

Abstract
We have previously demonstrated that oxidative stress increases in the inner ear of aging CBA/J mice and might contribute to the loss of function of the sensory system. We now investigate the activation of cell death pathways in the cochlea of these animals. Middle-aged (12 months) and old (18-26 months) mice with hearing deficits displayed outer hair cell nuclei with apoptotic and, to a lesser extent, necrotic features. Both intrinsic and extrinsic cell death pathways were activated by translocation or post-translational modification of proteins in the aging cochlea as compared to young (3 months) animals. Cytosolic cytochrome C increased, formed a complex with, and activated caspase 9. Endonuclease G translocated to the nuclei of aging outer hair cells suggesting its function as an apoptotic DNase. The cleaved (and hence active) forms of calpain I and calpain II increased while active cathepsin D was transiently elevated in middle-aged but not old animals. Finally, increases in the phosphorylation of p38 MAPK and JNK implicated the additional involvement of the MAPK pathway. The results suggest that multiple cell death pathways, all potentially linked to oxidative stress, are activated in hair cells of the auditory organ in aging mice.
Keywords: Cochlea, age-related hearing loss, apoptosis, oxidative stress, caspase, calpain
1. Introduction
Acquired hearing loss affects a large segment of the population worldwide. Among the diverse causes of hearing impairment developing over a life time, presbycusis (age-related hearing loss) holds a prominent place. Presbycusis can start in people in their forties and by age 70, approximately 50% suffer from a significant auditory deficit. Such declining auditory acuity can be associated with the failure of several tissues within the inner ear, leading to different clinical manifestations of hearing loss (Schuknecht, 1964). Frequently, pathology involves the loss of hair cells, the sensory cells in the inner ear. Unable to regenerate, death of these cells and subsequent degeneration of nerve fibers leads to permanent impairment of function in sensori-neural presbycusis.
Loss of sensory cells is well documented both in aging humans and in corresponding animal models (Sha et al., 2008). Oxidative stress might be one of the factors associated with cell death (Jiang et al., 2007), consistent with findings from a variety of organisms and tissues that support oxidant stress as a general factor in the pathology of aging (Lee et al., 1999; Sohal and Dubey 1994; Sohal and Weindruch 1996). In the cochlea, stress markers appear with age in CBA/J mice (Jiang et al., 2007) and an increase in mitochondrial deletions, indicative of oxidative damage, correlates with hearing loss in rats (Seidman 2000). Furthermore, age-related hearing loss developed earlier and was more severe in mice with deletions of superoxide dismutase genes, inferring a need to detoxify ROS in order to stave off presbycusis (McFadden et al., 1999; McFadden et al., 2001).
Noxious stimuli can induce both apoptosis and necrosis in a cell and mixed forms of cell death as well as autophagy have also been observed (Leist and Jäättelä 2001; Kelekar, 2006), In the context of oxidative stress, redox-regulated signaling has been linked to both extrinsic and intrinsic pathways, including the activation of caspases, protein translocations following mitochondrial pore transitions, JNK-initiated cascades and homeostatic modulation by BCl2-family proteins (Ryter et al., 2007; Trachootham et al., 2008). In particular, release of cytochrome C from mitochondria activates caspase-dependent apoptosis (Leist and Jäättelä 2001), while endonuclease G (Endo G) translocation into the nuclei indicates an initiation of caspase-independent cell death (Li et al., 2001). Active calpains and cathepsins participate in the regulation of both apoptosis and necrosis whereby calpains activate caspases as a major form of their action (Brunk et al., 1997; Tsukuba et al., 2000). The release of lysosomal proteases, such as the cysteine cathepsins B and L and the aspartyl cathepsin D, can lead to cellular autolysis and damage to neighboring cells under uncontrolled cellular stress, causing necrosis, apoptosis-like or necrosis-like cell death depending on the cell types and the stimuli (Roberg et al., 2002; Guicciardi et al., 2000). Finally, both the p38 MAP kinase and SAPK/JNK pathway are crucial for neuronal apoptosis in response to a variety of cellular stresses.
