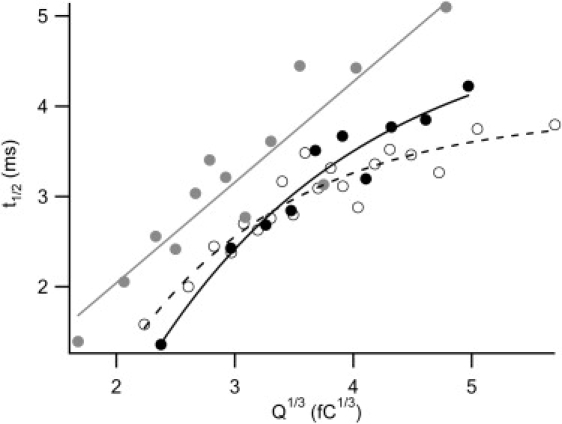
Relation between the halfwidth time of the amperometric spike (t1/2) and the cubic root of quantal size (Q1/3) is shown. These analyses are based on the amperometric signals obtained from the same experiments presented in Figs. 1 and 2. Each open circle represents the average of 80 vesicle fusion events in response to high K+ stimulation without MβCD treatment. These data points can be well fitted by an exponential (dashed curve). Each solid gray circle represents the average of 20-vesicle fusion in response to high K+ stimulation after a 30-min treatment with 5 mM MβCD. To indicate the trend, the gray circles are fitted by a line (gray line). Each solid circle represents the average of 40-vesicle fusion from the cells whose membrane cholesterol was reloaded after depletion. The data is fitted by an exponential (solid curve).
Exogenous cholesterol as secretagogue
Cholesterol not only makes exocytosis and vesicle fusion more efficient, it can also directly stimulate exocytosis after insertion into the exoplasmic leaflet of the plasma membrane. As demonstrated in Fig. 4 A, amperometric signals appeared after local superfusion with a cholesterol-containing bath solution (2 μM diluted from 2 mM cholesterol-ethanol stock) to cells via an application pipette. The vehicle control (0.1% v/v ethanol in bath) was not able to elicit any response (Fig. S4). The average increases in the accumulative spike number over time, in response to 2 min of cholesterol stimulation (solid curve; 22 cells) and high K+ stimulation in parallel experiments (gray curve; 10 cells), are depicted in Fig. 4 B. Notably, cholesterol stimulation is much less potent than high K+ stimulation.
Figure 4.
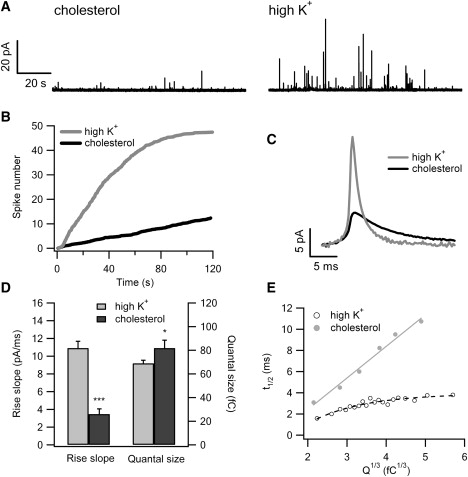
Free cholesterol triggers exocytosis. (A) Local application of 2 μM cholesterol to the cell for 1 min elicited amperometric responses (left). A high K+ triggered amperometric recording in a parallel experiment on a different cell is shown (right) for comparison. (B) The average increase of accumulative spike number over time, in response to cholesterol stimulation (solid curve; 22 cells) or high K+ stimulation (gray curve; 10 cells). (C) The average amperometric spikes from cholesterol stimulation (solid; 272 events) and high K+ (gray; 474 events). (D) The rise slope of cholesterol-induced vesicle fusion (solid column on the left; 3.46 ± 0.59 pA/ms) is smaller than that of high K+-induced fusion (gray column on the left; 10.90 ± 0.77 pA/ms), whereas the quantal size of cholesterol-induced fusion (solid column on the right; 81.82 ± 6.76 fC) is larger than that of high K+-induced fusion (gray column on the right; 68.91 ± 2.67 fC). (E) For cholesterol-induced vesicle fusion, the halfwidth time of the amperometric spike (t1/2) is linearly proportional to the cubic root of the quantal size (Q1/3; gray circles fitted by a line; each circle represents the average from 40 vesicles). For a given Q1/3 (vesicle size), cholesterol-triggered fusion takes longer to release compared to high K+-induced fusion (open circles fitted by an exponential; same data set as shown in Fig. 4). ∗p < 0.05, ∗∗∗p < 0.001.
