

Abstract
The origin of the biexponential fluorescence decay of Trp in ribonuclease T1 under mildly destabilizing conditions, such as increased pH or temperature, or the presence of detergent, is still not understood. We have performed two extended replica-exchange molecular dynamics simulations to obtain a detailed representation of the native state at two protonation states corresponding to a high and low pH. At high pH, the appearance of partially unfolded states is evident. We found that this pH-induced destabilization originates from increased global repulsion as well as reduced local favorable electrostatic interactions and reduced H-bonding strength of His27, His40, and His92. At high pH, alternative tryptophan rotamers appear and are linked to a distorted environment of the tryptophan, which also acts as a separate source of ground-state heterogeneity. The total population of these alternative conformations agrees well with the amplitude of the experimentally observed secondary fluorescence lifetime.
Introduction
The interpretation of the time-resolved fluorescence decay of tryptophan in proteins continues to challenge both experimentalists and theoreticians. The most often cited model, known as the rotamer model, provides a simple explanation for the ubiquitous multiplicity of fluorescence lifetimes (1–3). According to this model, each lifetime component is associated with a single rotamer state or a narrow distribution of rotamer states, which, upon excitation, decays at a characteristic exponential rate. Since the environment of each rotamer state can be very different, and since the Trp fluorescence quenching rate is very sensitive to the electrostatic field, different rotamers are expected to have different lifetimes. Spectral relaxation, another often-cited model, also relies on the sensitivity of Trp fluorescence to the electrostatic field (4). A redistribution of the electron charge upon electronic excitation causes a dielectric response of the protein environment (or the Trp side chain itself by a rotamer shift). A change of the dielectric field on the timescale of the excited-state lifetime (on the order of nanoseconds) in turn causes a change in the fluorescence quenching rate. The dielectric relaxation model and the rotamer model may jointly contribute to the nonexponential decay, as relaxation might occur within single rotamer states. Both models have gained their share of supporters over the past decades.
This work focuses mainly on a primary condition for the rotamer model, namely the existence of Trp rotamers in the electronic ground state. Recently, we reported an efficient equilibrium molecular dynamics (MD) simulation on the Bacillus caldolyticus cold shock protein using an adapted version of the replica-exchange method (REM) (5). To improve sampling efficiency, exchanges between replicas were performed at increased temperature and volume while the protein was restrained within the native state. The generated ensemble of protein structures was distributed between two long-lived Trp rotamers, the populations of which correlated well with fluorescence-lifetime amplitudes. The single Trp side chain of B. caldolyticus cold shock protein is relatively solvent-exposed (43% (5)), allowing for side-chain flexibility. The next logical step was to examine whether the Trp side chain may also adopt alternative conformations when buried inside the protein matrix, and how those alternative conformations are related to protein structure and dynamics.
Ribonuclease T1 (RNase T1) from the mold fungus Aspergillus oryzae contains a single tryptophan side chain, Trp59, which resides in a hydrophobic protein environment and is almost completely shielded from solvent. Only one Trp59 rotamer could be identified in all x-ray crystal and NMR structures. Earlier molecular mechanics and MD studies suggested that Trp59 resides in a conformationally restricted region (6). Its side chain conformational freedom is restrained within one rotamer state by the protein matrix, and rotamer interconversions are very unlikely (7,8).
The RNase T1 crystal structure (Fig. 1) contains one α-helix and two antiparallel β-sheets, A and B. Sheet A consists of two β-strands (βA and βB). Sheet B, composed of five β-strands (β1–β5), is strongly twisted to form an open barrel and harbors the RNase T1 active site. When the single-stranded RNA substrate is bound to the enzyme, it acts as the missing sixth strand to complete the β-barrel. The α-helix is tightly bound to sheet B to increase stability. Extra stability is provided by two disulfide bonds, Cys2-Cys10 and Cys6-Cys103. Trp59 is part of strand β2, which, it has been suggested, is part of the folding core (9). The indole ring of Trp59 is located between the helix at one side and the loops between helix-β1 and β2-β3 at the other side of the plane.
Figure 1.
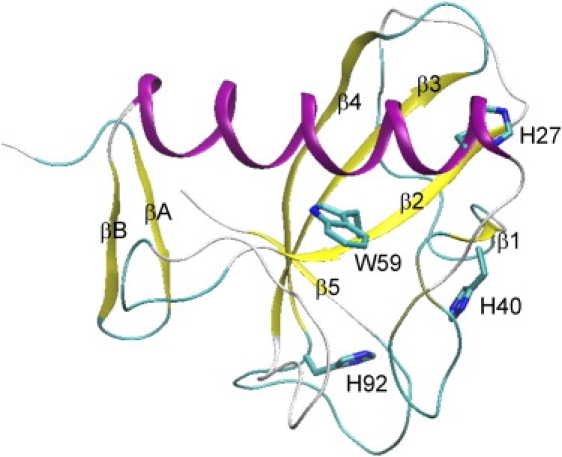
Ribbon diagram of the RNase T1 crystal structure (PDB code 9RNT).
RNase T1 cleaves the phosphodiester bond of single-stranded RNA at the 3′ position of guanosine nucleotides with high specificity. The two-step cleavage mechanism involves a transesterification to produce a 2′,3′-cyclic phosphate followed by hydrolysis to the 3′-phosphate. The affinity for the substrate is highest at the guanine binding site (residues 42–62 and 98), whereas the cleavage occurs at the catalytic site. Residues involved in catalytic activity are Tyr38, His40, Glu58, Arg77, and His92 (10).
The fluorescence intensity decay of RNase T1 is well described as a single-exponential process at pH ≤6, whereas at higher pH, two exponential terms are required (11–15). A biexponential decay has also been observed at pH 5.5 at increased temperature or in the presence of guanidine hydrochloride detergent (16).
In this work, we present the results of two extended simulations of RNase T1 using the same REM scheme as in our earlier work (5), with different protonation states, corresponding to high and low pH, aiming to elucidate the conformational states that might be responsible for the biexponential fluorescence decay. At the same time, the wealth of information acquired from the simulations provided us with a detailed description of the RNase T1 native-state dynamics and stability.
