

Abstract
Recent experimental studies show that carbon nanotubes impact the aggregation process of proteins associated with neurodegenerative diseases. However, the details of molecular interactions between proteins and carbon nanotubes are still not well understood. In this study, we investigate the initial adsorption features and dynamics of the Alzheimer's amyloid-β peptide spanning residues 25–35 (Aβ25–35) on a single-walled carbon nanotube (SWNT) surface using fully atomic molecular dynamics simulations (MD) in explicit solvent. The initial configurations of the Aβ25–35 peptides consist of two preformed bilayer β-sheets, each with four or five β-strands in parallel or mixed antiparallel-parallel orientations. Our simulations show, for what we believe is the first time, that two disjointed Aβ25–35 β-sheets with mixed antiparallel-parallel strands can assemble into β-barrels wrapping the SWNT. In contrast, both simulations of Aβ25–35 without SWNT, and simulations of SWNT−Aβ25–35 with purely parallel β-strands, lead to disordered aggregates. We find that Aβ25–35 β-barrel formation involves at least two steps: i), curving of the Aβ25–35 β-sheets as a result of strong hydrophobic interactions with carbon nanotube concomitantly with dehydration of the SWNT-peptide interface; and ii), intersheet backbone hydrogen bond formation with fluctuating intrasheet hydrogen bonds. Detailed analysis of the conversion shows that β-barrel formation on SWNT surface results from the interplay of dehydration and peptide-SWNT/peptide-peptide interactions. Implications of our results on amyloid fibril inhibition are discussed.
Introduction
Carbon nanotubes have drawn significant attention in the field of nanobiotechnology and nanomedicine due to their potential applications in biosensors (1), drug delivery (2), and templates for biomolecule assembly (3). Self-assembly of peptides is also attracting attention due to its applications in bio-nanomaterial design (4) and its implications in amyloidoses such as Alzheimer's disease (5,6). These diseases involve self-assembly of soluble proteins into insoluble fibrils through a nucleation-dependent polymerization process, during which the slow formation of a nucleus (nucleation phase) is followed by a very rapid elongation phase (7,8). There is experimental evidence that graphite sheets (9,10), collagen fibers (11), dichloromethane (12), nanogels (13), carbon nanotubes (14,15), and fullerenes (16,17) promote (prevent) amyloid formation depending on protein intrinsic stability by decreasing (increasing) the lag time for nucleation, but they all leave the elongation phase invariant, suggesting a surface-controlled nucleation mechanism (12,15,16,18). Although these experimental studies have greatly enhanced our understanding of the impact of surfaces on the kinetics of amyloid formation, the detailed adsorption dynamics remains, however, to be determined. Such a knowledge at an atomic level of detail has still not been explored by classical molecular dynamics (MD) simulations (19,20), recent MD focusing on folding and stability of α-helical peptides within single-walled carbon nanotube (SWNT) (21,22), encapsulation of various peptides into SWNT (23,24) and the conformational change of the human serum albumin protein for 2 ns on SWNT surfaces (20).
The goal of this work is to study the interaction of the low molecular weight (LMW) oligomers of the Alzheimer's amyloid-β peptide spanning residues 25–35 (Aβ25–35) with a SWNT of different diameters. This 11-residue fragment of sequence GSNKGAIIGLM, with a positively charged N-terminus and a hydrophobic C terminus, forms amyloid fibrils in vitro and retains the toxicity of the full-length Aβ peptide (25,26). Hydrogen/deuterium exchange NMR experiments indicate that Aβ25–35 amyloid fibrils display a cross-β core formed from K28 to M35, with residues I31 and I32 being the most protected, and suggest two fibril models with out-of-register antiparallel and in-register parallel β-strands (27).
Because the formation of β-rich LMW oligomers from random states is out of reach using all-atom explicit-solvent MD simulations at physiological temperature (28,29), and the equilibrium ensemble of an octamer consists of various bilayer β-sheets and amorphous states (30–33), we study, as a first step, the interaction between two preformed Aβ25–35 β-sheets and carbon nanotubes. By using all-atom MD simulations in explicit solvent, we show that two disjointed Aβ25–35 β-sheets with mixed antiparallel-parallel strands, can assemble into β-barrels wrapping the SWNT. In contrast, both simulations of Aβ25–35 without SWNT, and simulations of SWNT/Aβ25–35 with purely parallel β-strands, lead to disordered aggregates. The conversion process of two β-sheets into β-barrels is studied in detail and the implications of β-barrel formation on amyloid fibril inhibition are discussed.
Materials and Methods
Our initial Aβ25–35 states consist of two layers, each composed of four or five β-strands with the solid state NMR-derived parallel or mixed antiparallel-parallel out-of-register arrangements (27) and the N- and C-extremities charged for each chain (27). Unless specified and shown in Fig. 1, the two β-sheets are separated by intersheet Cα-Cα distances of at least 1.73 nm, i.e., free of any intersheet side chain-side chain contacts, are parallel to the central axis of SWNT (SWNT axis or z axis) and characterized by angles θ1 (between strands d and e) and θ2 (between strands a and h) varying between 90° and 180°. Note that most initial orientations are centered at 90° so that the system can freely evolve toward 0° and 180°. A series of armchair SWNT (n, n) are constructed with n = 3, 4, and 5 yielding the tube diameters of 0.404, 0.542, and 0.677 nm, respectively. All tube lengths are set to 4.25 nm to provide sufficient surface for Aβ25–35 peptides to adsorb. The SWNT-Aβ25–35 complexes are placed in a dodecahedron box of simple point charge water molecules (34) and the minimum distance between the oligomer and the water box wall is 1.2 nm.
Figure 1.