The average amperometric signals from 272 cholesterol-induced events and 474 high K+-induced events are plotted in Fig. 4 C. Compared to that induced by high K+, cholesterol-induced vesicle fusion manifested much slower kinetics. Specifically, the rise slope of cholesterol-induced fusion (3.47 ± 0.59 pA/ms) was much smaller than that of high K+-induced fusion (10.90 ± 0.77 pA/ms), indicating a slower rate of fusion pore expansion (Fig. 4 D). The quantal size of cholesterol-induced fusion, however, is larger than that of the high K+-induced fusion events (81.82 ± 6.76 vs. 68.91 ± 2.67 fC; Fig. 4 D). In the case of cholesterol stimulation, the fusion pore opening (t1/2) is linearly proportional to the vesicle size (Q1/3; Fig. 4 E), and for the same vesicle size, cholesterol-induced vesicle fusion took a much longer time (t1/2) to release as compared to high K+-stimulated fusion. Insertion of exogenous cholesterol by micropipette delivery of cholesterol-loaded MβCD also induced similar amperometric responses with slow kinetics (Fig. S6).
The ability of cholesterol to elicit vesicle fusion was confirmed by total internal reflection fluorescence microscopy (TIRFM; Movie S1). With selective evanescent illumination of the thin section (∼200 nm) just above the interface between the glass coverslip and the adhered cell, TIRFM can provide insights into dynamic events occurring at or close to the plasma membrane of living cells, with outstanding optical contrast and resolution. Subplasmalemmal secretory vesicles in PC12 cells, specifically labeled by neuropeptide Y conjugated with enhanced green fluorescent protein (NPY-EGFP), were individually visualized and tracked at 0.5 s intervals with the use of TIRFM.
Fig. 5 A presents a pair of TIRFM images of a PC12 cell immediately before (top) and 100 s after (bottom) the application of 2 μM cholesterol to the bath solution. Vesicles in the subplasmalemmal region can be clearly resolved. In this experiment, the total number of the original subplasmalemmal vesicles was reduced from 19 to four in <2 min. In the resting state, the total number of vesicles in the subplasmalemmal region stayed constant (open circles in Fig. 5 B; average from five cells). In contrast, after the application of cholesterol, the total vesicle number decreased over time (solid circles in Fig. 5 B; average from six cells). The delivery rate of newly arrived vesicles (solid triangles in Fig. 5 B) was enhanced in the presence of free cholesterol, presumably to compensate for vesicle release.
Figure 5.
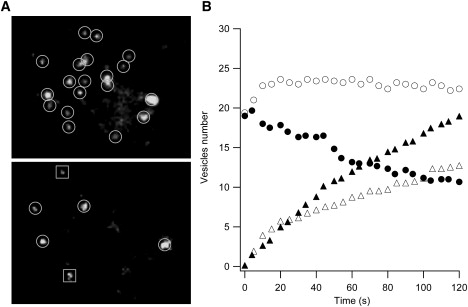
Cholesterol-stimulated vesicle release revealed by TIRFM. (A) NPY-EGFP-labeled subplasmalemmal vesicles are visualized by TIRFM before (top) and 100 s after (bottom) application of 2 μM cholesterol to the bath solution. The vesicles are highlighted by circles or squares. The squares indicate vesicles newly arrived from the cytosol. (B) The total number of visible subplasmalemmal vesicles decreased over time in the presence of cholesterol (solid circles; average from six cells), whereas it remained stable without cholesterol (open circles; average from five cells). The accumulative number of newly arrived vesicles was enhanced in the presence of cholesterol (solid triangles) compared to that without cholesterol (open triangles).
Cholesterol depletion impairs vesicle trafficking and docking
Secretory vesicles undertake constant lateral movements in the subplasmalemmal membrane region, where they explore, interact with various proteins, and eventually are released at exocytotic sites upon triggering. How vesicles move in the near membrane region directly relates to the dynamics of exocytosis (24). The lateral motion of individual vesicles was tracked, at 0.5 s intervals for 2 min, from their appearance at the beginning of imaging (for “predocked” vesicles) or from their arrival to the subplasmalemmal region (for newly arrived vesicles) until their retrieval back to the inner cytosol. Fig. 6 A presents the trajectories of vesicle lateral motion in a typical control (left) and an MβCD-treated PC12 cell (right). An example trajectory from the control cell is illustrated with an expanded scale (middle). From this representative trace, and as demonstrated in our previous study (34) and by others (35,36), it is clear that random vesicle lateral movement is not free Brownian motion; rather, it is confined as if the vesicle were “caged” by certain physical barriers or tethering interactions. Of interest, vesicle lateral motion was more severely restricted in the cholesterol-depleted cells (Movie S2) as compared to the control cells (Movie S3).
Figure 6.