Methods
Replica-exchange molecular dynamics
The details of the MD simulations and analysis are provided in the Supporting Material. The initial structure was obtained from the x-ray structure of RNase T1 (Protein Data Bank (PDB) code 9RNT). REM simulations were carried out based on the method of Paschek and García (17). Trial swaps were performed between replicas with different temperatures and volumes. The acceptance probability for replica exchange between two states i and j with corresponding temperatures Ti and Tj and box volumes Vi and Vj is given by (17)
| (1) |
where β = 1/kT and U(x)V is the potential energy of the state with coordinates x and volume V. The energy U(xi)Vj is approximated as
| (2) |
where M represents the number of molecules in the simulation box, and Pi′ and βi′ denote the instantaneous pressure and temperature. An analogous expression is used for U(xj)Vi. The justification (17) for Eq. 2 is based on the idea that only small volume changes occur between two neighboring replicas (0.00925 g cm−3 density change). Due to this energy approximation, however, the detailed balance condition is only approximately satisfied.
It was observed that, for efficient sampling of the Trp59 side chain, reduced densities and high temperatures were necessary. When the system was heated without reducing the density, χ2 transitions became very unlikely due to the high system pressure. Therefore, 40 replicas were distributed exponentially over a 300–645 K temperature range and evenly over a 1.02–0.65 g cm−3 density range. Temperature and volume exchange moves occurred simultaneously. Volume changes were performed such that only intermolecular distances were altered. To mitigate the small nonequilibrium effects that stem from Eq. 2, after the exchange was accepted, densities were allowed to change gradually over a time period of 0.15 ps, followed by a 0.15 ps equilibration time before data collection. In this time frame, the nonequilibrium effects caused by sudden volume changes disappeared exponentially.
For the generation of starting conformations, each replica was heated to its target temperature, changed gradually to the desired density and subsequently simulated for 1 ns without replica exchange moves. Replicas were simulated in the NVT ensemble. The time constant for temperature coupling was 0.5 ps. Exchanges were attempted every 0.5 ps. Acceptance probabilities varied between 0.13 and 0.26. Both REM simulations were carried out for 66 ns per replica, the last 17.5 ns of which were used to calculate the reported thermodynamic properties.
Dead-end elimination method
To investigate whether backbone flexibility is necessary to accommodate alternative Trp rotamers, the REM simulations were compared with a dead-end elimination (DEE) (18) analysis on the possible Trp conformations. The identification of the rotamer clusters occurred as previously described by Hellings et al. (19). A full-hydrogen model and energy-minimized x-ray structure (PDB code 9RNT) was used as the starting structure. Four internal crystallographic water molecules (WAT107, WAT108, WAT110, and WAT124) were included in the calculation and were allowed to rotate. The used rotamer library was an enhanced version of the library of De Maeyer et al. (20). The DEE algorithm has been implemented in the BRUGEL package (21) using an adapted version of the CHARMM22 force field (22).
Results
We have conducted extended REM simulations of RNase T1 in two different protonation states. In the first protonation state, all histidine residues (His27, His40, and His92) are doubly protonated, corresponding to pH 6. In the second protonation state, all His residues are singly protonated, corresponding to pH 8. His27 is deprotonated at Nδ1, and His40 and His92 are deprotonated at Nɛ2 (23).
The RNase T1 native state
Backbone deviations and fluctuations
The backbone root mean-square deviation (bRMSD) histograms at pH 6 (Fig. 2) reveal a flexible native state with most bRMSD values ranging between 1 and 3.3 Å, well below the restraint value of 4.5 Å. Three substates are evident: a major peak at 2.0 Å and two minor peaks at 1.4 and 2.5 Å. At pH 8, the protein deviates much more from the x-ray structure (Fig. 2). Here, we find large peaks at 2.2 Å and 3.1 Å and a shoulder at 3.3 Å. The small peaks at 4.0 Å and 4.3 Å contain alternative Trp59 conformations (vide infra). From the size of the error bars, it is clear that both simulations have not yet fully equilibrated.
Figure 2.
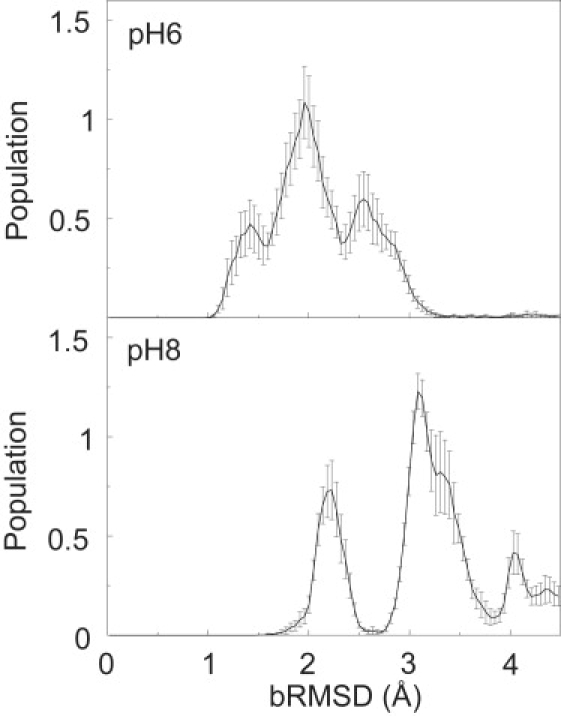
Normalized populations of the backbone RMSD from the x-ray structure for the simulations at pH 6 and pH 8.
Looking at the average bRMSD per residue (Fig. 3), deviations from the x-ray structure are found mainly in the loops. The highest divergence is located in the loops between strands β1 and β2 and between strands β4 and β5, which are both part of the guanine binding site. In both basic and acidic pH simulations, the catalytic residues remain close to the x-ray structure, with the exception of His92, located in the loop between β4 and β5. The configuration around Trp59 is highly preserved in solution, in agreement with deuterium exchange experiments (9). At pH 8, additional divergence from the x-ray structure is found at the N-terminus, in β-sheet A, at the N- and C-terminal ends of the helix, in the loop between the helix and strand β1, in strand β5, and at the C-terminus.
Figure 3.