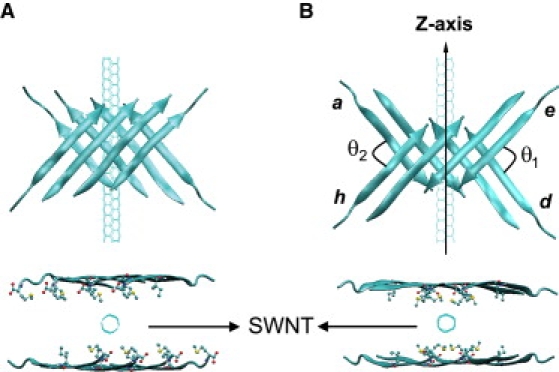
Top view and side view of the initial states. Cartoon representation of one of the starting disjointed Aβ25–35 bilayer β-sheets with eight (A) purely parallel and (B) mixed antiparallel-parallel β-strands and (θ1 and θ2) values at (98.8°, 100.5°), and line representation of SWNT (the central axis of SWNT is set to be z-axis). Upper: side view; lower: top view. The side chains of hydrophobic residues facing SWNT, i.e., I31 and M35, are shown by CPK representation in the top view. The θ1 and θ2 are the angles between chains e and d, between chains a and h, respectively. They are defined by arc cosine of the dot product of the normalized K28-to-M35 (Cα-Cα) unit vectors.
All MD simulations are carried out in the isothermal-isobaric (NPT) ensemble at neutral pH using the GROMACS software package (35) and GROMOS96 force field (36). The solute and solvent are separately coupled to external temperature and pressure baths. The temperature is maintained close to 300 K by weak coupling to an external temperature bath (37) with a coupling constant of 0.1 ps. The pressure is kept constant at 1 bar with a coupling time of 1 ps (37). The SETTLE algorithm (38) is used to constrain the bond lengths and bond angles of simple point charge molecules. The bond lengths of the peptides are constrained by LINCS (39). This allows an MD integration time step of 2 fs. A twin-range cutoff 0.9/1.4 nm is used for the nonbonded interactions and a reaction-field correction with dielectric permittivity ɛ = 78 is used to calculate the long-range electrostatic interactions. Carbon atoms of SWNT are uncharged in accordance with Hummer et al. (40) and the Lennard-Jones parameters for the protein-SWNT and water-SWNT interactions are obtained using the Lorentz-Berthelot rule (41).
Because the amino acids G25-S26-N27 are mostly random during our simulations and are excluded from the cross-β core of the Aβ25–35 fibrils (27), we use, unless specified, all amino acids for the potential energy analysis, but only the amino acids from K28 to M35 for all other analysis. These include the 2D free energy surface, backbone root mean-square deviation (RMSD) of the Aβ oligomer from the final β-barrel structure, the number of intersheet and intrasheet backbone H-bonds, and the total radius of gyration (Rg) (including side chain and backbone). The hydrophobic residues radius gyration (Rg-HP) uses only the I31 and M35 side chains because the A30, I32, and Leu34 side chains mostly point to water bulk during the simulations.
In this work, the radius of gyration of Aβ25–35 peptides is calculated with respect to the central axis of SWNT, unless specified. One H-bond is considered formed if the distance between N and O is <3.5 Å and the angle of N-H…O is >120° (36). We also calculate the angles θ1 and θ2. An open (closed) β-barrel is considered formed if there are ≥3 H-bonds between strands e and d and zero (≥3) H-bonds between strands a and h, or vice versa. For simplicity, we rename the β-sheets as octamer or decamer according to the total number of strands and we use Aβ for Aβ25–35. All the systems are displayed using the VMD program (42).
Results
The setup details of 86 MD runs totaling 900 ns starting from two disjointed β-sheets are shown in Table 1. For each system, we give the number of runs and simulation time, the initial values of the angles θ1 and θ2, and the number of MD runs in which a closed or open β-barrel is observed. Although these simulations do not test all possible (θ1, θ2) couples, binding of Aβ oligomers with initial purely parallel β-strands on SWNT surfaces and interaction of Aβ oligomers in the absence of SWNT both lead to disordered aggregates within 10–30 ns. By contrast, adsorption of Aβ octamer and decamer with mixed antiparallel-parallel strands onto different diameter of carbon nanotubes often leads to β-barrel formation, independently of the starting (θ1, θ2) values.
Table 1.
Setup details of all simulations
| System | MD runs | Simulation time | θ1, θ2 (°) |
|---|---|---|---|
| SWNT (3,3) + octamer (P) | run 1-run 8 | 10 ns × 8 (0) | 98.8, 100.5 |
| SWNT (3,3) + octamer (m A + P) | run 9-run 53 | 10 ns × 35 (9)10 ns × 5 (1)10 ns × 5 (1) | 98.8, 100.5120.6, 118.7180.0, 180.0 |
| SWNT (4,4) + octamer (P) | run 54-run 57 | 10 ns × 4 (0) | 116.7, 115.9 |
| SWNT (4,4) + octamer (m A + P) | run 58-run 62 | 10 ns × 5 (2) | 116.7, 115.9 |
| SWNT (4,4) + decamer (P) | run 63-run 67 | 30 ns × 1 (0)10 ns × 4 (0) | 117.8, 118.9 |
| SWNT (4,4) + decamer (m A + P) | run 68-run 75 | 30 ns × 1 (1)10 ns × 7 (3) | 117.8, 118.9 |
| SWNT (5,5) + decamer (m A + P) | run 76-run 78 | 10 ns × 3 (1) | 117.8, 118.9 |
| Octamer (m A + P) without SWNT | run 79-run 83 | 10 ns × 5 (0) | 98.8, 100.5 |
| Decamer (m A + P) without SWNT | run 84-run 86 | 10 ns × 3 (0) | 117.8, 118.9 |
Letters in parenthesis of the first column give the β-strand character (P, parallel; m A + P,: mixed antiparallel-parallel) of Aβ sheets, and the number in parenthesis of the third column gives the number of MD runs in which a β-barrel is observed. Note that the runs starting from a given (θ1, θ2) pair use different initial velocities, and only the runs 49–53 start from antiparallel β-sheets.
Binding of Aβ oligomer with purely parallel β-strands on SWNT surface leads to disordered aggregates
The runs (1–8, 54–57, and 63–67) show that neither open nor closed β-barrel forms on SWNT surface starting from parallel β-strand arrangements of Aβ. This is independent of the dimension of the carbon nanotube (SWNT (3,3), SWNT(4,4)) or the Aβ oligomer size (octamer and decamer). Looking at the SWNT(4,4)-octamer trajectories, we observe the two β-sheets quickly adsorb on the SWNT (4,4) surface; however the global structure of the octamer is disordered with two to three peptides in fully random coils after 10–30 ns. The Aβ assembly at 10 ns and the time evolution of the β-sheet content of each peptide in run 1 are shown in Fig. S1 in the Supporting Material. These results indicate that parallel β-strand arrangements do not favor β-barrel formation in a mixed hydrophobic-polar environment, in agreement with our survey of soluble and transmembrane protein structures (see http://www.rcsb.org/pdb/home/).