Redox imbalance as a potential cause of hair cell death has also been invoked for hearing loss induced by noise trauma and drugs such as the aminoglycoside antibiotics and cisplatin (Henderson et al., 2008; Rybak et al., 2008). In these pathologies, which can effectively be replicated in animal models, a variety of cell death pathways can be observed, leading to both apoptosis and necrosis (Hu et al., 2002; Yamashita et al., 2004; Jiang et al., 2006). A recent study on gene expression in the CBA/CaJ mouse (Tadros et al., 2008) also suggested that apoptosis in age-related hearing loss is induced by multiple pathways. Based on these considerations, we investigate here intrinsic and extrinsic apoptotic as well as necrotic pathways that have frequently been associated with oxidative stress and other inner ear pathologies. We focus on an investigation at the protein level because the regulation of signaling pathways is frequently associated with post-translational modifications that are not revealed in gene expression. We analyze here the activation of proteins by proteolytic cleavage or phosphorylation, and changes in their downstream targets by intracellular translocations.
The animal of choice is the CBA/J mouse in which we have previously characterized age-related auditory pathology and oxidant stress (Jiang et al., 2007; Sha et al., 2008). Loss of outer hair cells and high-frequency hearing in this strain commences between the ages of 12 and 18 months and increases thereafter. The current study focuses on hair cells since strial degeneration is essentially absent in this aging strain (Sha et al., 2008), and compares middle-aged and old mice with auditory deficits to young animals with normal auditory thresholds.
2. Materials and Methods
2.1 Animals
Male CBA/J mice were purchased from Harlan Sprague-Dawley Co. (Indianapolis, IN) through the National Institute on Aging two weeks prior to the intended age for the experiments. The animals were kept at 22 1°C under a 12h:12h light-dark cycle, and had free access to water and a regular mouse diet (Purina 5025, St. Louis, MO). Animal care was supervised by the University of Michigan's Unit for Laboratory Animal Medicine and all experimental protocols were approved by the University of Michigan Committee on Use and Care of Animals.
2.2 Materials
ECL (enhanced chemiluminescence) reaction kits for antibody detection on Western blots were purchased from Amersham Pharmacia Biotech (Piscataway, NJ); anti-endonuclease G rabbit polyclonal antibody from Chemicon International (Temecula, CA); anti-calpain I (μ-calpain) and anti-calpain II (m-calpain) rabbit polyclonal antibodies, anti-phospho-p38 MAPK antibody, anti-phospho-JNK1/2 rabbit polyclonal antibody and anti-cleaved caspase-3 rabbit polyclonal antibody from Cell Signaling Technology (Beverly, MA); monoclonal anti-cytochrome c (denatured) and rabbit polyclonal anti-caspase-9 from BD Biosciences Pharmingen (San Diego, CA); and anti-cathepsin D from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Secondary antibodies for Western blotting were purchased from Jackson Immunoresearch (West Grove, PA) and secondary fluorescence antibodies (Alexa 488 and Alexa 546), rhodamine phalloidin, Hoechst 33342 and propidium iodide (PI) from Molecular Probes (Eugene, OR). Complete™ mini EDTA free protease inhibitor cocktail tablets were purchased from Roche Diagnostic (Mannheim, Germany). All other reagents were obtained from Sigma Chemical (St. Louis, MO).
2.3 Auditory brain stem response (ABR)
Visual inspection of the tympanic membrane for middle ear infections was performed through otoscopic examination at the time of each physiological measurement and again at the time of euthanasia. Animals with middle ear infections were excluded from the study.
