

Abstract
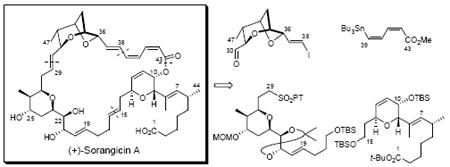
The final synthetic challenges associated with (+)-sorangicin A have been overcome, thus leading to the first total synthesis of this complex macrolide antibiotic. Highlights of the highly convergent synthesis comprised two Julia-Kocienski olefinations to unite three advanced fragments with high E-stereoselectivity. Critical to the final-stage success was the use of a carefully defined Stille coupling, a Mukaiyama macrolactonization, as well as Lewis and protic acid promoted deprotections to suppress E/Z isomerization and/or destruction of the delicate (Z,Z,E)-trienoate linkage.
Myxobacteria Sorangium cellulosum is a rich source of secondary metabolites possessing important biological profiles. Perhaps most notable, the epothilone class of natural products, analogues of which are currently in clinical use for the treatment of solid tumors resistant to alternative treatments (i.e., Taxol), were isolated by Höfle and co-workers in 1996 from the Sorangium cellulosum strain So ce 90.1 Earlier (1985), the same research group reported the isolation of members of another family of architecturally complex macrolides from the strain So ce 12, termed the sorangicins.2 Importantly, (+)-sorangicin A (1), the most potent and prevalent congener, demonstrated remarkable antibiotic activity against a broad spectrum of both Gram-positive (MIC 0.01–0.3 μg/mL) and Gram-negative bacteria (MIC 3–25 μg/mL). The mechanism of action was subsequently determined to entail inhibition of RNA-polymerase in both E. coli and S. aureus, while leaving eukaryotic cells unaffected.3 Elegant cross-resistance studies by the Jansen and Darst groups on (+)-sorangicin A and rifampicin, the latter a clinically used ansamycin antibiotic, also revealed that although both bind the same β-subunit pocket of RNA-polymerase, (+)-sorangicin A displays an advantageous profile against rifampicin resistant microbial mutants, a result proposed to arise from increased conformational flexibility, leading to better adaption to mutational changes in the binding pocket.4
The structure of (+)-sorangicin A (1, Scheme 1)5 comprises a signature dioxabicyclo[3.2.1]octane skeleton in conjunction with a rare (Z,Z,E)-trienoate linkage, both inscribed within a highly unsaturated 31-membered macrolactone ring. The initial structural assignment was based on extensive 1- and 2-D NMR experiments, along with high resolution mass spectrometry.2 Confirmation of both the connectivity and absolute configurations of the 15 stereogenic centers was subsequently secured by X-ray analysis.6 The structural features, in particular the (Z,Z,E)-trienoate linkage affixed to the bicyclic ether core provide the major source of the reported instability of the natural product towards a variety of reagents (e.g., fluoride ion, DDQ, and the dissolving metal sodium amalgam),7 thus presenting a serious test to the current state-of-the-art of organic synthesis. In addition, two polar structural elements, the C(1) carboxyl group and C(21, 22 and 25)-triol moiety of (+)-sorangicin A associate through aegis of a hydrogen bond to generate a hydrophilic region of this amphiphilic natural product. This interaction is particularly sensitive to solvent and pH environments, thereby leading to significant dependence of the 1H and 13C NMR spectra on the exact experimental variants.6
Scheme 1.

Not surprisingly the intricate architecture of sorangicin A (1), in conjunction with the important biological properties, has stimulated considerable interest within the synthetic community. Indeed, significant progress has been registered by the Schinzer,8 Crimmins,9 and Lee laboratories,10 in addition to ours.11 Although successful preparations of mono- and bicyclic subtargets have been described, the assembly of the highly sensitive (Z,Z,E)-trienoate linkage has been reported only in a simple model system.11c Moreover the total synthesis of 1 has remained an elusive goal. Herein, we describe the development of chemistry that addresses the synthetic challenges associated with 1, thus leading to the first total synthesis of this intriguing natural product.