Adsorption of Aβ oligomer with mixed antiparallel-parallel β-strands on SWNT surface leads to β-barrel formation
For all Aβ systems with mixed antiparallel-parallel β-strands, we observe the formation of open or closed β-barrels wrapping SWNT in 18 of 61 runs (three representative β-barrels are shown in Fig. 2, A–C). The remaining 43 runs either lead to disordered aggregates or assemblies not satisfying our criteria of open or closed β-barrels. We start the analysis with the simulations of an Aβ octamer with a SWNT (3,3) surface. Among a total of 45 independent MD runs (runs 9–53) starting from different (θ1, θ2) orientations or the same β-sheet orientation with various random seeds for velocity generation, 11 MD runs (runs 9–19) show a fast conversion of β-sheets into β-barrels.
Figure 2.
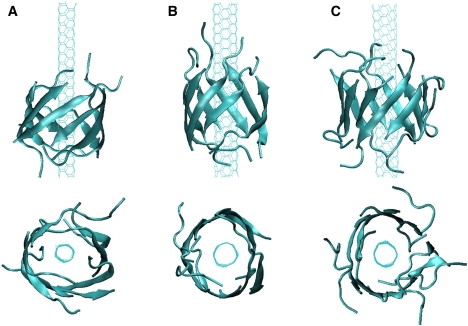
Cartoon representation of the MD-generated SWNT-β-barrels. (Upper) Side view; (lower) top view at t = 10 ns for three different SWNT-oligomer systems: (A) SWNT(3,3)-octamer in run 9. (B) SWNT(4,4)-octamer in run 58. (C) SWNT(4,4)-decamer in run 68.
Using the runs 9–19 of Aβ-octamer-SWNT (3,3), we plot the free energy landscape as a function of the number of intersheet backbone H-bonds and the Rg-HP (Fig. 3). Note that our calculation does not provide the full free energy surface of Aβ-SWNT, because the Aβ peptides are not initially in randomly chosen conformations and orientations. We observe a first delocalized, continuous basin spanning (H-bond, Rg-HP) values of (0, 0.7 nm)–(6, 0.7 nm) and a second smaller basin centered at (11, 0.65 nm). These basins correspond to compact assemblies and open β-barrels, and closed β-barrels of Aβ, respectively. The overall “L” shape of the free energy map with the structural description of the minima indicates that β-barrel formation is driven first by strong hydrophobic interactions of Aβ with SWNT and then by intersheet H-bond formation. This is further illustrated in Fig. 4 by the time evolution of three structural properties averaged over all runs 9–19 (Fig. 4, red lines) or in the single run 9 (Fig. 4, black lines): Rg and Rg-HP (Fig. 4 A), and the number of intersheet H-bonds (Fig. 4 B). It is noted that run 19, which starts from θ1 and θ2 ∼180°, is excluded from Fig. 4, E and F. In all simulations, the rapid reduction in Rg and Rg-HP is accompanied with a curving of the two β-sheets (see the snapshot at t = 0.2 ns in Fig. 4 G) and a small decrease of the angles θ1 and θ2 ∼85° (Fig. 4, E and F). Subsequently, the number of intersheet H-bonds increases and the open β-barrel forms concomitantly with a high plasticity of the two β-sheets (Fig. 4 C). An extreme plasticity is observed in run 9 (Fig. 4 C, black line) where the number of intrasheet H-bonds drops from 31 to 22 in the first 4 ns (indicating a partial disruption of the initial H-bond network) followed by an increase from 22 to 27 H-bonds. Note that the final H-bond network differs from the initial one in 3 of 11 runs as seen in Fig. 5. Finally, we see a gradual increase of the angles θ1 and θ2 up to 140–160°, indicating an antiparallel arrangement of the newly joined strands, and a constant decrease in the RMSD (Fig. 4 D) as a function of time.
Figure 3.
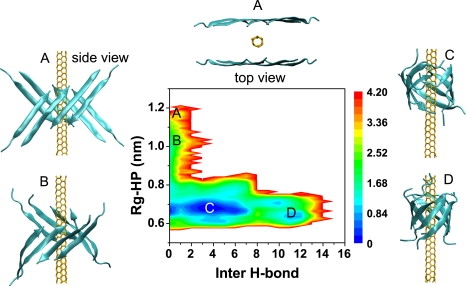
Free energy landscape of the Aβ25–35 octamer on a SWNT (3,3) surface along with the structures of SWNT−Aβ25–35 at four different locations. (A) One of the initial state (top view and side view). (B) SWNT-curved β-sheet. (C) SWNT-open β-barrel. (D) SWNT-closed β-barrel. The free energy (in Kcal/mol) is projected onto the number of intersheet backbone H-bonds and Rg-HP. The data are constructed using 11 independent MD runs leading to β-barrel formation.
Figure 4.