For ABR measurements, mice were anesthetized with an intra-peritoneal injection of xylazine (7 mg/kg), ketamine (65 mg/kg), and acepromizine (2 mg/kg) and body temperatures were monitored and maintained near 37°C with a warm-water heating pad. Acoustic stimuli were delivered in a sound-isolated and electrically-shielded booth (Acoustic Systems, Austin, TX) to the left ear via a Beyer earphone mated to a customized plastic speculum inserted into the ear canal. Sub-dermal needle electrodes were inserted at the vertex, under the left ear and under the right ear (ground). Tucker Davis Technology (Alachua, FL) and SigGen/Biosig software were used to present the stimuli (15 ms duration tone bursts with 1 ms rise-fall time) and record the response. The sound driver was calibrated with a B&K 1/8 inch condenser microphone (cartridge type 4138) in a volume approximating that of a mouse ear canal, and a Stanford Research Systems lock-in amplifier (model SR530). Up to 1024 responses were averaged for each stimulus level. Thresholds were determined for each frequency by reducing the sound intensity in 10 dB increments and then in 5 dB steps near the threshold. Thresholds were estimated to lie between the lowest stimulus level where an ABR response in wave V was observed and the next lowest level without response. All evaluations were carried out by an observer blinded to the experimental conditions.
2.3 Immunoprecipitation and Western blotting
For the extraction of proteins, cochleae were rapidly removed and dissected in ice-cold 10 mM phosphate buffered saline (PBS) at pH 7.4. To obtain total protein, tissue from one mouse cochleae was homogenized in ice-cold RIPA lysis buffer (Upstate, Waltham, MA, cat# 20-188) by using a glass/glass micro Tissue Grind pestle and vessel for 30 sec. After 30 min on ice, tissue debris was removed by centrifugation at 12,000 × g at 4°C for 10 min and the supernatants were kept as total protein. To obtain cytosolic protein, cochleae from four mice were pooled and homogenized in a glass tube with teflon pestle in cold lysis buffer (10 mM sodium HEPES, pH 7.9, containing 10 mM KCl, 1 mM EDTA, 1 mM EGTA, 5 mM dithiothreitol, 10 mM sodium fluoride, 10 mM sodium β-glycerophosphate, 1 mM phenylmethylsulfonyl fluoride, 1mM sodium orthovanadate and 1 tablet of the protease inhibitor cocktail/10 ml buffer; Igepal CA-630 was added for a final concentration of 0.6%). The homogenates were centrifuged at 750 × g at 4°C for 10 min and a cytosolic fraction with mitochondria was obtained by removing nuclei. This supernatant was further centrifuged at 15,000 × g for 10 min at 4°C to yield a “mitochondria-free fraction”. Extracts were stored at −80°C until analysis. Protein concentrations were determined using the Bio-Rad Protein Assay dye reagent (Bio-Rad, Hercules, CA) with bovine serum albumin as a protein standard.
For Immunoprecipitation, immunoglobulin proteins were pre-cleared from 150 – 300 μg of total cochlear protein by incubation with 0.25 μg of the control IgG and 20 μl of agarose-conjugated protein A at 4°C for 30 min. After centrifugation at 1,000 × g for 30 sec at 4°C to remove the bead-bound IgG, the cleared lysate was incubated with the primary antibody against cytochrome C (2 μg) for 6 hr at 4°C, and then 20 μl of agarose-conjugated protein A was added. After incubation at 4°C on a rotating device overnight, the pellets were collected by centrifugation at 1,000 × g for 1 min at 4°C. The pellets were washed 3 times with lysis buffer and resuspended in 20 μl of electrophoresis buffer (0.5 M Tris-HCl, 4% SDS, 20% glycerol, 0.02% bromphenol blue and 10% -mercaptoethanol). Subsequently, Western blotting for anti-caspase 9 was performed.
For Western blot analysis, protein samples (50 μg) were separated by SDS-PAGE. After electrophoresis, the proteins were transferred onto a nitrocellulose membrane (Pierce, Rockford, IL) and blocked with 5% nonfat dry milk in PBS with 0.1% Tween 20 (PBS-T). The membranes were incubated with primary antibodies, anti-cytochrome C (1:500), anti-caspase 9 (1:1,000), anti-calpain II (1:1,000), or anti-cathepsin D (1:500), overnight at 4°C, and then washed three times (10 min each) with PBS-T. Membranes were incubated with an appropriate secondary antibody at a concentration of 1:10,000 for 1 hr. Following extensive washing of the membrane, the immunoreactive bands were visualized by enhanced chemiluminescence using FluorChem SP instrumentation (Alpha Innotech, San Leandro, CA). Western blots were analyzed using AlphaEase software SpotDenso tool (Alpha Innotech, San Leandro, CA). Briefly, the band densities were first analyzed and normalized to background. Next, the band density of middle- or old-age samples was normalized to the control (young animal sample) run on the same gel. Finally, the relative densities of the age groups from four individual assays were averaged and analyzed for changes.