Although scalable routes to access bicyclic aldehyde (–)-2,11a,11c tetrahydropyran (–)-3,11b,12 and dihydropyran (–)-10-epi-611b,13 had been developed by our research group, the stereogenicity at C(10) in (–)-10-epi-6 required revision due to some confusion relating to this stereogenic center.5 Towards this end, we outline in Scheme 2 the requisite inversion of the C(10) stereogenic center. Global desilylation of (–)-10-epi-6, followed by chemoselective silylation,14 furnished allylic alcohol (–)-7. Initial attempts to invert the C(10) center with Mitsunobu methods failed due to the predominance of an SN2’ pathway, favored by the steric nature of the C(1)–C(8) side chain. Recourse was thus made to a Ley oxidation15/Luche reduction16 sequence, which generated the desired allylic alcohol (+)-8 as a single diastereomer. The high selectivity can be attributed to the same bulky side chain. Formal transposition of the TBS group, followed by standard protocols to introduce the sulfone unit furnished fragment (+)-10 in good yield (46% for the 8 steps).
Scheme 2.

With all major subtargets in hand, we were set to achieve their union. Towards this end, we envisaged construction of both the C(29)–C(30) and C(15)–C(16) trans double bonds via Julia-Kocienski olefinations, followed in turn by Stille union of 4 and macrolactonization to complete the overall carbon skeleton. The first Julia-Kocienski olefination however raised an unexpected challenge. Utilizing conditions developed by the Jacobsen group (LiHMDS in DMF/HMPA),17 E-olefin (–)-11 was obtained albeit in low yield (24%), along with substantial recovery of both coupling partners (–)-2 (36%) and (–)-3 (63%, Scheme 3). Switching the base counter-ion to Na increased the yield (40%) but the E/Z selectivity (3.6:1) decreased. Use of the traditional E-selective conditions (cf. KHMDS in DME)18 provided the highest yield (54%), but with the lowest E/Z selectivity (2.0:1). Barbier conditions19 or the corresponding benzothiazole sulfone (BT-sulfone) did not improve the situation. In the end, the most reliable conditions proved to be use of t-BuLi in aprotic polar solvents (DMF/HMPA), which on a 300 mg scale furnished the E-configured product in 39% yield. Several recycles permitted advance of material to (–)-11 in upwards of 65% yield.
Scheme 3.

The second Julia-Kocienski olefination involving sulfone (–)- 12, obtained from (–)-11 and aldehyde (–)-13, derived from (–)-10-epi-6, also proved problematic. In this case, the E/Z-selectivity trend was reversed, with the best selectivity obtained employing KHMDS in DME. Interestingly, no selectivity was observed with LiHMDS in DMF/HMPA (Scheme 3). In addition, both sets of conditions suffered from low conversion. At this point, we surmised that the low efficiency in both olefinations might arise vis-à-vis the nature of sulfone fragments [i.e., (–)-3 and (–)-12)], that is difficulty with either the initial deprotonation and/or intramolecular coordination of the resulting metalated species. To test these ideas, we reversed the coupling partners, preparing aldehyde (–)-15 from (–)-11 in two steps (Scheme 4) and sulfone (+)-10 as outlined in Scheme 2. Gratifyingly, employing the conditions of KHMDS in DME, the union proceeded both with complete conversion and with excellent stereocontrol, to provide E-olefin (+)-16 in 86% yield after desilylation,20 possessing the correct stereogenicity at C(10).21
Scheme 4.