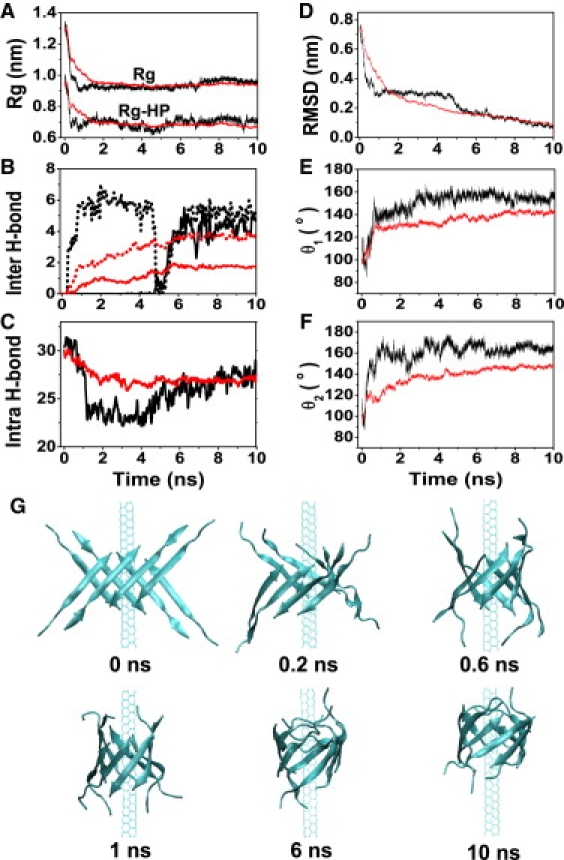
Detailed analysis of the assembly process of two tetrameric β-sheets into β-barrel induced by SWNT (3,3) surface. Black curves display run 9, whereas red curves online (or gray curves in print) are the averaged properties using runs 9–19 for A–D and runs 9–18 for E and F. Time evolution of: (A) total Rg and Rg-HP of the octamer; (B) number of intersheet backbone H-bonds (solid line: between strands e and d; dotted line: between strands a and h); (C) number of intrasheet backbone H-bonds; (D) backbone RMSD of the octamer with respect to the β-barrel structure generated at 10 ns; (E) angle θ1; and (F) angle θ2. (G) Six representative snapshots of SWNT(3,3)-Aβ octamer in run 9.
Figure 5.
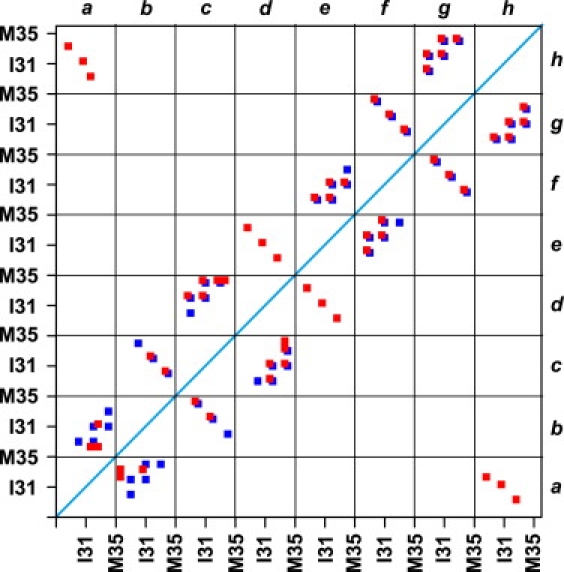
Backbone H-bond network map involving residues from K28 to M35 in the structures at t=0 (blue squares online or black squares in print) and t=10 ns (red squares online or gray squares in print) in run 9. The letters a–d are the labels of the four strands in one β-sheet and e–h the labels of the four strands in the other β-sheet. The two hydrophobic residues I31 and M35 in each strand are labeled. The red squares online (or gray squares in print) along the diagonal (from left top to right bottom) are the intersheet H-bond network and the others intrasheet H-bond network.
The conversion of two tetrameric β-sheets into a β-barrel on SWNT (3,3) surface can also be monitored by counting as a function of time the number of water molecules within successive cylindrical shells of 0.5 nm height (z direction) and 1.0 nm radius from z axis. In Fig. 6, we show the results of run 9, but a similar picture emerges from runs 10–19. The number of water molecules at the SWNT/Aβ interface in the region of z = −1.0–1.0 nm is ≥15 at 0 ns and decreases gradually with time reaching approximately zero at 1.0 ns (Fig. 6 A). The four snapshots at time of 0, 0.24, 0.48, and 1 ns in Fig. 6 B show a clear dehydration process of SWNT-Aβ interface.
Figure 6.
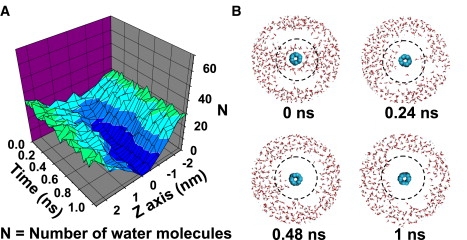
Dehydration process of SWNT-Aβ octamer interface observed in run 9. (A) Time evolution of the number of water molecules within each cylinder along SWNT-axis. The cylinder is 1.0-nm thick from SWNT-axis and 0.5 nm high. (B) Four snapshots of SWNT(3,3)-octamer-water (view parallel to SWNT-axis) generated within the first 1 ns. To visualize the dehydration process clearly, a radius of 1 nm from the SWNT-axis is drawn in dotted line, the Aβ octamer is omitted and only the water molecules within 2 nm from the SWNT-axis are displayed.
To understand further the physical driving forces underlying the conversion of two tetrameric β-sheets into a β-barrel, we plot the time evolution of the potential energy contribution of SWNT-water, SWNT-Aβ, Aβ-water, Aβ-Aβ, and water-water interactions from run 9 (Fig. 7). Given the fact that the simulation timescales are too short to estimate the entropic contributions, our analysis is only qualitative and indicative. We see that the SWNT-Aβ, Aβ-Aβ, and water-water interactions suffice to compensate the energy cost to repel water from the SWNT-Aβ interface and the energy lost associated with SWNT-water and Aβ-water interactions. Because simulations without SWNT do not lead to β-barrels (octamer: runs 79–83, decamer: runs 84–86; see Fig. S2), our energy decomposition along with the observation of dehydration at the SWNT(3,3)-Aβ octamer interface suggest that Aβ β-barrel formation on SWNT surface results from an interplay of dehydration, Aβ-SWNT, and Aβ-Aβ interactions.
Figure 7.
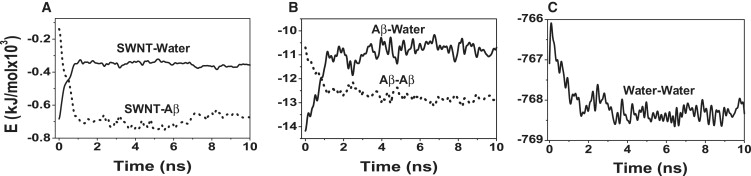
Time evolution of the potential energy components in Run 9. (A) SWNT-water and SWNT-Aβ. (B) Aβ-water and Aβ-Aβ. (C) Water-water.