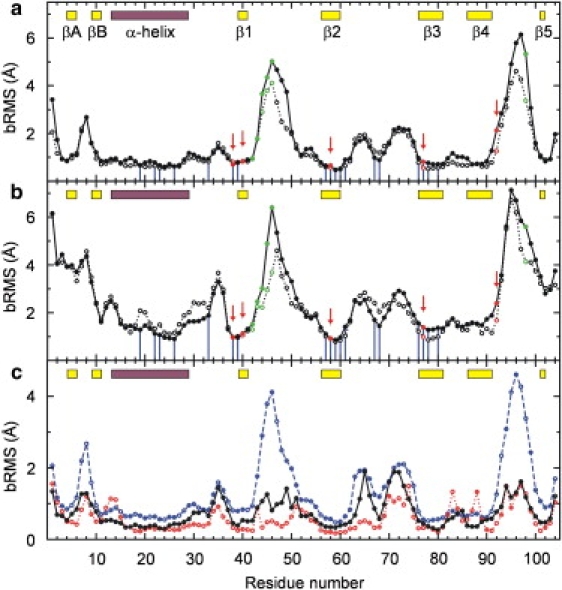
Average bRMS fluctuation (open circles) and bRMSD (solid circles) from the x-ray structure per residue at (a) pH 6 and (b) pH 8. Arrows (red) indicate residues involved in catalytic activity; light circles (green) indicate residues located in the guanine binding site; vertical droplines (blue) indicate residues located within a 4.5-Å sphere around Trp59. Color bars at top indicate the β-strands (yellow) and α-helix (purple) of the x-ray structure. (c) bRMS fluctuations for the pH 6 simulation (dashed line) and the NMR structures 1IYY (25) (solid line) and 1YGW (26) (dotted line).
The bRMS fluctuations are very similar to the average bRMSD values (Fig. 3, a and b) at both pH values. Thus, the protein fluctuates around its x-ray conformation, supporting a funnel-like shape of the RNase T1 energy surface (24).
The shape of the bRMS fluctuation curve at pH 6 is strikingly similar to the NMR-derived data of the Gln25 isozyme of RNase T1 at pH 5.5 (PDB code 1IYY) (Fig. 3 c) (25). The higher flexibility for the pH 6 simulation in the loops βA-βB, β1-β2, and β4-β5 can be expected, considering that NMR structures are generated using distance restraints and dihedral angles. Whereas the NMR signal is ensemble-averaged, this signal is usually imposed onto single structures during NMR refinement, thereby possibly biasing the resulting structural ensemble. The flexibility in such an “NMR ensemble” may therefore not be representative of fluctuations that take place in solution.
The overall fold was also very similar to the NMR structure of Lys25-RNase T1 at pH 5.5 (PDB code 1YGW) (26), but some marked differences are apparent (Fig. 3 c). Compared to our simulation data and 1IYY, increased disorder of residues 12–14, 83, and 88, and decreased disorder of residues 44–50 and 64–66 was observed in 1YGW. Lys25 is H-bonded with either Glu28 or Asp29, and all three residues are located on the α-helix and are not involved in any other local interaction. Therefore, we don't expect the Lys25Gln mutation to have a large effect on the global protein conformation, despite a 0.9 kcal mol−1 decrease of stability (27).
Different protocols were used for the structure determinations of 1IYY and 1YGW. 1IYY was determined by the hybrid distance geometry/simulated annealing protocol based on distance and dihedral angle constraints from nuclear Overhauser effect intensities and 3J coupling constants. The final set of structures was selected on the basis of minimum total energy and restraint violations. The 1YGW structures were generated using target function calculations based on nuclear Overhauser effect intensities, 3J coupling constants, and amide proton exchange rates. The final structure ensemble was selected comparing the global precision of structures with order parameters derived from 15N relaxation time measurements. Note that 15N spin relaxation techniques monitor the reorientation of 15N-H bond vectors on the picosecond to nanosecond timescale (28), serving as a lower limit of the actual degree of disorder.
Secondary structure
The average bRMSD and bRMS fluctuations are related to loss of secondary structure (Fig. S1). Most structural elements of the x-ray structure are preserved in the pH 6 simulation, apart from the H-bonded turns between the loops βA-βB, β1-β2, and β4-β5. In contrast, at pH 8 the secondary structure content is strongly reduced for strands βA, βB, and β5, the β-bridge formed by residues 33 and 38, and at the C- and N-terminal sides of the α-helix.
Protein substates
In Fig. 4, the free energy as a function of bRMSD and the fraction of native contacts (Q) is shown. At pH 6, most conformations retain >74% of the native contacts and are located within one of three free-energy basins separated by low barriers. The pH 8 simulation spans a much broader free-energy surface. Many substates are visible and Q values range between 0.46 and 0.85.
Figure 4.
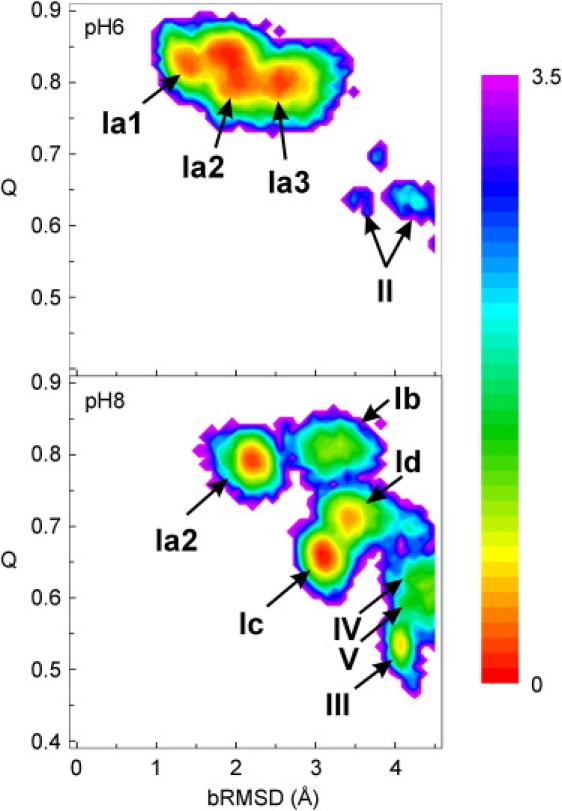
Free energy (kcal mol−1) as a function of bRMSD and Q for the REM simulations at pH 6 and pH 8. Trp59 rotamers are indicated with roman numerals I–V, as defined in Figs. 6 and 7.
Some representative RNase T1 conformations for the most stable substates shown in Fig. 4 are depicted in Fig. 5. Substates Ia1, Ia2, Ia3, and Ib largely preserve the x-ray fold and differ only in the loops. In state Ic, sheet A, the N-terminal end of the helix and strand β5 are broken. In state Id, strand β5 is broken and large displacements are observed in sheet A and loop β1-β2 and at the N- and C-termini.
Figure 5.