Having installed both the C(29)–C(30) and C(15)–C(16) trans olefins with high stereo-precision, all that remained to complete the synthesis of (+)-sorangicin A (1) was construction of the C(38)-C(39) and C(43)-O σ bonds, followed by global deprotection. Initially, we envisioned that the labile (Z,Z,E)-trienoate moiety could be introduced in protected form comprising a dienyne that would mask the C(39)–C(40) Z-olefin as an alkyne. Although the dienyne moiety could be introduced into a simple model bicyclic vinyl iodide employing known alkynyl stannane 4,22 subsequent semihydrogenation suffered extensive E/Z isomerization upon purification from excess hydrogen scavengers present in the reaction mixture. A solution to this issue entailed the use of an alternative linker, stannyl dienoate 5,23 which proved to be surprisingly stable under standard laboratory conditions. Again in a model system, we demonstrated the effective use of 5 in a Stille union employing excess Ph2PO2NBu4 (6 equiv) to suppress the E/Z isomerization.11c With (+)-16, however, even a larger excess of Ph2PO2NBu4 (12 equiv) was required to deliver geometrically pure (Z,Z,E)-trienoate (+)-17 in good yield (88%). Equally important, the subsequent hydrolysis of trienoate (+)-17 with LiOH in aqueous THF furnished the corresponding trieno-acid 18 without noticeable geometric isomerism (Scheme 4). Acid 18 did however prove to be extremely unstable, and therefore was employed in the next step without full characterization.
In the transformations leading up to the critical macro-cyclization, we fully anticipated the possibility of significant isomerization during activation of the trieno-acid. This indeed proved to be the case! However, after a careful survey, two sets of mild conditions were found to deliver the desired (Z,Z,E)-configured macrolactone (+)-19 as the major product: the Yonemitsu modification of the Yamaguchi conditions24 involving direct introduction at room temperature of DMAP at the outset without pre-formation of the mixed anhydride, and the Evans modified Mukaiyama protocol,25 employing NaHCO3 and 20 at room temperature (Scheme 5). Notwithstanding our initial success in solving the challenging macrolactonization, (+)-19 was contaminated with minor amounts of other geometric isomers, which proved difficult to separate. The problem appears to lie with the reversibility of a Michael addition of DMAP or iodide to the activated trieno-acid during the lactonization process. Another issue associated with Mukaiyama reagent 20 is halogen exchange to give 21, which is known to be nonreactive as an activating agent for carboxylic acid coupling.26 To address these issues, modified Mukaiyama reagent 2227 was adopted, which possessed a non-nucleophilic counterion (i.e., tetrafluoroborate), thus mitigating the undesired Michael addition, as well as the inactivation pathway. Pleasingly, reagent 22 delivered macrolide (+)-19 in 85% yield, with minimum isomerization (ca. <4%).
Scheme 5.

Clearly aware that our late-stage intermediates were exceedingly prone to isomerization and/or decomposition due to the delicate (Z,Z,E)-trienoate moiety, we were now compelled to identify mild conditions, albeit sufficiently potent to remove the MOM, acetonide, and t-butyl protecting groups. This task proved non-trivial! We first took the lead of the Höfle group, who had employed TFA in aqueous THF at 85 °C to remove the acetonide group in their synthesis of an extensive library of sorangicin analogues.28 Product yields however were highly substrate dependent, varying from 20% to 70%. Application of these conditions to the fully protected macrolide (+)-19 led only to decomposition. We therefore initiated an extensive series of deprotection studies on available individual fragments, which in the end led to the observation that although the MOM and acetonide groups could be removed under aqueous protic acidic conditions (at 85 and 45 °C, respectively), hydrolysis of the t-butyl ester was far from efficient (only 50% conversion at 85 °C for 3 h). A more efficient protocol to remove the t-butyl group had to be devised. Use of TFA in anhydrous CH2Cl2 led to destruction of the trienoate moiety. Turning to Lewis acids, B-bromocatecholborane29 rapidly removed both the MOM and acetonide groups, but removal of the t-butyl group was quite slow. Initially, TMSOTf proved able to remove the MOM and t-butyl moieties efficiently on individual fragments, but with the fully protected macrolide (+)-19, only decomposition occurred. Clearly TMSOTf was too reactive. Eventually we learned that the t-butyl ester could be cleanly transformed to the TBS ester employing the milder TBSOTf reagent, buffered with 2,6-lutidine, without compromising the delicate (Z,Z,E)-trienoate linkage. Without further purification the TBS ester was then treated with 4N HCl in THF at room temperature for 24 hr to produce (+)-sorangicin A (1) in 70% yield for the 2 steps (Scheme 5). Totally synthetic (+)-sorangicin (1) proved identical in all respects (e,g., 1H, 13C, HRMS and HPLC-LRMS) to an authentic natural sample provided by Dr. Jansen,30 including chiroptic properties {[α]D19 +56 (c 0.06, MeOH); lit.6 [α]D22 +60.9 (c 0.7, MeOH)}.