2.4 Surface preparations of the organ of Corti
Temporal bones were removed immediately after euthanasia of the mice and were intrascalarly perfused with cold fixative containing 4% paraformaldehyde in PBS, and kept in this medium overnight at 4°C. Cochleae were then rinsed in PBS and decalcified in 4% sodium EDTA/HCl (pH 7.4) for 48 h at 4°C. Following decalcification, the softened otic capsule, stria vascularis, Reissner's membrane, and tectorial membrane were removed. The epithelium of the organ of Corti (base to apex) was lifted off the modiolus in three sections, corresponding to apical, middle and basal portions of the cochlea. The hook region was generally not recovered. The epithelium of the organ of Corti was evaluated using the procedure of immunofluorescent assessment.
2.4 Immunohistochemistry
Following removal of the temporal bones, cochleae were fixed immediately in 4% paraformaldehyde overnight at 4°C and then rinsed in PBS and decalcified in 4% sodium EDTA/HCl, pH 7.4 for 48 h at 4°C. Cryostat sections of 8 μm were incubated in 0.5% Triton X-100 in PBS for 15 min at room temperature. The sections were then washed three times with PBS and blocked with 10% goat serum for 30 min at room temperature, followed by incubation with the primary antibodies for anti-Endo G (1:500), anti-cleaved caspase 3 (1:200), anti-calpain I (1:100), anti-phospho-p38 MAPK (1:100) and anti-phospho-JNK (1:100) at 4°C for 48 hr. After 3 rinses in PBS, the sections were incubated with the secondary antibody (Alexa 488 or Alexa 546 conjugated; 1:500) at 4°C overnight in darkness. After 3 washes with PBS, the sections were incubated with propidium iodide (2 μg/ml in PBS) or Hoechst 33342 (2 μg/ml in PBS) for 30 min at room temperature. After a final wash with PBS, the slides were mounted. Control incubations were routinely processed without primary antibody. Preparations were observed with a Zeiss Axioplan microscope and imaged with a Zeiss laser confocal microscope (Zeiss LSM 510; Carl Zeiss Microimaging, Thornwood, NY).
Immunostaining of phospho-p38 MAPK was quantified from confocal images taken under identical conditions and equal setting parameters using ImageJ software (National Institute of Health, Bethesda, MD). The fluorescence intensity of nuclei of outer hair cells was measured in 0.212-mm segments of the basal portion of the cochlea from a surface preparation containing about 70 outer hair cells. The “region of interest” was outlined with a circle covering approximately 85% of the nucleus (20 μm2 area per outer hair cell nucleus). The average fluorescence intensity per nucleus was then calculated, and the relative fluorescence intensity was quantified by normalizing the ratio of average fluorescence intensity of cells in old animals to the average fluorescence intensity of cells in young animals. Individual preparations from three animals per condition were examined and data subjected to statistical analysis.
2.5 Statistical analysis
Results reported are representative of at least three independent assays, as indicated in the legends. Data were statistically evaluated for significance (p < 0.05) using Primer of Biostatistics software (McGraw-Hill Software, New York, NY). Western blot band densities were tested for statistical significance with either a one- or two-tailed one-sample t-test using GraphPad Software (GraphPad Software, San Diego, CA).
3. Results
3.1 Hearing loss in aging CBA/J mice
The aging CBA/J mouse develops elevated auditory thresholds at high frequencies, as assessed by evoked auditory brain stem responses (ABR), beginning at an age of about 12 months (Sha et al., 2008). For the current study, animals were divided into three age groups: young controls (3 months), middle-aged (12 months) and old (18 to 26 months) animals. In order to be included into the study, young mice had to have normal thresholds while middle-aged and old mice had to meet the criterion of a significant (p < 0.01) auditory deficit, defined as a threshold at 24 kHz above 30 dB SPL (middle-aged animals) or 40 dB (old animals). Thresholds at 24 kHz in the three age groups were 18 ± 5 dB (3 mo, n = 15), 45 ± 13 (12 mo, n = 5), and 72 ± 28 (18-26 mo, n = 27).