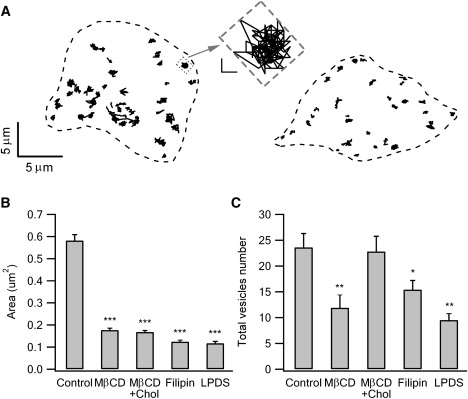
Vesicle trafficking and docking is cholesterol-dependent. (A) The motion trajectories of the subplasmalemmal vesicles in a control cell (left) and a cell depleted of cholesterol by 5 mM MβCD (right). The cell contours are outlined by dashed lines. An example trajectory from the control cell is illustrated with an expanded scale (the horizontal and vertical scale bars indicate 200 nm). The smallest rectangle that just encases all the vesicle footprints (gray and dashed rectangle) gives the first-order estimation of the coverage area of vesicle motion. (B and C) The statistics (mean ± SE) of the vesicle motion area and the number of subplasmalemmal vesicles in control and differently treated cells (see text). ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
The area of the smallest rectangle that just encases all the vesicle footprints gives a first-order estimation of vesicle motion coverage, as indicated in Fig. 6 A (middle). In the cholesterol-depleted cells, the average area of motion coverage was 0.18 ± 0.01 μm2 (466 vesicles in 16 cells), i.e., only ∼31% of that in control cells (0.58 ± 0.03 μm2, averaged from 509 vesicles in 10 cells; Fig. 6 B). The analyses included all “predocked” and newly arrived vesicles. Vesicles also transit vertically between the inner cytosol and the subplasmalemmal region, such that the vesicles near the membrane are recycled and replenished. However, the total vesicle number in the subplasmalemmal region remains stable (Fig. 5 B, open circles) due to the balance between vesicle arrival and retrieval. As also revealed by TIRFM, the number of subplasmalemmal vesicles was significantly smaller in the MβCD-treated cells than in the control cells (23.6 ± 2.7 vs. 11.9 ± 2.6; Fig. 6 C), indicating that the removal of cholesterol impaired vesicle tethering and docking. Adding cholesterol back into the cholesterol-depleted cells restored the number of subplasmalemmal vesicles (22.8 ± 3.0, 16 cholesterol-reloaded cells; Fig. 6 C), implying the specificity of the cholesterol effect. On the other hand, replenishment of membrane cholesterol was unable to rectify the MβCD effect on vesicle motion (Fig. 6 B). The motion coverage of the cholesterol-replenished cells was 0.17 ± 0.01 μm2 (523 vesicles in 16 cells), similar to that in the MβCD treated cells. The reason for this is unknown, but it could be due to damage to the cholesterol-protein assemblies, after cholesterol depletion, that cannot be reversed by replacement of cholesterol.
As an alternative to MβCD treatment, the availability of membrane cholesterol was reduced by sequestration of cholesterol with filipin (37), which binds specifically to membrane cholesterol. The filipin treatment also led to a significant reduction in the motion area of the vesicles (0.12 ± 0.01 μm2, 328 vesicles in 17 cells) and the number of subplasmalemmal vesicles (15.4 ± 1.8). Furthermore, when cholesterol was depleted metabolically (by LPDS medium), the cells also manifested severely restricted vesicle motion (0.12 ± 0.01 μm2, 286 vesicles in 24 cells) and a reduced number of subplasmalemmal vesicles (9.5 ± 1.3), similar to the MβCD-treated and filipin-treated cells. The reduced vesicle motion was not correlated to the change in the vesicle dwell time. The average vesicle dwell times in the subplasmalemmal region for the untreated cells, the MβCD-treated cells, the cholesterol-reloaded cells, the filipin-treated cells, and the cells after metabolic deprivation of cholesterol were 36.8 ± 2.0 s, 25.5 ± 1.8 s, 44.3 ± 1.9 s, 62.6 ± 2.4 s, and 59.9 ± 2.6 s, respectively. Therefore, the reduced motion coverage is due to a reinforced confinement of vesicle movement imposed by certain cholesterol-dependent mechanisms.
The similar observations from three different protocols to decrease the availability of membrane cholesterol corroborate the notion that membrane cholesterol is critical for vesicle docking and trafficking. This could also provide an explanation for the observed effects of cholesterol depletion on the frequency and fusion kinetics of the exocytotic events.
Discussion
As a key constituent of the cell membranes and a precursor to steroid hormones, cholesterol is implicated in numerous structural and functional capacities. Therefore, cholesterol levels are highly regulated at both body and cellular levels. Abnormalities in its homeostasis can lead to various diseases, such as developmental abnormalities, neurodegenerative disease, or hormone disorders (8,38). It is becoming increasingly clear that cholesterol is critically involved in membrane fusion and exocytosis (10,16,21). In this study, we carefully examined and demonstrated the importance of cholesterol for various aspects of vesicular exocytosis in PC12 cells by 1), comparing exocytotic responses from the same cell before and after MβCD-induced cholesterol depletion to avoid the typically large cell-to-cell variation; 2), addressing concerns about the specificity of MβCD by replenishing cholesterol in the cell membrane and using alternative strategies for cholesterol deprivation; 3), analyzing in detail the effects of cholesterol on quantal vesicle fusion; 4), performing TIRFM imaging of vesicle docking and trafficking, which is directly related to the exocytotic competence and dynamics (24); and 5), demonstrating the ability of exogenous cholesterol to induce vesicle fusion.