In summary, the first total synthesis of the structurally complex macrolide (+)-sorangicin A (1) has been achieved in a highly convergent fashion. Late stages of the synthetic venture featured the use of two Julia-Kocienski olefinations to unite three complex advanced fragments with high E-stereoselectivity. The final steps of the synthesis then involved a modified Stille union and Mukaiyama macrolactonization, as well as Lewis and protic acid promoted deprotections, employing carefully defined conditions required to suppress E/Z isomerization and/or destruction of the sensitive (Z,Z,E)-trienoate linkage.
Supplementary Material
Acknowledgments
Support was provided by the National Institutes of Health (Institute of General Medical Sciences) through Grant No. GM-29028. We thank Drs. George Furst and Rakesh Kohli (University of Pennsylvania) for assistance in obtaining NMR spectra and high-resolution mass spectra, respectively. We also thank Dr. Rolf Jansen (Helmholtz Center for Infection Research, Braunschweig, Germany) for an authentic sample of (+)-sorangicin A.
Footnotes
Supporting Information Available: Experimental procedures and spectroscopic and analytical data for all transformations and new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.(a) Höfle G, Bedorf N, Steinmetz H, Schomburg D, Gerth K, Reichenbach H. Angew Chem Int Ed. 1996;35:1567. [Google Scholar]; (b) Gerth K, Bedorf N, Höfle G, Irschik H, Reichenbach H. J Antibiotics. 1996;49:560. doi: 10.7164/antibiotics.49.560. [DOI] [PubMed] [Google Scholar]
- 2.Jansen R, Wray V, Irschik H, Reichenbach H, Höfle G. Tetrahedron Lett. 1985;26:6031. [Google Scholar]
- 3.Irschik H, Jansen R, Gerth K, Höfle G, Reichenbach H. J Antibiot. 1987;40:7. doi: 10.7164/antibiotics.40.7. [DOI] [PubMed] [Google Scholar]
- 4.Campbell EA, Pavlova O, Zenkin N, Leon F, Irschik H, Jansen R, Severinov K, Darst SA. The EMBO Journal. 2005;24 doi: 10.1038/sj.emboj.7600499. and references cited therein.
- 5.The stereocenter at C(10) in (+)-sorangicin A (1), as confirmed by Dr. R. Jansen (Helmholtz Center for Infection Research, Braunschweig, Germany) is S as originally reported in reference 6 and not R as depicted in reference 7. We thank Dr. R. Jansen for this clarification.
- 6.Jansen R, Irschik H, Reichenbach H, Schomburg D, Wray V, Höfle G. Liebigs Ann Chem. 1989:111. [Google Scholar]
- 7.Schummer D, Irschik H, Höfle G. Liebigs Ann Chem. 1993:293. [Google Scholar]
- 8.Schinzer D, Schulz C, Krug O. Synlett. 2004;15:2689. [Google Scholar]
- 9.Crimmins MT, Haley MW. Org Lett. 2006;8:4223. doi: 10.1021/ol061339e. [DOI] [PubMed] [Google Scholar]
- 10.Park SH, Lee HW. Bull Korean Chem Soc. 2008;29:1661. [Google Scholar]
- 11.(a) Smith AB, III, Fox RJ. Org Lett. 2004;6:1477. doi: 10.1021/ol049644s. [DOI] [PubMed] [Google Scholar]; (b) Smith AB, III, Fox RJ, Vanecko JA. Org Lett. 2005;7:3099. doi: 10.1021/ol051119l. [DOI] [PubMed] [Google Scholar]; (c) Smith AB, III, Dong S. Org Lett. 2009;11:1099. doi: 10.1021/ol802942j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brenneman, J. B. unpublished results, University of Pennsylvania.