3.2 Necrosis and apoptosis contribute to outer hair cell death
Nuclei suggesting either apoptosis or necrosis were present in outer hair cells of mice with hearing deficits (fig. 1). In young animals, outer hair cells in the basal turn of the cochlea exhibited a normal nuclear morphology in surface preparations of the organ of Corti. In the middle-aged group, the orderly outline of three rows of outer hair cells was still maintained but both nuclear swelling, considered to be indicative of necrosis (fig. 1, arrowheads), and nuclear condensation, considered to be indicative of apoptosis (fig. 1, arrows) were evident. A quantitative determination of the relative contributions of apoptosis and necrosis to cell death at different ages was not attempted. In old animals, hair cells were frequently missing and lumpy chromatin condensation indicated impending apoptotic cell death (arrow); necrotic nuclei were essentially absent in the old age group.
Figure 1. Apoptotic and necrotic hair cell death in the aging cochlea.
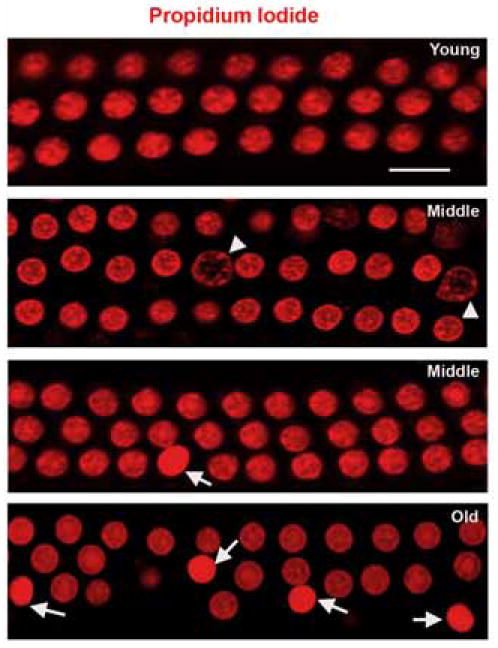
Surface preparations of the basal turn of the cochlear neuroepithelium were prepared as described in ‘Methods’. Outer hair cells of the organ of Corti of a young animal were orderly arranged and showed normal morphology of propidium iodide-stained nuclei. In middle-aged and old animals, nuclei suggesting apoptosis (arrows) were observed. Occasionally, nuclei indicative of necrosis (arrowheads) appeared in the middle-aged groups. The figure is representative of 5 individual preparations at young and old ages, and 3 individual preparations at middle age. Scale bar = 10 μm.
3. Caspase 3-dependent cell death pathways are activated with aging
Release of cytochrome C from mitochondria into the cytosol can activate caspase-dependent signaling cascades leading to apoptotic cell death. To detect cytochrome C release, we extracted a cytosolic fraction from tissue homogenates by removal of nuclear and mitochondrial material. Western blotting of this mitochondria-free preparation showed elevated cytosolic cytochrome C in old but not in young animals (fig. 2A).
Figure 2. Cytochrome C complexes with and activates caspase 9.