Several lines of evidence have suggested the existence of membrane microdomains enriched with cholesterol and sphingolipids (39–42). These microdomains are usually called “lipid rafts” because they exist in a less fluid and more ordered state than glycerophospholipid-rich domains of the membrane. Small cholesterol molecules are thought to intercalate between hydrocarbon chains of sphingolipids, acting as a dynamic glue to stabilize the raft assembly. The ability of lipid rafts to concentrate specific proteins and exclude others makes them ideal functional platforms for spatially organizing cellular events with high efficiency on the plasma membrane. In view of the raft theory (42), it is plausible that a reduction of membrane cholesterol can disrupt raft structures and change the biological functions mediated by raft proteins. Cholesterol-rich microdomains have been proposed to be the sites of vesicle docking and fusion because proteins such as syntaxin and SNAP-25, which are critically implicated in exocytosis, have been demonstrated to associate with these domains (15,43), and cholesterol depletion dissociates SNAREs from rafts, leading to severe inhibition of exocytosis (9,16). Our observations that deprivation of membrane cholesterol by different means significantly reduced the number of membrane-tethered or -docked vesicles (Fig. 6 C) and the extent of Ca2+-triggered exocytosis (Fig. 1) lend further support to this hypothesis.
Depletion of membrane cholesterol led to markedly reduced lateral motion of the subplasmalemmal vesicles. This may partly account for the inhibition in exocytosis caused by cholesterol depletion, because the constant movement of the subplasmalemmal vesicles directly relates to the dynamics of exocytosis (24,34). Studies have demonstrated that depletion of membrane cholesterol by MβCD causes the formation of solid-like membrane domains and consequently impedes the diffusion of membrane proteins and lipids (44,45). The hindered protein diffusion may be related to the reduction in vesicle lateral motion, since subplasmalemmal vesicles are presumably tethered to the plasma membrane through a set of membrane-associated proteins (e.g., SNAREs) and move together with these protein complexes as a drifting raft. The ability of cholesterol to facilitate vesicle motion may help vesicles to explore sufficiently near the plasma membrane and become fully assembled to fusion competence. Since vesicle motion depends on the cortical actin network (46), cholesterol depletion could exert its effect by modulating actin polymerization (22). However, fluorescence staining and imaging of the cortical actin network using TIRFM and confocal imaging did not show any obvious differences between MβCD-treated and untreated cells (data not shown). Nonetheless, it is noteworthy that any possible alterations in the nanostructured actin meshwork with a size below the diffraction limit (∼150 nm) cannot be resolved by optical imaging.
It has been hypothesized that, before fusion, the outer leaflet of the vesicle membrane and the inner leaflet of the plasma membrane merge first while the distal membrane leaflets remain separated (47). This intermediate hemifusion structure is delicately stabilized by the proteins and particular lipid species until the final thrust, probably provided by twisting of the SNARE complex upon triggering, causes the formation of a small aqueous fusion pore (∼1.5 nm). Soon after formation, the fusion pore dilates rapidly to release the vesicular contents. The reduction in exocytosis by cholesterol depletion may be partly attributable to an inhibition of hemifusion formation, and thus decrease in the number of releasable vesicles.
In a previous study on PC12 cells (16), it was reported that cholesterol deprivation by MβCD reduced the extent of exocytosis but did not affect the quantal release. Here, we carefully revisited that issue by comparing the exocytotic events before and after MβCD treatment in the same cell. We found that depletion of cholesterol caused slower fusion kinetics and a smaller quantal size (Fig. 2). This is in accordance with the notion that negatively curved cholesterol (48) facilitates the formation and expansion of the fusion pore. The percentage and size of the foot signal were reduced by cholesterol depletion, indicating that the initial opening of the fusion pore was hindered. It is consistent with the view that depletion of cholesterol impairs the efficiency of the Ca2+-dependent fusion process (10). In addition, vesicles took a longer time to discharge their contents after cholesterol depletion (Fig. 3). In particular, the molecular machine that has been postulated to facilitate the fusion of large vesicles was disabled by cholesterol depletion. The roles of cholesterol in fusion therefore appear to be threefold, as it facilitates the formation of hemifusion structure, fusion pore expansion, and the assembly of the “additional” fusion machines in large vesicles.