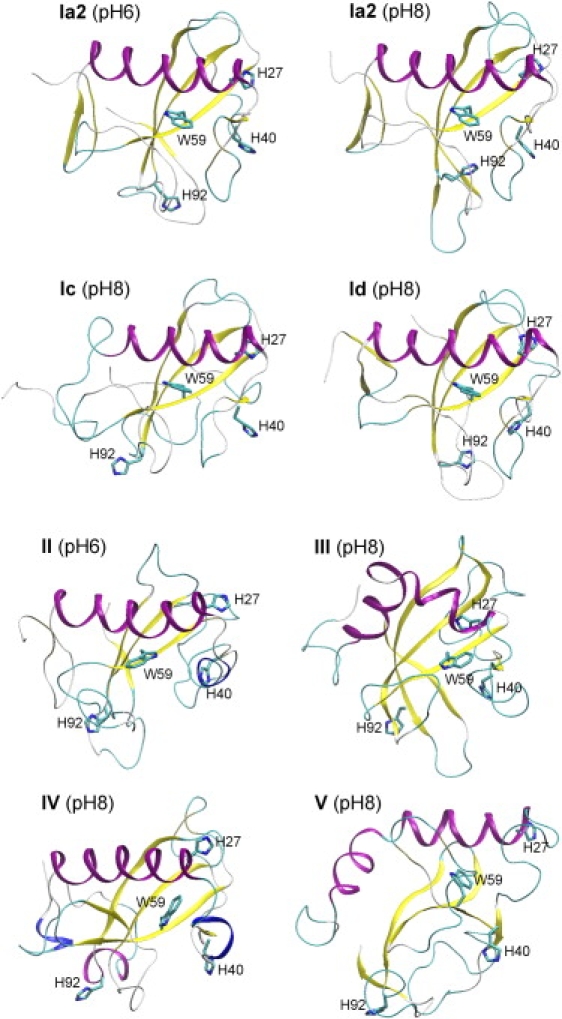
Representative RNase T1 conformations of the most stable substates and alternative Trp59 rotamers at pH 6 and pH 8, with numbering according to Figs. 4, 6, and 7. Stereo figures are available in Fig. S2.
States II–V (Figs. 4 and 5) contain more severely distorted secondary structural features and correspond to alternative Trp59 conformations, as verified by bRMSD-Q plots for each of the Trp59 states. These will be discussed in more detail (vide infra).
Trp59 H-bonds
In the RNase T1 x-ray crystal structure, the Trp59 indole is H-bonded via the indole nitrogen Nɛ1-Hɛ1 with a structural water molecule (WAT107) that is buried inside the protein. This H-bond is conserved in both pH 6 and pH 8 simulations (−1.21 and −1.23 kcal mol−1, respectively), and is present only when Trp59 is in its x-ray rotamer state (vide infra). The H-bonded water stabilizes loop β2-β3 through additional H-bonds with Tyr68 N-H and Pro60 O, and forms a chain of four H-bonded internal water molecules (WAT107-WAT108-WAT110-WAT124) located between loop β2-β3 and the helix, connecting the bulk water with the protein interior.
Histidine H-bonds
To uncover the origin of the reduced stability in the pH 8 simulation, we examined in detail the H-bonds that are formed by the histidines. A list of the H-bonds involving the histidines is summarized in Table S7. His27, located at the C-terminal end of the helix, forms an H-bond via the Hɛ2 of the imidazole nitrogen Nɛ2 with the Glu82 carboxylate oxygen (Oɛ1 or Oɛ2), strengthening the interaction between the helix and the turn between β3 and β4. This H-bond is conserved at pH 6, but largely lost at pH 8. At high pH, Glu82 mostly orients away from His27 toward the solvent. When His27 is H-bonded to Glu82, its rotamer state is fixed at (χ1 = 173°, χ2 = 63°); otherwise, the imidazole ring rotates freely around χ2. H-bonds of His40 with Asn36 and Ser37 stabilize the loop between the helix and β1. His40 occupies four rotamer states that are favorable for H-bonding. 1), In the x-ray rotamer state (χ1 = 52°, χ2 = −120°), His40 Nδ1-Hδ1 is H-bonded with Asn36 main-chain O and/or Ser37 main-chain O. The doubly protonated form is extra stabilized by an H-bond between Nɛ2-Hɛ2 and Glu58 Oɛ1/ɛ2. 2), Rotamer (χ1 = 176°, χ2 = −110°) forms an H-bond with Glu58 Oɛ1/ɛ2 via Nδ1-Hδ1. 3), Rotamer (χ1 = −53°, χ2 = 134°) H-bonds with Glu58 Oɛ1/ɛ2 via Nδ1-Hδ1 and is strengthened at pH 6 by H-bonds with Asn36 main-chain O and/or Asn36 Oδ1 via Nɛ2-Hɛ2. 4), Rotamer (χ1 = 62°, χ2 = 76°) facilitates H-bonding between Nδ1-Hδ1 and Lys41 main-chain O. Here, the REM results do not agree well with the NMR data. In 1IYY, rotamers (χ1 = 20°, χ2 = 41°) and (χ1 = −3°, χ2 = −70°) are present for His40. In 1YGW, only rotamer (χ1 = 69°, χ2 = 80°) is observed. His92 is located at the start of the very flexible loop β4-β5, allowing the formation of many rotamers and H-bonds. The difference in total His92 H-bonding interaction energy between the two pH simulations is relatively small.
Thermal unfolding
At pH 7, RNase T1 is most stable at 267 K (29). The experimentally determined midpoint of thermal unfolding, Tm, ranges between 321.1–323.8 K at pH 7 and 327 K at pH 5.5 (29–31). RNase T1 unfolding is well described by a two-state process between a native (N) and unfolded (U) state (31,32).
Fig. S3 shows the bRMSD populations at various temperatures between 300 K and 331 K. At pH 6, substates Ia1 and Ia2 gradually decrease with concomitant population increase of substate Ia3, and the region with bRMSD > 3.5 Å. Between 312 K and 331 K, the population of substate Ia3 stays constant. At 3.5 Å bRMSD, the population is very low for all plotted temperatures, indicating that the unfolding transition state is located at this bRMSD value. Note that for the majority of protein structures (82%), the bRMSD is <3.5 Å at 331 K. In contrast, at pH 8, the protein appears much more sensitive to a temperature increase. The peaks below 3.8 Å decrease at a much faster rate, whereas above this bRMSD value, the population increases markedly. At 331 K, most structures (73%) have bRMSD values >3.8 Å. Unfortunately, we were unable to compare absolute values with experimentally determined Tm values, because bRMSD values >4.5 Å were not allowed in our simulations.