β-barrel formation with larger Aβ oligomers and SWNT surfaces
To determine whether β-barrel formation occurs for various SWNT-Aβ sizes, we carry out MD simulations of an Aβ octamer with SWNT (4,4) and SWNT (5,5) and simulations of an Aβ decamer with SWNT (5,5). Although all MD simulations of Aβ oligomers with parallel strands (octamer: runs 54–57 and decamer: runs 63–67) result in amorphous aggregates, seven among 15 runs with mixed antiparallel-parallel strands lead to β-barrels, indicating a significant effect of β-sheets with mixed antiparallel-parallel strands on the formation of β-barrels. Fig. 8 and Fig. S3 follow the formation of open and closed β-barrels in run 58 of SWNT(4,4)-Aβ octamer and in run 68 of SWNT(4,4)-Aβ decamer. The time evolution similarity of Rg and the number of intersheet and intrasheet H-bonds between SWNT(4,4)-Aβ octamer (Fig. 8), SWNT(4,4)-Aβ decamer (Fig. S3) and SWNT(3,3)-Aβ octamer (Fig. 4) is striking. Taken together with the dehydration process in SWNT(3,3)-Aβ octamer (Fig. 6) and SWNT(4,4)-Aβ decamer (Fig. S4), we can assess that the formation of open/closed β-barrels involves two steps, independently of the SWNT diameter and Aβ oligomer size: 1), curving of the Aβ β-sheets as a result of strong hydrophobic interactions of peptides with SWNT and dehydration of the SWNT-peptide interface; and 2), formation of intersheet backbone H-bonds along with the disruption and reformation of intrasheet H-bonds.
Figure 8.
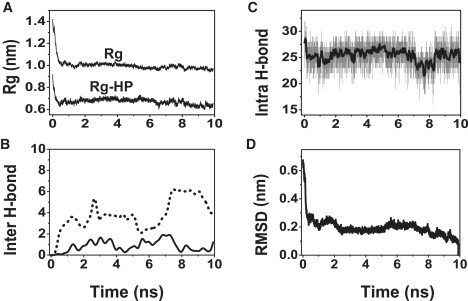
Analysis of the assembly process of two tetrameric β-sheets into a 8-stranded β-barrel structure on a SWNT (4,4) surface observed in run 58. Time evolution of: (A) total Rg and Rg-HP of the octamer; (B) number of intersheet backbone H-bonds; (C) number of intrasheet backbone H-bonds (the black curve is a smoothed line over 20 data points); and (D) backbone RMSD of the octamer with respect to the β-barrel structure generated at 10 ns.
Discussion and Conclusions
The formation of Aβ25–35 β-barrels from two four- and five-stranded β-sheets with mixed parallel/antiparallel strands is not systematic and we find that 30% of the simulations lead to β-barrel surrounding the carbon nanotube. To explore the impact of the initial configuration of SWNT-Aβ β-sheets on β-barrel formation, we carry out five additional MD simulations starting from a different orientation of the two β-sheets relative to SWNT-axis (Fig. S5). Again, we observe the formation of β-barrel around SWNT. To determine the stability of the β-barrel, we also extend the MD run 10 to 80 ns. In Fig. S6 we see that all the conformations explored after t = 1 ns deviate by <3.0 Å RMSD from the β-barrel formed at 80 ns. Taken together, these results indicate that the β-barrel is likely to be a true free energy minimum for Aβ25–35 in the presence of carbon nanotubes. However, replica exchange molecular dynamics are needed to get the β-barrel populations with high accuracy.
Similarly, further studies are needed to understand the dependence of β-barrel formation on SWNT diameter and peptide amino acid sequence. We expect, however, based on our findings that strong hydrophobic interactions of Aβ with SWNT and water expulsion dominate in the early steps of β-barrel formation, and the rate of conversion can be improved by using ideal amino acid patterns with high β-sheet preferences (43). Alternatively, there exists certainly a critical ratio of hydrophobic to hydrophilic, charged to neutral amino acids or unfavorable amino acid patterns, at which the binding of amyloid peptides to the carbon nanotube surface is very weak, therefore hindering the formation of β-barrel.
An important finding of this study is that initial structures composed of parallel β-strands do not form β-barrels in a mixed polar/apolar environment. To understand the microscopic reasons of the orientation preference of the chains, we carry out five runs starting from an idealized barrel containing only parallel β-strands (P-barrel) and five runs starting from the perfect barrel with antiparallel-parallel β-strands generated in run 10 (AP-barrel) at 300 K for 10 ns.
The time evolution of the backbone RMSD, Rg and percentage of initial inner Aβ-Aβ side chain-side chain contacts, and Aβ-Aβ potential energy averaged over the five runs are shown in Fig. 9. Although the RMSD of AP-barrel remains <0.2 nm during 10 ns, the RMSD of P-barrel increases to 0.7 nm after 1 ns (Fig. 9 A). Similarly, the Rg of AP-barrel remains at 0.63 nm, whereas the Rg of P-barrel fluctuates ∼1.1 nm and this comes mainly from the rapid increase of the M35 Rg (Fig. 9 B). The percentage of initial inner (M35-M35, N27-N27, I31-I31) contacts in Fig. 3 C decreases to (15%, 30%, and 95%) in P-barrel versus (80%, 90%, and 95%) in AP-barrel, indicating that the geometric constraints imposed by the bulky M35 side chains play a dominant role in the collapse of P-barrel, although the contribution of N27 cannot be ignored. Finally the substantial Aβ-Aβ potential energy difference between AP-barrel and P-barrel in Fig. 9 D, which cannot be compensated by the other potential energy terms involving water and SWNT, explains why our simulations do not form P-barrels.
Figure 9.