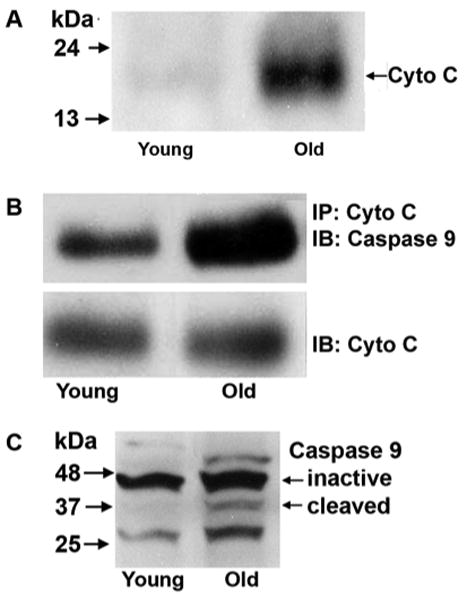
A: Cytochrome C was detected in the cytosolic fraction of the cochlea obtained by the removal of the mitochondrial fraction from the homogenate using Western blotting. The cytochrome C band in old animals was consistently stronger than in young animals. The panel is representative of 3 individual preparations for each age group.
B: Total cochlear homogenate was assayed for a complex between cytochrome C and caspase 9 by an immunoprecipitation assay as described in ‘Methods’. Following precipitation with cytochrome C, caspase 9 was detected by immunoblot. The band of the complex, stained by anti-caspase 9, was consistently stronger in old than in young animals. Cytochrome C was stained on the same gels and remained unchanged. A total of three assays were conducted on two pools of tissue. Each pool was derived from ten cochleae.
C: Inactive and activated (cleaved) caspase 9 were detected in cochlear homogenates by Western blotting. The band of activated caspase 9 increased with age. The panel is representative of 3 individual preparations at each age.
Next, we analyzed total cochlear homogenates in order to detect whether cytochrome C associated with caspase 9. Immunoprecipitation of cytochrome C co-precipitated caspase 9 as determined by subsequent immunoblotting, consistent with the formation and age-dependent increase of a complex between cytochrome C and caspase 9 (fig. 2B). In contrast, the total amount of cytochrome C (cytosolic and mitochondrial) remained unchanged, indicating that the complex arose from cytochrome C transition into the cytosol. Consequently, activated caspase 9 increased in old animals (fig. 2C).
3.4 Caspase-independent cell death
To investigate the potential contribution of mitochondria-mediated but caspase-independent pathways, we examined the translocation of the mitochondrial protein endonuclease G. Endo G was essentially absent from nuclei in young animals but appeared in the condensed nuclei of some outer hair cells (fig. 3, arrows) of middle-aged and old animals, suggesting that it can act as an apoptotic nuclear DNase in the aging cochlea.
Figure 3. Endonuclease G is translocated to condensed nuclei of outer hair cells.
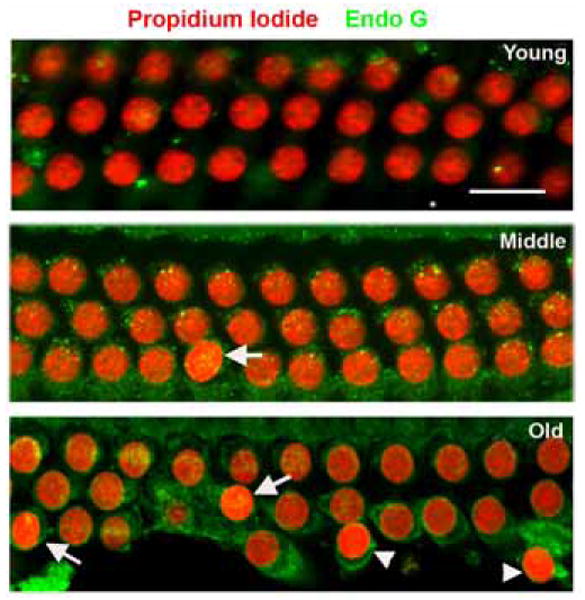
Endo G was translocated into some (arrows) but not all (arrowheads) condensed nuclei of outer hair cells in the basal turn of middle-aged and old animals. Red: PI staining for nuclei; green: staining for Endo G; nuclear Endo G appears as yellow (merged red and green) spots. The figure is representative of 5 individual preparations of the organ of Corti at each age. Scale bar = 10 μm.