Because the large dense core vesicles in neuroendocrine cells, and synaptic vesicles in neurons are small (<100 nm) and their vesicular fusion occurs rapidly (on the millisecond scale) with involvement of various protein and lipid factors, it is difficult to identify their hemifusion structure and determine how it transits to full fusion upon triggering. Other model systems have been utilized to study membrane fusion and the regulatory roles of cholesterol. Hemifused intermediate was identified in artificial liposomes by means of microspectrofluorometry (49). Garcia et al. (50) found that cholesterol stabilizes hemifused phospholipid bilayer vesicles. The same study, however, suggested that cholesterol inhibits transition from hemifusion to full fusion, contradicting our observation that removal of cholesterol slowed the fusion pore expansion. Homotypic fusion between cortical vesicles, which possess protein machinery that is critical for exocytosis and are much larger than large dense core vesicles and synaptic vesicles, has been used as a reduced model system for studying vesicular fusion (51,52). Churchward et al. (10,53) demonstrated that cholesterol, by virtue of its intrinsic negative curvature, promotes the formation of a highly curved fusion intermediate and Ca2+-triggered fusion pore formation and expansion. Fusion between cell membranes catalyzed by minimal fusion proteins (viral protein Hemagglutinin) is instrumental in investigating the roles of various lipids, such as cholesterol (54). A recent study (21) based on this technique corroborated the hypothesis that cholesterol acts during two stages in membrane fusion: before fusion pore opening and during fusion pore expansion.
It has been theoretically demonstrated that membrane tension created by adding cholesterol molecules to the exoplasmic leaflet of the plasma membrane may provide a direct driving force to cause full expansion of the hemifusion diaphragm (55). It explains our observation that the addition of free cholesterol to the bath solution triggered vesicle fusion, albeit with less potency compared to high K+ stimulation (Fig. 4 B). Although cholesterol-induced fusion is distinctly different from native Ca2+-triggered vesicular fusion, the results suggest that the semistable hemifusion structure is in a delicate energetic balance, and the addition of negatively curved cholesterol favors the transition from hemifusion to “spontaneous” full fusion. Not unexpectedly, cholesterol-induced quantal release, without the driven force provided by secretory proteins, manifested a much slower expansion of the fusion pore and longer fusion time for a given vesicle size (Fig. 4, C–E). When subjected to the same uniform driving force (membrane stress introduced by the addition of cholesterol), t1/2 scales linearly with Q1/3 for both small and large vesicles, i.e., larger vesicles take a longer time to release their contents. The comparison between lipid-driven fusion and protein-driven fusion may allow us to decipher the energetic actions provided by the fusion proteins at different stages of fusion.
Functional studies of membrane cholesterol have relied mostly on pharmacological agents (β-cyclodextrins, particularly MβCD) as cholesterol acceptors to remove membrane cholesterol, or cholesterol donors to load cell membranes with cholesterol. Although this is regarded as the most popular and effective method to manipulate membrane cholesterol content, nonspecific effects of cyclodextrin have also been reported, and hence one should use caution when interpreting the data (29). We used different methods to deplete cholesterol to confirm the specificity of cholesterol. In addition, we showed that cholesterol depletion did not affect cell viability (Fig. S2), morphology, or adhesion (Fig. S3). Furthermore, the replenishment of cholesterol was able to completely rescue the fusion kinetics (i.e., the amplitude, rise slope, quantal size, and fusion time) and vesicle docking. On the other hand, MβCD-induced inhibition of exocytosis and confinement of vesicle motion could not be reversed by replenishment of membrane cholesterol. However, it is unlikely that the observed phenomena resulted from nonspecific MβCD actions, because additional application of cholesterol-loaded MβCD did not cause further suppression (Figs. 1 and 6). Instead, it is conceivable that disruptions (e.g., on cholesterol-protein assemblies or membrane microdomains) caused by cholesterol depletion that are relevant to vesicle competence and movement cannot be reversed by simply adding cholesterol back into the plasma membrane.
In conclusion, the data presented in this study establish that, in addition to its known effect in maintaining the extent of exocytosis, cholesterol plays multiple roles in facilitating exocytosis. The efficiency of exocytosis depends on the availability of membrane cholesterol, which is important for maintaining vesicle fusion/docking structures and facilitating vesicle trafficking. Cholesterol also ensures the efficiency of quantal vesicle fusion by promoting the formation of negatively curved fusion structures and by stabilizing fusion machines.
Supporting Material
Materials and methods, six figures, and references are available at http://www.biophysj.org/biophysj/supplemental/S0006-3495(09)01164-3.
Supporting Material
Acknowledgments
This study was supported by an Academic Research Fund Tier 2 grant (T206B3220) from the Ministry of Education (Singapore) and a Start-Up Grant from Nanyang Technological University.
Footnotes
Jing Zhang and Renhao Xue contributed equally to this work.