Tryptophan rotamers
Dead-end elimination
The result of the DEE calculation, based on 9RNT is shown in Fig. 6. The major rotamer cluster I (χ1 = 170°, χ2 = −120°) corresponds to the x-ray conformation of Trp59. The minor cluster II (χ1 = −180°, χ2 = 90°) is much less stable. The nonbonded energy difference of Trp59 with the protein environment, ETrpnb(II) − ETrpnb(I), equals 11.1 kcal mol−1. Rotamer I is stabilized by H-bonding of indole Nɛ1-Hɛ1 with WAT107. Simultaneously, rotamer II is destabilized by interactions between Trp59 Cζ3-Hζ3 and the water oxygen. This result is consistent with the single fluorescence lifetime data at low pH 6. The DEE plot of 8RNT, crystallized at pH 5.2, was very similar (although minor energy differences were noted), confirming its high structural similarity with 9RNT, which crystallized at pH 7.
Figure 6.
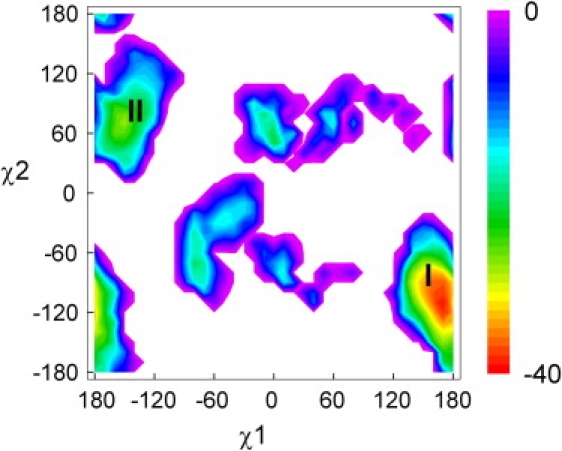
DEE result of the nonbonded interaction energy (kcal mol−1) of Trp59 with the surrounding protein in the function of χ1 and χ2.
Rotamer populations from replica exchange
The REM free energy plots of the Trp59 χ1 × χ2 map at pH 6 (Fig. 7) strongly resembles the DEE plot (Fig. 6). With 0.994 ± 0.005 of the population within rotamer state I, rotamer II is negligible at 300 K. As the pH increases and/or the temperature increases, the population of rotamer I decreases and several additional rotamers emerge (Fig. 7 and Fig. S4).
Figure 7.
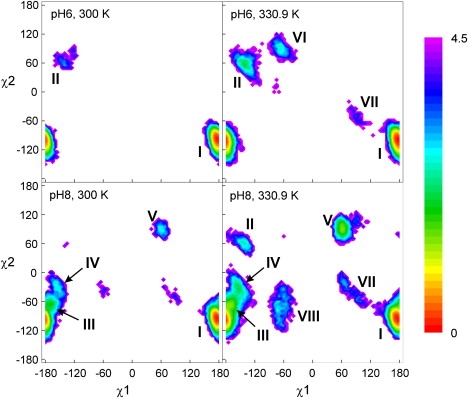
Free energy plotted as a function of Trp59χ1 and χ2 from the REM simulations at 300 K and 330.9 K for pH 6 and pH 8.
In Fig. 8, the equilibration of the four highest Trp59 rotamer populations over time is shown for the pH 8 simulation. Accurate ratios of the alternative rotamers (III, IV, and V) could not be obtained, but the population of the dominant rotamer (I) seems well equilibrated. At pH 8, we found a rotamer I population of 0.91 ± 0.02. The second highest rotamer (III) has a population of ∼0.05. Note that in the χ1 − χ2 plot, it would be difficult to distinguish rotamers I, III, and IV. The distinction becomes apparent when the free energy is plotted against Q and the bRMSD and χ2 (Fig. 4) for each rotamer.
Figure 8.
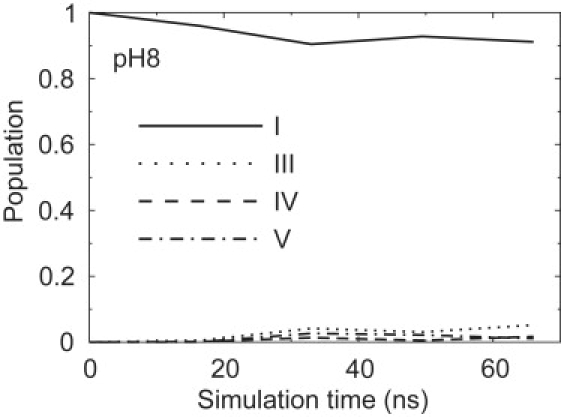
Evolution of the main Trp59 rotamer populations at 300 K as a function of simulation time (numbering according to Fig. 7).
As is evident in Figs. 4 and 5, and Fig. S6, a and c, all protein conformations with an alternative Trp59 rotameric conformation strongly deviate from the native state. Severe disruption of the secondary structure is evident, most notably sheet A and parts of the helix and sheet B, whereas many contacts between the helix and sheet B have disappeared. In conjunction with the global backbone deviations, the immediate surroundings of the alternative rotamers diverge strongly (Fig. S6, b and d). In contrast, most rotamer I structures keep their Trp59 environment very close to the x-ray structure. For the alternative rotamers, deviations are located mainly in the loop helix-β1 and β2-β3. Relative to rotamer I, the strained, nonrotameric conformational states, III and IV, are partially rotated about χ2 (Fig. 7) by steric interaction with the loop helix-β1. Rotamer III is in close contact with the Tyr38 carbonyl and Cα atoms; rotamer IV makes close contact with the Pro39 ring. Both rotamers III and IV kept their respective rotamer state during a 2-ns MD simulation.
Solvent accessibility
The solvent accessibility of Trp59 is related to the backbone deviations. Compared to rotamer I, higher average solvent-accessible surface area (SASA) values and standard deviations are observed for all the alternative rotamers (Table 1). However, the limited number of data points does not allow a statistical error estimation for the alternative rotamers.
Table 1.