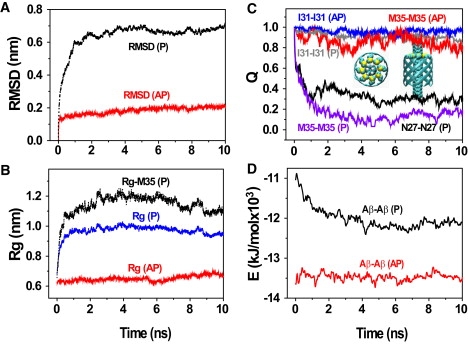
Analysis of the destabilization of an ideal β-barrel with purely parallel β-strands (P-barrel) coaxial with a SWNT (3,3) averaged over five independent 10-ns MD runs. Time evolution of: (A) backbone RMSD of all residues; (B) Rg of the side chains located in the inner side of the β-barrel (i.e., N27, I31, and M35) and Rg of the bulky Met35 side chains; (C) fraction of the initial inter-side chain contacts located in the inner side of β-barrel; and (D) the Aβ-Aβ potential energy. For comparison, the same analyses are reported for mixed antiparallel-parallel β-barrel (AP-barrel). We also show in C the top view and side view of the constructed perfect P-barrel with the bulky side chains of M35 in VDW representation.
Recent experimental studies showed that the influence of nanoparticles on fibrillation is protein-dependent and varies with particle characteristics. Copolymeric NiPAM/BAM nanoparticles (18) and fullerene (16) retard full-length Aβ1–40 fibrillation, whereas NiPAM/BAM nanoparticles and carbon nanotubes accelerate the nucleation phase of the 99-residue human β2m-microglobulin (15). The different effects on fibrillation were attributed to the intrinsic stability of the systems: β2m-microglobulin is a partially unfolded protein, whereas Aβ1–40 is mostly unstructured peptide in solution (18). By using surface plasmon resonance experiments, Linse and collaborators (15) could not distinguish between binding of monomeric and oligomeric Aβ1–40 forms on NiPAM/BAM nanoparticles (18).
Although our simulations do not start from disordered Aβ peptides and do not explore how carbon nanotubes influence the nucleation process, they provide strong evidence that Aβ25–35 oligomeric species with β-barrel and more disordered characteristics form easily around SWNT. These newly Aβ LMW aggregates therefore block backbone amide sites for further monomer or oligomer addition with β-sheet structure and reduce the population of monomers and small oligomers available for the next steps of aggregation. Whether there are multiple binding sites between full-length peptides and carbon nanotubes remains to be determined, Aβ1–40/1–42 also contains two other hydrophobic cores spanning residues 17–21 and 36–40/42. However, the propensity of the residues 28–35 to form barrels around SWNT is likely to reduce the loop or bent formation of the region Ser26-Ala30 proposed in the Aβ1–42 fibrillar (44) and Aβ17–42 annular (45) models, and this would also enhance the lag time for Aβ nucleation in the presence of SWNT.
The construction of molecules inhibiting Aβ aggregation is a very active field. The designed green fluorescent protein variant P13H with a β-barrel structure was found to inhibit Aβ1-42 fibrillation (46). Although the interaction sites between Aβ25–35–SWNT and Aβ1-42–P13H are different, Aβ1–42 and Aβ25–35 both bind to a cylindrical substrate (P13H or SWNT) through noncovalent interactions. Recent studies also showed that fullerene C60 inhibited the fibrillation of Aβ1-40 (16) and Aβ25–35 (17), and carbon nanotubes prevented the aggregation of protein (14). Based on the previous experimental studies (14,16,17,27) and the finding of the β-barrel formation of Aβ25–35 oligomers on SWNT surfaces, we propose that carbon nanotubes are likely to be potent inhibitors of Aβ25–35 fibril formation. Whether the Aβ25–35–SWNT aggregates and β-barrels are toxic remains to be determined, but a recent study suggests that SWNTs of short lengths <5–10 μm (i.e., larger than our SWNT length) are not (47).
In summary, we have investigated the interactions of two Aβ25–35 oligomeric β-sheets with a SWNT in explicit solvent. Our all-atom MD simulations of multiple SWNT-water-Aβ and water-Aβ oligomer systems show, for what we believe is the first time, that adsorption of Aβ25–35 peptides with mixed antiparallel-parallelβ-strands on carbon nanotubes can lead to Aβ25–35 open/closed β-barrels wrapping the SWNT. Our 2D free energy surface and MD trajectories indicate that β-barrel formation is driven first by strong hydrophobic interactions of Aβ with SWNT simultaneously with water expulsion and then the formation of intersheet hydrogen bonds. Note that this first step was also observed during the assembly of Aβ16–22 protofilaments in solution (48). We also find that the prestructured oligomers display substantial intrasheet H-bond fluctuation during the conversion process, in agreement with simulations on monomer addition to preformed assemblies (49). By contrast, both simulations of Aβ25–35 without SWNT, and simulations of SWNT/Aβ25–35 with purely parallel β-strands, lead to disordered aggregates. Detailed analysis of the interactions between Aβ oligomers, water and SWNT show that β-barrel formation on the SWNT surface results from an interplay of dehydration, Aβ-SWNT, and Aβ-Aβ interactions. Collectively, these results help understand the interactions between Aβ25–35 oligomers and SWNT at an atomic level of detail. The formation of β-barrels may offer the prospect of using carbon nanotubes as a new type of therapeutic agent to retard/prevent Aβ25–35 fibrillation.
Acknowledgments
We thank Dr. Buyong Ma for helpful discussion and Dr. Ruhong Zhou for careful reading and constructive comments on the manuscript.
This work is funded by the National Science Foundation of China (grant 10674029) and supported by the Program for New Century Excellent Talents in University (NCET-08-0125). P.D. acknowledges financial support from CNRS, the University of Paris Diderot, and a fellowship from Fudan University. Simulations were carried out at the Shanghai Supercomputing Center and the National High Performance Computing Center of Fudan University.