References
- 1.Burgoyne R.D., Morgan A. Secretory granule exocytosis. Physiol. Rev. 2003;83:581–632. doi: 10.1152/physrev.00031.2002. [DOI] [PubMed] [Google Scholar]
- 2.Jahn R., Scheller R.H. SNAREs—engines for membrane fusion. Nat. Rev. Mol. Cell Biol. 2006;7:631–643. doi: 10.1038/nrm2002. [DOI] [PubMed] [Google Scholar]
- 3.Rohrbough J., Broadie K. Lipid regulation of the synaptic vesicle cycle. Nat. Rev. Neurosci. 2005;6:139–150. doi: 10.1038/nrn1608. [DOI] [PubMed] [Google Scholar]
- 4.Bankaitis V.A., Morris A.J. Lipids and the exocytotic machinery of eukaryotic cells. Curr. Opin. Cell Biol. 2003;15:389–395. doi: 10.1016/s0955-0674(03)00076-0. [DOI] [PubMed] [Google Scholar]
- 5.Wenk M.R., De Camilli P. Protein-lipid interactions and phosphoinositide metabolism in membrane traffic: insights from vesicle recycling in nerve terminals. Proc. Natl. Acad. Sci. USA. 2004;101:8262–8269. doi: 10.1073/pnas.0401874101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tang N., Ong W.Y., Zhang E.M., Chen P., Yeo J.F. Differential effects of ceramide species on exocytosis in rat PC12 cells. Exp. Brain Res. 2007;183:241–247. doi: 10.1007/s00221-007-1036-7. [DOI] [PubMed] [Google Scholar]
- 7.Belmonte S.A., Lopez C.I., Roggero C.M., De Blas G.A., Tomes C.N. Cholesterol content regulates acrosomal exocytosis by enhancing Rab3A plasma membrane association. Dev. Biol. 2005;285:393–408. doi: 10.1016/j.ydbio.2005.07.001. [DOI] [PubMed] [Google Scholar]
- 8.Brunham L.R., Kruit J.K., Verchere C.B., Hayden M.R. Cholesterol in islet dysfunction and type 2 diabetes. J. Clin. Invest. 2008;118:403–408. doi: 10.1172/JCI33296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chintagari N.R., Jin N., Wang P.C., Narasaraju T.A., Chen J.W. Effect of cholesterol depletion on exocytosis of alveolar type II cells. Am. J. Respir. Cell Mol. Biol. 2006;34:677–687. doi: 10.1165/rcmb.2005-0418OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Churchward M.A., Rogasevskaia T., Hofgen J., Bau J., Coorssen J.R. Cholesterol facilitates the native mechanism of Ca2+-triggered membrane fusion. J. Cell Sci. 2005;118:4833–4848. doi: 10.1242/jcs.02601. [DOI] [PubMed] [Google Scholar]
- 11.Pike L.J., Miller J.M. Cholesterol depletion delocalizes phosphatidylinositol bisphosphate and inhibits hormone-stimulated phosphatidylinositol turnover. J. Biol. Chem. 1998;273:22298–22304. doi: 10.1074/jbc.273.35.22298. [DOI] [PubMed] [Google Scholar]
- 12.Wasser C.R., Ertunc M., Liu X.R., Kavalali E.T. Cholesterol-dependent balance between evoked and spontaneous synaptic vesicle recycling. J. Physiol. 2007;579:413–429. doi: 10.1113/jphysiol.2006.123133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gondre-Lewis M.C., Petrache H.I., Wassif C.A., Harries D., Parsegian A. Abnormal sterols in cholesterol-deficiency diseases cause secretory granule malformation and decreased membrane curvature. J. Cell Sci. 2006;119:1876–1885. doi: 10.1242/jcs.02906. [DOI] [PubMed] [Google Scholar]
- 14.Wang Y.Z., Thiele C., Huttner W.S. Cholesterol is required for the formation of regulated and constitutive secretory vesicles from the trans-Golgi network. Traffic. 2000;1:952–962. doi: 10.1034/j.1600-0854.2000.011205.x. [DOI] [PubMed] [Google Scholar]
- 15.Chamberlain L.H., Burgoyne R.D., Gould G.W. SNARE proteins are highly enriched in lipid rafts in PC12 cells: implications for the spatial control of exocytosis. Proc. Natl. Acad. Sci. USA. 2001;98:5619–5624. doi: 10.1073/pnas.091502398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lang T., Bruns D., Wenzel D., Riedel D., Holroyd P. SNAREs are concentrated in cholesterol-dependent clusters that define docking and fusion sites for exocytosis. EMBO J. 2001;20:2202–2213. doi: 10.1093/emboj/20.9.2202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lang T. SNARE proteins and ‘membrane rafts’. J. Physiol. 2007;585:693–698. doi: 10.1113/jphysiol.2007.134346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Epand R.M. Cholesterol and the interaction of proteins with membrane domains. Prog. Lipid Res. 2006;45:279–294. doi: 10.1016/j.plipres.2006.02.001. [DOI] [PubMed] [Google Scholar]
- 19.Barenholz Y. Cholesterol and other membrane active sterols: from membrane evolution to “rafts”. Prog. Lipid Res. 2002;41:1–5. doi: 10.1016/s0163-7827(01)00016-9. [DOI] [PubMed] [Google Scholar]