Trp59 solvent accessibilities and indole-carbonyl distances
| Rotamer | SASA (SD) (Å2) | Indole-carbonyl distance∗ (Å) | ||
|---|---|---|---|---|
| I | (9RNT) | 3.3 | 4.9 | |
| I | (pH 6) | 4.0 ± 0.2 | (3.2) | 4.7 |
| I | (pH 8) | 5.7 ± 0.3 | (4.9) | 4.6 |
| II | (pH 8) | 23.3 | (12.3) | 4.6 |
| III | (pH 8) | 5.9 | (7.7) | 4.3 |
| IV | (pH 8) | 10.8 | (10.0) | 4.8 |
| V | (pH 8) | 15.2 | (7.3) | 4.9 |
Average distance between the center of the indole phenyl ring and the nearest carbonyl carbon atom.
Indole-carbonyl distances
For comparison with the experimental fluorescence lifetime components, we calculated the distance between the center of the indole phenyl ring and the nearest carbonyl carbon (Table 1). For rotamer I, the nearest carbonyl was most often Ala22 (78%). Markedly shorter indole-carbonyl distances were found for rotamer III, due to the carbonyl groups of Tyr38 (65%) and Leu26 (30%). The same trend was observed when the distance between the nearest phenyl carbon and the nearest carbonyl carbon was considered.
Discussion
Sources of protein (de)stabilization
We have performed two extended REM simulations on RNase T1, each with different histidine protonation states, corresponding to pH 6 and pH 8. Our data indicate a strongly reduced stability and increased flexibility at higher pH. This result is consistent with experiment. RNase T1 is most stable at pH 4.5 (33). The conformational stability increases more than 4 kcal mol−1 from pH 9 to pH 5 (34). The increased stability at low pH can be explained by reduced global electrostatic repulsion. In the pH 8 simulation, the protein has a total charge of −9 e. These repulsions are, however, partly screened by the counterions. Another stabilizing effect is caused by local charge—charge interactions and H-bonds between cationic histidines and carboxylate anions. Deprotonation of His27 at Nδ1 causes a weakening of the H-bond interaction with Glu82, and a consequent weakening of the tight interaction between turn β4-β5 and the C-terminal end of the helix. The importance of the turn β4-β5 for folding and stability has been demonstrated by Garrett et al. from circularly permuted variants of RNase T1 (35), and by Mullins et al., who observed high protection against deuterium exchange of the backbone amide hydrogens in this region (9). H-bond interactions of His40 with Asn36 and Ser37, which stabilize the secondary structure of the loop between the helix and β1, are reinforced in the doubly protonated form of His40 by the formation of additional H-bonds, especially with Glu58. The effect of this stabilization is clearly seen in Fig. 3 and Fig. S1. Both stabilizing factors also account for the increased pKa values for the three histidines (7.08–7.3, 7.75–7.9, and 7.31–7.8 for His27, His40, and His92, respectively (23,34)). As a reference, for a histidine in the unfolded state, a pKa value of 6.6 would be expected.
Tryptophan fluorescence and ground-state heterogeneity
Studies of the RNase T1 fluorescence properties have been thoroughly discussed in the literature. The high-fluorescence quantum yield of 0.31 (36) is similar to that of 3-methylindole in various solvents (37). The blue-shifted fluorescence maximum at 320 nm and the fine structure of the fluorescence spectrum are characteristic for the hydrophobic environment (38,39). RNase T1 undergoes a time-dependent spectral shift to longer emission wavelengths characterized by a single dipolar relaxation time of 0.56–0.696 ns, but the spectral shift is very small (∼44 cm−1, ∼0.5 nm) (13,39). The relatively high activation energy for acrylamide quenching (8 kcal mol−1, compared to 3.7 kcal mol−1 for indole derivatives in water (40)) suggested that the protein undergoes fluctuations on the nanosecond timescale, allowing acrylamide molecule diffusion into the protein core (15). No fast-rotational correlation time is associated with anisotropy measurements, suggesting that Trp59 is practically immobilized inside the protein matrix (16).
In conjunction with the DEE results, the simulation data suggest that Trp59 side-chain flexibility is strongly related to protein stability. Alternative Trp59 rotamers are found by our simulation under mildly destabilizing conditions, such as pH or temperature increase. This observation correlates remarkably well with fluorescence decay properties. A single decay with fluorescence lifetime (τ = 4.0 ns) is observed at low pH and temperature, whereas a double-exponential decay (τ1 = 3.9 ns; τ2 = 0.8–1.2 ns) with corresponding amplitudes (α1 = 0.84–0.9; α2 = 0.16–0.1) is observed at high pH or high temperature (8,11). Above pH 7, the amplitude fraction of the major (long) lifetime component coincides well with the population of the dominant rotamer I. Given the high similarity of the Trp59 environments for the majority of rotamer I conformations, we assign a single lifetime to this rotamer state. Although the dihedral angle diagram of the fluorophore (Fig. 7) discloses the presence of alternative structures, a full deconvolution of these structures can only be realized by adding extra dimensions of bRMSD and Q. Although the limited sampling of these alternative structures does not provide conclusive evidence, we suspect that their combined heterogeneity is responsible for the minor (short) lifetime component. The short distance between the indole ring of rotamer III and the Tyr38 carbonyl might allow for fast electron transfer quenching, which could explain the short lifetime of the minor decay component. Note also that the rotamers observed at pH 8 and 300 K differ strongly from the ones at pH 6 and 331 K (Fig. 7), suggesting different unfolding pathways in both destabilizing conditions (high temperature or high pH), as is also seen in Fig. S3.
At pH 7.4, acrylamide quenches the minor lifetime component ∼5 times faster than it does the major component, whereas the amplitudes of the two components are approximately independent of quencher concentration, suggesting two Trp conformations (11). Our simulations confirm the rotamer interpretation of the quenching experiments. All the alternative structures have higher SASA values and SASA deviations than rotamer I (Table 1).
Besides multiple Trp conformations (11), several other sources of heterogeneous fluorescence decay of RNase T1 have been suggested in the literature, including multiple protein states (16), multiple excited-state energy transfer acceptors (41), multiple histidine protonation states, and multiple neighboring side-chain isomers (42). According to our simulation data, multiple rotamer states, each associated with a different protein state, are responsible for the lifetime heterogeneity. In the case of rotamers I and III, which are very close in the χ1 × χ2 space (Fig. 7), the protein environment plays a dominant role.