Supporting Material
References
- 1.Chen R.J., Bangsaruntip S., Drouvalakis K.A., Wong Shi Kam N., Shim M. Noncovalent functionalization of carbon nanotubes for highly specific electronic biosensors. Proc. Natl. Acad. Sci. USA. 2003;100:4984–4989. doi: 10.1073/pnas.0837064100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bianco A., Kostarelos K., Prato M. Applications of carbon nanotubes in drug delivery. Curr. Opin. Chem. Biol. 2005;9:674–679. doi: 10.1016/j.cbpa.2005.10.005. [DOI] [PubMed] [Google Scholar]
- 3.Thauvin C., Rickling S., Schultz P., Celia H., Meunier S. Carbon nanotubes as templates for polymerized lipid assemblies. Nat. Nanotechnol. 2008;3:743–748. doi: 10.1038/nnano.2008.318. [DOI] [PubMed] [Google Scholar]
- 4.Cherny I., Gazit E. Amyloids: not only pathological agents but also ordered nanomaterials. Angew. Chem. Int. Ed Engl. 2008;47:4062–4069. doi: 10.1002/anie.200703133. [DOI] [PubMed] [Google Scholar]
- 5.Goedert M., Spillantini M.G. A century of Alzheimer's disease. Science. 2006;314:777–781. doi: 10.1126/science.1132814. [DOI] [PubMed] [Google Scholar]
- 6.Roberson E.D., Mucke L. 100 years and counting: prospects for defeating Alzheimer's disease. Science. 2006;314:781–784. doi: 10.1126/science.1132813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lomakin A., Teplow D.B., Kirschner D.A., Benedek G.B. Kinetic theory of fibrillogenesis of amyloid β-protein. Proc. Natl. Acad. Sci. USA. 1997;94:7942–7947. doi: 10.1073/pnas.94.15.7942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fay N., Inoue Y., Bousset L., Taguchi H., Melki R. Assembly of the yeast prion Ure2p into protein fibrils: thermodynamic and kinetic characterization. J. Biol. Chem. 2003;278:30199–30205. doi: 10.1074/jbc.M303000200. [DOI] [PubMed] [Google Scholar]
- 9.Kowalewski T., Holtzman D.M. In situ atomic force microscopy study of Alzheimer β-amyloid peptide on different substrates: new insights into mechanism of β-sheet formation. Proc. Natl. Acad. Sci. USA. 1999;96:3688–3693. doi: 10.1073/pnas.96.7.3688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yang G., Woodhouse K.A., Yip C.M. Substrate-facilitated assembly of elastin-like peptides: studies by variable-temperature in situ atomic force microscopy. J. Am. Chem. Soc. 2002;124:10648–10649. doi: 10.1021/ja027302g. [DOI] [PubMed] [Google Scholar]
- 11.Relini A., Canale C., De Stefano S., Rolandi R., Giorgetti S. Collagen plays an active role in the aggregation of β2-microglobulin under physiopathological conditions of dialysis-related amyloidosis. J. Biol. Chem. 2006;281:16521–16529. doi: 10.1074/jbc.M513827200. [DOI] [PubMed] [Google Scholar]
- 12.Ruschak A.M., Miranker A.D. Fiber-dependent amyloid formation as catalysis of an existing reaction pathway. Proc. Natl. Acad. Sci. USA. 2007;104:12341–12346. doi: 10.1073/pnas.0703306104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ikeda K., Okada T., Sawada S.-i., Akiyoshi K., Matsuzaki K. Inhibition of the formation of amyloid β-protein fibrils using biocompatible nanogels as artificial chaperones. FEBS Lett. 2006;580:6587–6595. doi: 10.1016/j.febslet.2006.11.009. [DOI] [PubMed] [Google Scholar]
- 14.Ghule A.V., Kathir K.M., Suresh Kumar T.K., Tzing S.-H., Chang J.-Y. Carbon nanotubes prevent 2,2,2 trifluoroethanol induced aggregation of protein. Carbon. 2007;45:1586–1589. [Google Scholar]
- 15.Linse S., Cabaleiro-Lago C., Xue W.F., Lynch I., Lindman S. Nucleation of protein fibrillation by nanoparticles. Proc. Natl. Acad. Sci. USA. 2007;104:8691–8696. doi: 10.1073/pnas.0701250104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kim J.E., Lee M. Fullerene inhibits β-amyloid peptide aggregation. Biochem. Biophys. Res. Commun. 2003;303:576–579. doi: 10.1016/s0006-291x(03)00393-0. [DOI] [PubMed] [Google Scholar]
- 17.Podolski I.Y., Podlubnaya Z.A., Kosenko E.A., Mugantseva E.A., Makarova E.G. Effects of hydrated forms of C60 fullerene on amyloid 1-peptide fibrillization in vitro and performance of the cognitive task. J. Nanosci. Nanotechnol. 2007;7:1479–1485. doi: 10.1166/jnn.2007.330. [DOI] [PubMed] [Google Scholar]
- 18.Cabaleiro-Lago C., Quinlan-Pluck F., Lynch I., Lindman S., Minogue A.M. Inhibition of amyloid β protein fibrillation by polymeric nanoparticles. J. Am. Chem. Soc. 2008;130:15437–15443. doi: 10.1021/ja8041806. [DOI] [PubMed] [Google Scholar]
- 19.Yeh I.-C., Hummer G. Nucleic acid transport through carbon nanotube membranes. Proc. Natl. Acad. Sci. USA. 2004;101:12177–12182. doi: 10.1073/pnas.0402699101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shen J.-W., Wu T., Wang Q., Kang Y. Induced stepwise conformational change of human serum albumin on carbon nanotube surfaces. Biomaterials. 2008;29:3847–3855. doi: 10.1016/j.biomaterials.2008.06.013. [DOI] [PubMed] [Google Scholar]
- 21.Sorin E.J., Pande V.S. Nanotube confinement denatures protein helices. J. Am. Chem. Soc. 2006;128:6316–6317. doi: 10.1021/ja060917j. [DOI] [PubMed] [Google Scholar]
- 22.O'Brien E.P., Stan G., Thirumalai D., Brooks B.R. Factors governing helix formation in peptides confined to carbon nanotubes. Nano Lett. 2008;8:3702–3708. doi: 10.1021/nl8019328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Trzaskowski B., Jalbout A.F., Adamowicz L. Molecular dynamics studies of protein-fragment models encapsulated into carbon nanotubes. Chem. Phys. Lett. 2006;430:97–100. [Google Scholar]