- 13.The clarification of (+)-sorangicin A structure (S, not R for C(10) configuration) was achieved in 2008. At the beginning of this project, 10-epi-sorangicin A was targeted, leading to the preparation of (–)-10-epi-6 in 2005.
- 14.Chaudhary SK, Hernandez O. Tetrahedron Lett. 1979:99. [Google Scholar]
- 15.Ley SV, Norman J, Griffith WP, Marsden SP. Synthesis-Stuttgart. 1994:639. [Google Scholar]
- 16.Luche JL. J Am Chem Soc. 1978;100:2226. [Google Scholar]
- 17.Liu P, Jacobsen EN. J Am Chem Soc. 2001;123:10772. doi: 10.1021/ja016893s. [DOI] [PubMed] [Google Scholar]
- 18.Blakemore PR, Cole WJ, Kocienski PJ, Morley A. Synlett. 1998:26. [Google Scholar]
- 19.Under Barbier conditions, LiHMDS was added to a mixture of sulfone (–)-3 and aldehyde (–)-2 in DMF/HMPA (3:1), leading to a less than 24% yield of (–)-11, along with recovery of starting materials.
- 20.For the first Julia-Kocienski olefination, we envisioned that the reversal of coupling partners might be not without risk due to the proclivity of β-elimination of the bicyclic sulfone derived from (–)-2.
- 21.Earlier studies had led to the construction of (–)-10-epi-16.
- 22.Rossi R, Bellina F, Catanese A, Mannina L, Valensin D. Tetrahedron. 2000;56:479. [Google Scholar]
- 23.Franci X, Martina SLX, McGrady JE, Webb MR, Donald C, Taylor RJK. Tetrahedron Lett. 2003;44:7735. Stannyl dienoate 5 was prepared in an analogous fashion according to procedures described therein.
- 24.(a) Inanaga J, Hirata K, Saeki H, Katsuki T, Yamaguchi M. Bull Chem Soc Jpn. 1979;52:1989. [Google Scholar]; (b) Hikota M, Sakurai Y, Horita K, Yonemitsu O. Tetrahedron Lett. 1990;31:6367. [Google Scholar]
- 25.(a) Mukaiyama T, Usui M, Saigo K. Chem Lett. 1976:49. [Google Scholar]; (b) Evans DA, Starr JT. J Am Chem Soc. 2003;125:13531. doi: 10.1021/ja037643+. [DOI] [PubMed] [Google Scholar]
- 26.(a) Oh SH, Cortez GS, Romo D. J Org Chem. 2005;70:2835. doi: 10.1021/jo050024u. [DOI] [PubMed] [Google Scholar]; (b) Bradlow HL, Vanderwerf CA. J Org Chem. 1951;16:1143. [Google Scholar]
- 27.Xu J-C, Li P. Tetrahedron. 2000;56:8119. For advantages of using modified Mukaiyama reagents, see: Folmer JJ, Acero C, Thai DL, Rapoport H. J Org Chem. 1998;63:8170.
- 28.Jansen R, Schummer D, Irschik H, Höfle G. Liebigs Ann Chem. 1990:975. [Google Scholar]
- 29.Boeckman RK, Jr, Potenza JC. Tetrahedron Lett. 1985;26:1411. [Google Scholar]
- 30.NMR spectra of both natural and synthetic samples are sensitive to NMR experimental conditions. More specifically, in 1H NMR spectra, the resonances for protons of the C(1)–C(8) side chain are dependent on pH and solvent environments, most apparently for H(2), H(7), and H(44). In 13C NMR spectra, the chemical shifts and intensity for both C(1) and C(2) vary significantly in different solvent and pH environments.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.