Based on folding studies of an RNase T1 mutant with a single cis peptide bond at Pro39, Kiefhaber et al. (38) suggested that the second lifetime component is related to a residual trans-Pro39 folding intermediate. Since Pro39 is in close contact with Trp59 in the native state, a trans ω Tyr38-Pro39 dihedral angle might influence the fluorescence lifetime. With the exception of the highest temperature replicas, we did not observe any trans-Pro39 conformation. However, we cannot exclude the possibility of a trans-Pro39 substate beyond a bRMSD value of 4.5 Å.
The findings presented here may be validated experimentally using point mutations that alter rotamer distributions and fluorescence properties. For instance, further destabilization of the folded state, e.g., by introducing residues that decrease the net protein charge, might raise the alternative rotamers' populations.
In conclusion, we have reached a detailed understanding of the ground-state heterogeneity of a buried Trp in a protein matrix. Deprotonation of the three His groups allows for partial unfolding and appearance of several Trp conformations by destabilizing the native state through reduction of global and local electrostatic interactions. The instability gives rise to multiple ground-state conformations, which can only partly be characterized as different rotamer states, but also show distinctions between degrees of distortion of the protein environment for states that otherwise belong to the same rotamer basin. A temperature increase also destabilizes the native state, but the nature of the partially unfolded states and rotamer states differs from those observed at increased pH.
Acknowledgments
Financial support from the Fonds voor Wetenschappelijk Onderzoek-Vlaanderen is gratefully acknowledged.
Supporting Material
References
- 1.Moors S.L.C., Jonckheer A., De Maeyer M., Engelborghs Y., Ceulemans A. How do rotameric conformations influence the time-resolved fluorescence of tryptophan in proteins? A perspective based on molecular modeling and quantum chemistry. Curr. Protein Pept. Sci. 2008;9:427–446. doi: 10.2174/138920308785915236. [DOI] [PubMed] [Google Scholar]
- 2.Donzel B., Gauduchon P., Wahl P. Study of the conformation in the excited state of two tryptophanyl diketopiperazines. J. Am. Chem. Soc. 1974;96:801–808. [Google Scholar]
- 3.Gauduchon P., Wahl P. Pulsefluorimetry of tyrosyl peptides. Biophys. Chem. 1978;8:87–104. doi: 10.1016/0301-4622(78)85026-1. [DOI] [PubMed] [Google Scholar]
- 4.Lakowicz J.R. On spectral relaxation in proteins. Photochem. Photobiol. 2000;72:421–437. doi: 10.1562/0031-8655(2000)072<0421:OSRIP>2.0.CO;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Moors S.L.C., Hellings M., De Maeyer M., Engelborghs Y., Ceulemans A. Tryptophan rotamers as evidenced by X-ray, fluorescence lifetimes, and molecular dynamics modeling. Biophys. J. 2006;91:816–823. doi: 10.1529/biophysj.106.085100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fushman D., Ohlenschlager O., Ruterjans H. Determination of the backbone mobility of ribonuclease-T1 and its 2′GMP complex using molecular-dynamics simulations and NMR relaxation data. J. Biomol. Struct. Dyn. 1994;11:1377–1402. doi: 10.1080/07391102.1994.10508074. [DOI] [PubMed] [Google Scholar]
- 7.Elofsson A., Nilsson L. A 1.2 ns molecular dynamics simulation of the ribonuclease T-1–3′-guanosine monophosphate complex. J. Phys. Chem. 1996;100:2480–2488. [Google Scholar]
- 8.Ababou A., Bombarda E. On the involvement of electron transfer reactions in the fluorescence decay kinetics heterogeneity of proteins. Protein Sci. 2001;10:2102–2113. doi: 10.1110/ps.05501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mullins L.S., Pace C.N., Raushel F.M. Conformational stability of ribonuclease T1 determined by hydrogen-deuterium exchange. Protein Sci. 1997;6:1387–1395. doi: 10.1002/pro.5560060702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gohda K., Oka K., Tomita K., Hakoshima T. Crystal-structure of RNase T1 complexed with the product 3′-GMP. Structural evidence for direct interaction of histidine 40 and glutamic acid 58 with the 2′-hydroxyl group of the ribose. J. Biol. Chem. 1994;269:17531–17536. doi: 10.2210/pdb1rls/pdb. [DOI] [PubMed] [Google Scholar]
- 11.Chen L.X.Q., Longworth J.W., Fleming G.R. Picosecond time-resolved fluorescence of ribonuclease T1. A pH and substrate analogue binding study. Biophys. J. 1987;51:865–873. doi: 10.1016/S0006-3495(87)83414-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Alcala J.R., Gratton E., Prendergast F.G. Interpretation of fluorescence decays in proteins using continuous lifetime distributions. Biophys. J. 1987;51:925–936. doi: 10.1016/S0006-3495(87)83420-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mei G., Di Venere A., De Matteis F., Rosato N. The recovery of dipolar relaxation times from fluorescence decays as a tool to probe local dynamics in single tryptophan proteins. Arch. Biochem. Biophys. 2003;417:159–164. doi: 10.1016/s0003-9861(03)00351-5. [DOI] [PubMed] [Google Scholar]
- 14.Johnson J.L., Raushel F.M. Influence of primary sequence transpositions on the folding pathways of ribonuclease T1. Biochemistry. 1996;35:10223–10233. doi: 10.1021/bi953026p. [DOI] [PubMed] [Google Scholar]
- 15.Eftink M.R., Ghiron C.A. Frequency-domain measurements of the fluorescence lifetime of ribonuclease-T1. Biophys. J. 1987;52:467–473. doi: 10.1016/S0006-3495(87)83235-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gryczynski I., Eftink M., Lakowicz J.R. Conformation heterogeneity in proteins as an origin of heterogeneous fluorescence decays, illustrated by native and denatured ribonuclease T-1. Biochim. Biophys. Acta. 1988;954:244–252. doi: 10.1016/0167-4838(88)90079-9. [DOI] [PubMed] [Google Scholar]