- 24.Kang Y., Wang Q., Liu Y.-C., Wu T., Chen Q. Dynamic mechanism of collagen-like peptide encapsulated into carbon nanotubes. J. Phys. Chem. B. 2008;112:4801–4807. doi: 10.1021/jp711392g. [DOI] [PubMed] [Google Scholar]
- 25.Walencewicz-Wasserman A.J., Kosmoski J., Cribbs D.H., Glabe C.G., Cotman C.W. Structure-activity analyses of β-amyloid peptides: contributions of the β25–35 region to aggregation and neurotoxicity. J. Neurochem. 1995;64:253–265. doi: 10.1046/j.1471-4159.1995.64010253.x. [DOI] [PubMed] [Google Scholar]
- 26.Yang D.-S., Yip C.M., Huang T.H.J., Chakrabartty A., Fraser P.E. Manipulating the amyloid-beta aggregation pathway with chemical chaperones. J. Biol. Chem. 1999;274:32970–32974. doi: 10.1074/jbc.274.46.32970. [DOI] [PubMed] [Google Scholar]
- 27.Ippel J.H., Olofsson A., Schleucher J., Lundgren E., Wijmenga S.S. Probing solvent accessibility of amyloid fibrils by solution NMR spectroscopy. Proc. Natl. Acad. Sci. USA. 2002;99:8648–8653. doi: 10.1073/pnas.132098999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ma B., Nussinov R. The stability of monomeric intermediates controls amyloid formation: Aβ25–35 and its N27Q mutant. Biophys. J. 2006;90:3365–3374. doi: 10.1529/biophysj.105.075309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ma B., Nussinov R. Simulations as analytical tools to understand protein aggregation and predict amyloid conformation. Curr. Opin. Chem. Biol. 2006;10:445–452. doi: 10.1016/j.cbpa.2006.08.018. [DOI] [PubMed] [Google Scholar]
- 30.Lopez de la Paz M., de Mori G.M., Serrano L., Colombo G. Sequence dependence of amyloid fibril formation: insights from molecular dynamics simulations. J. Mol. Biol. 2005;349:583–596. doi: 10.1016/j.jmb.2005.03.081. [DOI] [PubMed] [Google Scholar]
- 31.Melquiond A., Boucher G., Mousseau N., Derreumaux P. Following the aggregation of amyloid-forming peptides by computer simulations. J. Chem. Phys. 2005;122:174904. doi: 10.1063/1.1886725. [DOI] [PubMed] [Google Scholar]
- 32.Melquiond A., Mousseau N., Derreumaux P. Structures of soluble amyloid oligomers from computer simulations. Proteins. 2006;65:180–191. doi: 10.1002/prot.21100. [DOI] [PubMed] [Google Scholar]
- 33.Lu Y., Derreumaux P., Guo Z., Mousseau N., Wei G. Thermodynamics and dynamics of amyloid peptide oligomerization are sequence dependent. Proteins. 2009;75:954–963. doi: 10.1002/prot.22305. [DOI] [PubMed] [Google Scholar]
- 34.Berendsen H.J.C., Postma J.P.M., von Gunsteren W.F., Hermans J. D. Reidel Publishing; Dordrecht, The Netherlands: 1981. Intermolecular Forces: Interaction Models for Water in Relation to Protein Hydration. [Google Scholar]
- 35.Lindahl E., Hess B., van der Spoel D. GROMACS 3.0: a package for molecular simulation and trajectory analysis. J. Mol. Model. 2001;7:306–317. [Google Scholar]
- 36.Van Gunsteren, W.F., S.R. Billeter, A.A. Eising, P.H. Hunenberger, P. Kruger, et al. 1996. Biomolecular Simulation: The GROMOS96 Manual and User Guide. Vdf Hochschulverland, ETH, Zurich, Switzerland.
- 37.Berendsen H.J.C., Postma J.P.M., von Gunsteren W.F., DiNola A., Haak J.R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984;81:3684–3690. [Google Scholar]
- 38.Miyamoto S., Kollman P.A. Settle an analytical version of the shake and rattle algorithm for rigid water models. J. Comput. Chem. 1992;13:952–962. [Google Scholar]
- 39.Hess B., Bekker H., Berendsen H.J.C., Fraaije J. A linear constraint solver for molecular simulations. J. Comput. Chem. 1997;18:1463–1472. [Google Scholar]
- 40.Hummer G., Rasaiah J.C., Noworyta J.P. Water conduction through the hydrophobic channel of a carbon nanotube. Nature. 2001;414:188–190. doi: 10.1038/35102535. [DOI] [PubMed] [Google Scholar]
- 41.Hirschfelder J.O., Curtiss C.F., Brid R.B. John Wiley and Sons; New York: 1954. Molecular Theory of Gases and Liquids. [Google Scholar]
- 42.Humphrey W., Dalke A., Schulten K. VMD: Visual molecular dynamics. J. Mol. Model. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 43.Hecht M.H. De novo design of beta-sheet proteins. Proc. Natl. Acad. Sci. USA. 1994;91:8729–8730. doi: 10.1073/pnas.91.19.8729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Luhrs T., Ritter C., Adrian M., Riek-Loher D., Bohrmann B. 3D structure of Alzheimer's amyloid-beta(1–42) fibrils. Proc. Natl. Acad. Sci. USA. 2005;102:17342–17347. doi: 10.1073/pnas.0506723102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zheng J., Jang H., Ma B., Nussinov R. Annular structures as intermediates in fibril formation of Alzheimer Aβ17–42. J. Phys. Chem. B. 2008;112:6856–6865. doi: 10.1021/jp711335b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Takahashi T., Mihara H. Peptide and protein mimetics inhibiting amyloid β-peptide aggregation. Acc. Chem. Res. 2008;41:1309–1318. doi: 10.1021/ar8000475. [DOI] [PubMed] [Google Scholar]
- 47.Kostarelos K. The long and short of carbon nanotube toxicity. Nat. Biotechnol. 2008;26:774–776. doi: 10.1038/nbt0708-774. [DOI] [PubMed] [Google Scholar]
- 48.Krone M.G., Hua L., Soto P., Zhou R., Berne B.J. Role of water in mediating the assembly of Alzheimer amyloid-beta Abeta16–22 protofilaments. J. Am. Chem. Soc. 2008;130:11066–11072. doi: 10.1021/ja8017303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nguyen P.H., Li M.S., Stock G., Straub J.E., Thirumalai D. Monomer adds to preformed structured oligomers of Abeta-peptides by a two-stage dock-lock mechanism. Proc. Natl. Acad. Sci. USA. 2007;104:111–116. doi: 10.1073/pnas.0607440104. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.