- 20.Byfield F.J., Aranda-Espinoza H., Romanenko V.G., Rothblat G.H., Levitan I. Cholesterol depletion increases membrane stiffness of aortic endothelial cells. Biophys. J. 2004;87:3336–3343. doi: 10.1529/biophysj.104.040634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Biswas S., Yin S.R., Blank P.S., Zimmerberg J. Cholesterol promotes hemifusion and pore widening in membrane fusion induced by influenza hemagglutinin. J. Gen. Physiol. 2008;131:503–513. doi: 10.1085/jgp.200709932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chadda R., Howes M.T., Plowman S.J., Hancock J.F., Parton R.G. Cholesterol-sensitive Cdc42 activation regulates actin polymerization for endocytosis via the GEEC pathway. Traffic. 2007;8:702–717. doi: 10.1111/j.1600-0854.2007.00565.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Girao H., Pereira P., Ramalho J., Quinlan R., Prescott A. Cholesterol oxides mediated changes in cytoskeletal organisation involves Rho GTPases. Exp. Cell Res. 2003;291:502–513. doi: 10.1016/j.yexcr.2003.08.002. [DOI] [PubMed] [Google Scholar]
- 24.Holz R.W., Axelrod D. Secretory granule behaviour adjacent to the plasma membrane before and during exocytosis: total internal reflection fluorescence microscopy studies. Acta Physiol. (Oxf). 2008;192:303–307. doi: 10.1111/j.1748-1716.2007.01818.x. [DOI] [PubMed] [Google Scholar]
- 25.Christian A.E., Haynes M.P., Phillips M.C., Rothblat G.H. Use of cyclodextrins for manipulating cellular cholesterol content. J. Lipid Res. 1997;38:2264–2272. [PubMed] [Google Scholar]
- 26.Chen P., Gillis K.D. The noise of membrane capacitance measurements in the whole-cell recording configuration. Biophys. J. 2000;79:2162–2170. doi: 10.1016/S0006-3495(00)76464-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen P., Hwang T.C., Gillis K.D. The relationship between cAMP, Ca2+, and transport of CFTR to the plasma membrane. J. Gen. Physiol. 2001;118:135–144. doi: 10.1085/jgp.118.2.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Xue R.H., Zhao Y.Y., Chen P. Involvement of PKC α in PMA-induced facilitation of exocytosis and vesicle fusion in PC12 cells. Biochem. Biophys. Res. Commun. 2009;380:371–376. doi: 10.1016/j.bbrc.2009.01.105. [DOI] [PubMed] [Google Scholar]
- 29.Zidovetzki R., Levitan I. Use of cyclodextrins to manipulate plasma membrane cholesterol content: evidence, misconceptions and control strategies. Biochim. Biophys. Acta. 2007;1768:1311–1324. doi: 10.1016/j.bbamem.2007.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hao M.M., Mukherjee S., Maxfield F.R. Cholesterol depletion induces large scale domain segregation in living cell membranes. Proc. Natl. Acad. Sci. USA. 2001;98:13072–13077. doi: 10.1073/pnas.231377398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lasa M., Perezcaballero F.J., Usera F., Chiloeches A., Montes A. Cholesterol cell content affects prolactin but not growth-hormone release in Gh4c1 cells. Endocrinology. 1993;132:1701–1706. doi: 10.1210/endo.132.4.8462470. [DOI] [PubMed] [Google Scholar]
- 32.Schroeder T.J., Borges R., Finnegan J.M., Pihel K., Amatore C. Temporally resolved, independent stages of individual exocytotic secretion events. Biophys. J. 1996;70:1061–1068. doi: 10.1016/S0006-3495(96)79652-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.San Tang K., Wang N., Tse A., Tse F.W. Influence of quantal size and cAMP on the kinetics of quantal catecholamine release from rat chromaffin cells. Biophys. J. 2007;92:2735–2746. doi: 10.1529/biophysj.106.088997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang E.M., Xue R.H., Soo J., Chen P. Effects of phorbol ester on vesicle dynamics as revealed by total internal reflection fluorescence microscopy. Pflugers Arch. 2008;457:211–222. doi: 10.1007/s00424-008-0485-6. [DOI] [PubMed] [Google Scholar]
- 35.Johns L.M., Levitan E.S., Shelden E.A., Holz R.W., Axelrod D. Restriction of secretory granule motion near the plasma membrane of chromaffin cells. J. Cell Biol. 2001;153:177–190. doi: 10.1083/jcb.153.1.177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Oheim M., Stuhmer W. Tracking chromaffin granules on their way through the actin cortex. Eur. Biophys. J. 2000;29:67–89. doi: 10.1007/s002490050253. [DOI] [PubMed] [Google Scholar]