- 17.Paschek D., Garcia A.E. Reversible temperature and pressure denaturation of a protein fragment: a replica exchange molecular dynamics simulation study. Phys. Rev. Lett. 2004;93:238105. doi: 10.1103/PhysRevLett.93.238105. [DOI] [PubMed] [Google Scholar]
- 18.De Maeyer M., Desmet J., Lasters I. The dead-end elimination theorem: mathematical aspects, implementation, optimizations, evaluation, and performance. Methods Mol. Biol. 2000;143:265–304. doi: 10.1385/1-59259-368-2:265. [DOI] [PubMed] [Google Scholar]
- 19.Hellings M., De Maeyer M., Verheyden S., Hao Q., Van Damme E.J.M. The dead-end elimination method, tryptophan rotamers, and fluorescence lifetimes. Biophys. J. 2003;85:1894–1902. doi: 10.1016/s0006-3495(03)74617-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.De Maeyer M., Desmet J., Lasters I. All in one: a highly detailed rotamer library improves both accuracy and speed in the modelling of sidechains by dead-end elimination. Fold. Des. 1997;2:53–66. doi: 10.1016/s1359-0278(97)00006-0. [DOI] [PubMed] [Google Scholar]
- 21.Delhaise P., Bardiaux M., De Maeyer M., Prevost M., Vanbelle D. The Brugel package: toward computer-aided design of macromolecules. J. Mol. Graph. 1988;6:219. [Google Scholar]
- 22.MacKerell A.D., Bashford D., Bellott M., Dunbrack R.L., Evanseck J.D. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B. 1998;102:3586–3616. doi: 10.1021/jp973084f. [DOI] [PubMed] [Google Scholar]
- 23.Spitzner N., Lohr F., Pfeiffer S., Koumanov A., Karshikoff A. Ionization properties of titratable groups in ribonuclease T-1-I. pK(a) values in the native state determined by two-dimensional heteronuclear NMR spectroscopy. European Biophysics Journal with Biophysics Letters. 2001;30:186–197. doi: 10.1007/s002490100138. [DOI] [PubMed] [Google Scholar]
- 24.Leopold P.E., Montal M., Onuchic J.N. Protein folding funnels: a kinetic approach to the sequence structure relationship. Proc. Natl. Acad. Sci. USA. 1992;89:8721–8725. doi: 10.1073/pnas.89.18.8721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hatano K., Kojima M., Suzuki E., Tanokura M., Takahashi K. Determination of the NMR structure of Gln25-ribonuclease T1. Biol. Chem. 2003;384:1173–1183. doi: 10.1515/BC.2003.130. [DOI] [PubMed] [Google Scholar]
- 26.Pfeiffer S., Karimi-Nejad Y., Rüterjans H. Limits of NMR structure determination using variable target function calculations: ribonuclease T1, a case study. J. Mol. Biol. 1997;266:400–423. doi: 10.1006/jmbi.1996.0784. [DOI] [PubMed] [Google Scholar]
- 27.Shirley B.A., Stanssens P., Steyaert J., Pace C.N. Conformational stability and activity of ribonuclease-T1 and mutants. Gln25-Lys, Glu58-Ala, and the double mutant. J. Biol. Chem. 1989;264:11621–11625. [PubMed] [Google Scholar]
- 28.Kay L.E. Protein dynamics from NMR. Nat. Struct. Biol. 1998;5:513–517. doi: 10.1038/755. [DOI] [PubMed] [Google Scholar]
- 29.Pace C.N., Heinemann U., Hahn U., Saenger W. Ribonuclease-T1: structure, function, and stability. Angew. Chem. Int. Ed. Engl. 1991;30:343–360. [Google Scholar]
- 30.Thurlkill R.L., Grimsley G.R., Scholtz M., Pace C.N. Hydrogen bonding markedly reduces the pK of buried carboxyl groups in proteins. J. Mol. Biol. 2006;362:594–604. doi: 10.1016/j.jmb.2006.07.056. [DOI] [PubMed] [Google Scholar]
- 31.Matsuura H., Shimotakahara S., Sakuma C., Tashiro M., Shindo H. Thermal unfolding of ribonuclease T1 studied by multidimensional NMR spectroscopy. Biol. Chem. 2004;385:1157–1164. doi: 10.1515/BC.2004.149. [DOI] [PubMed] [Google Scholar]
- 32.Kiefhaber T., Quaas R., Hahn U., Schmid F.X. Folding of ribonuclease-T1. 2. Kinetic models for the folding and unfolding reactions. Biochemistry. 1990;29:3061–3070. doi: 10.1021/bi00464a024. [DOI] [PubMed] [Google Scholar]
- 33.Pace C.N., Laurents D.V., Thomson J.A. pH dependence of the urea and guanidine hydrochloride denaturation of ribonuclease A and ribonuclease T1. Biochemistry. 1990;29:2564–2572. doi: 10.1021/bi00462a019. [DOI] [PubMed] [Google Scholar]
- 34.McNutt M., Mullins L.S., Raushel F.M., Pace C.N. Contribution of histidine residues to the conformational stability of ribonuclease T1 and mutant Glu-58-Ala. Biochemistry. 1990;29:7572–7576. doi: 10.1021/bi00485a005. [DOI] [PubMed] [Google Scholar]
- 35.Garrett J.B., Mullins L.S., Raushel F.M. Are turns required for the folding of ribonuclease T1? Protein Sci. 1996;5:204–211. doi: 10.1002/pro.5560050203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.James D.R., Demmer D.R., Steer R.P., Verrall R.E. Fluorescence lifetime quenching and anisotropy studies of ribonuclease T1. Biochemistry. 1985;24:5517–5526. doi: 10.1021/bi00341a036. [DOI] [PubMed] [Google Scholar]
- 37.Meech S.R., Phillips D., Lee A.G. On the nature of the fluorescent state of methylated indole derivatives. Chem. Phys. 1983;80:317–328. [Google Scholar]
- 38.Kiefhaber T., Schmid F.X., Willaert K., Engelborghs Y., Chaffotte A. Structure of a rapidly formed intermediate in ribonuclease T1 folding. Protein Sci. 1992;1:1162–1172. doi: 10.1002/pro.5560010910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lakowicz J.R. Kluwer Academic; New York: 1999. Principles of Fluorescence Spectroscopy. [Google Scholar]
- 40.Eftink M.R., Ghiron C.A. Dynamics of a protein matrix revealed by fluorescence quenching. Proc. Natl. Acad. Sci. USA. 1975;72:3290–3294. doi: 10.1073/pnas.72.9.3290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bajzer Z., Prendergast F.G. A model for multiexponential tryptophan fluorescence intensity decay in proteins. Biophys. J. 1993;65:2313–2323. doi: 10.1016/S0006-3495(93)81325-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Haydock, C., and F. G. Prendergast. 1993. Fluorescence intensity decay of ribonuclease T1 tryptophan-59: minimum perturbation mapping study of neighbor side-chain isomerization. 1993 Biophyical Society Meeting Abstracts. Biophys J., Supplement, 64:A54, Abstract, 180-Pos.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.