- 37.Ferraro J.T., Daneshmand M., Bizios R., Rizzo V. Depletion of plasma membrane cholesterol dampens hydrostatic pressure and shear stress-induced mechanotransduction pathways in osteoblast cultures. Am. J. Physiol. Cell Physiol. 2004;286:C831–C839. doi: 10.1152/ajpcell.00224.2003. [DOI] [PubMed] [Google Scholar]
- 38.Simons K., Ehehalt R. Cholesterol, lipid rafts, and disease. J. Clin. Invest. 2002;110:597–603. doi: 10.1172/JCI16390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Simons K., Ikonen E. Functional rafts in cell membranes. Nature. 1997;387:569–572. doi: 10.1038/42408. [DOI] [PubMed] [Google Scholar]
- 40.Allen J.A., Halverson-Tamboli R.A., Rasenick M.M. Lipid raft microdomains and neurotransmitter signalling. Nat. Rev. Neurosci. 2007;8:128–140. doi: 10.1038/nrn2059. [DOI] [PubMed] [Google Scholar]
- 41.Hanzal-Bayer M.F., Hancock J.F. Lipid rafts and membrane traffic. FEBS Lett. 2007;581:2098–2104. doi: 10.1016/j.febslet.2007.03.019. [DOI] [PubMed] [Google Scholar]
- 42.Salaun C., James D.J., Chamberlain L.H. Lipid rafts and the regulation of exocytosis. Traffic. 2004;5:255–264. doi: 10.1111/j.1600-0854.2004.0162.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Salaun C., Gould G.W., Chamberlain L.H. Lipid raft association of SNARE proteins regulates exocytosis in PC12 cells. J. Biol. Chem. 2005;280:19449–19453. doi: 10.1074/jbc.M501923200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nishimura S.Y., Vrljic M., Klein L.O., McConnell H.M., Moerner W.E. Cholesterol depletion induces solid-like regions in the plasma membrane. Biophys. J. 2006;90:927–938. doi: 10.1529/biophysj.105.070524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Vrljic M., Nishimura S.Y., Moerner W.E., McConnell H.M. Cholesterol depletion suppresses the translational diffusion of class II major histocompatibility complex proteins in the plasma membrane. Biophys. J. 2005;88:334–347. doi: 10.1529/biophysj.104.045989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Trifaro J.M., Gasman S., Gutierrez L.M. Cytoskeletal control of vesicle transport and exocytosis in chromaffin cells. Acta Physiol. (Oxf). 2008;192:165–172. doi: 10.1111/j.1748-1716.2007.01808.x. [DOI] [PubMed] [Google Scholar]
- 47.Zimmerberg J., Chernomordik L.V. Membrane fusion. Adv. Drug Deliv. Rev. 1999;38:197–205. doi: 10.1016/s0169-409x(99)00029-0. [DOI] [PubMed] [Google Scholar]
- 48.Coorssen J.R., Rand R.P. Effects of cholesterol on the structural transitions induced by diacylglycerol in phosphatidylcholine and phosphatidylethanolamine bilayer systems. Biochem. Cell Biol. 1990;68:65–69. doi: 10.1139/o90-008. [DOI] [PubMed] [Google Scholar]
- 49.Lei G.H., MacDonald R.C. Lipid bilayer vesicle fusion: intermediates captured by high-speed microfluorescence spectroscopy. Biophys. J. 2003;85:1585–1599. doi: 10.1016/S0006-3495(03)74590-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Garcia R.A., Pantazatos S.P., Pantazatos D.P., MacDonald R.C. Cholesterol stabilizes hemifused phospholipid bilayer vesicles. Biochim. Biophys. Acta. 2001;1511:264–270. doi: 10.1016/s0005-2736(01)00283-8. [DOI] [PubMed] [Google Scholar]
- 51.Coorssen J.R., Blank P.S., Tahara M., Zimmerberg J. Biochemical and functional studies of cortical vesicle fusion: the SNARE complex and Ca2+ sensitivity. J. Cell Biol. 1998;143:1845–1857. doi: 10.1083/jcb.143.7.1845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zimmerberg J., Coorssen J.R., Vogel S.S., Blank P.S. Sea urchin egg preparations as systems for the study of calcium-triggered exocytosis. J. Physiol. 1999;520:15–21. doi: 10.1111/j.1469-7793.1999.00015.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Churchward M.A., Rogasevskaia T., Brandman D.M., Khosravani H., Nava P. Specific lipids supply critical negative spontaneous curvature—an essential component of native Ca2+-triggered membrane fusion. Biophys. J. 2008;94:3976–3986. doi: 10.1529/biophysj.107.123984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kaplan D., Zimmerberg J., Puri A., Sarkar D.P., Blumenthal R. Single cell-fusion events induced by influenza hemagglutinin—studies with rapid-flow, quantitative fluorescence microscopy. Exp. Cell Res. 1991;195:137–144. doi: 10.1016/0014-4827(91)90509-s. [DOI] [PubMed] [Google Scholar]
- 55.Hed G., Safran S.A. Initiation and dynamics of hemifusion in lipid bilayers. Biophys. J. 2003;85:381–389. doi: 10.1016/S0006-3495(03)74482-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.