3.5 Activation of calpain/cathepsin pathways
Calpains and cathepsin are activated by increased cellular calcium and can participate in the regulation of both apoptosis and necrosis. Calcium binding leads to the autocatalytic cleavage of the catalytic and the regulatory subunits of calpain to smaller peptides which can be assayed as a measure of activation. Staining for processed calpain I by immunohistochemistry increased in outer and inner hair cells, supporting cells, and spiral ganglion cells with old age (fig. 4A). Higher levels of calcium (usually in the millimolar range) are required for the activation of calpain II as compared to calpain I, and the proteolytically cleaved activated form of calpain II was likewise significantly elevated, as seen by Western blotting analysis (fig. 4B).
Figure 4. Calpain increases in the aging cochlea.
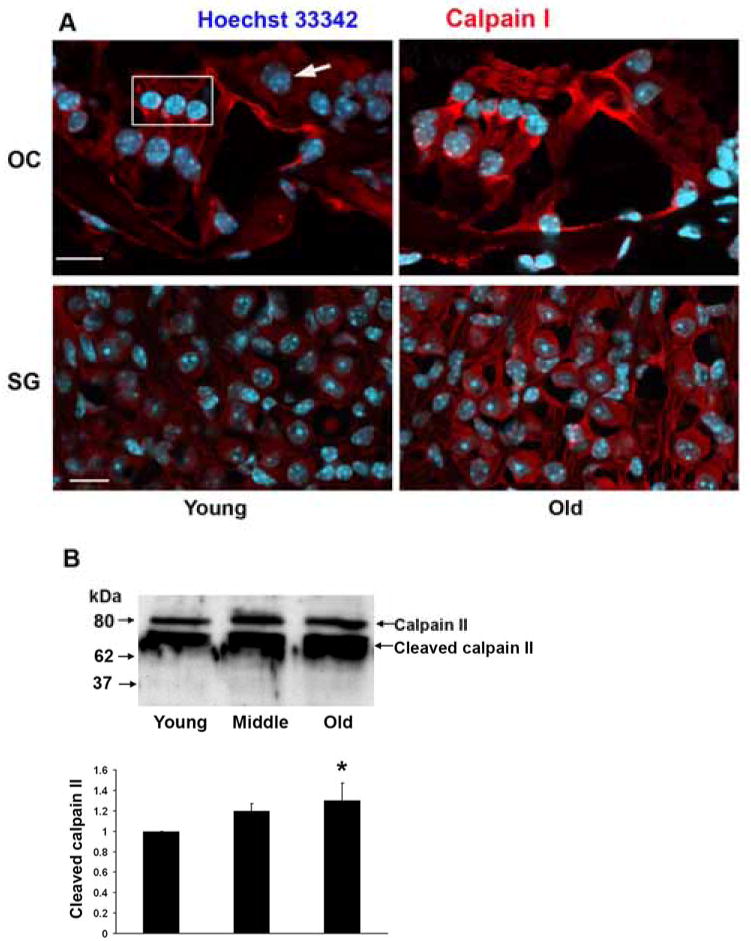
A: Fluorescent immunocytochemical staining for calpain I increased in outer and inner hair cells of the basal turn of the cochlea, supporting cells and spiral ganglion cells with aging. Red, calpain I; blue, Hoechst 33342 staining for nuclei. The box in the upper left panel outlines the region of outer hair cells, the arrow indicates an inner hair cell. The figure is representative of 3 individual preparations from the basal turn of the cochlea of young and old animals. Scale bar = 10 μm.
B: Cochlear homogenates were assayed for calpain II by Western blot and band density was analyzed with GraphPad software. Because conventional controls for protein load of the gels (such as GAPDH or actin) might change with aging percentile changes between young, middle aged and old animals are compared with standard deviation set to “zero” in the young group. Data are presented as means ± s.d. *p < 0.05; n = 3 in young and middle aged animals and n = 4 in old age.
A different temporal pattern of activation was observed for cathepsin D (fig. 5). The activated proteolysis products at 42 kDa and 31 kDa increased in middle-aged animals while old animals reverted to levels similar to those in young animals. This transient change was specific for the activated cathepsin D peptide, as the inactive precursor (52 kDa) did not show this pattern.
Figure 5. Cathepsin D is cleaved and activated in middle-aged cochleae.
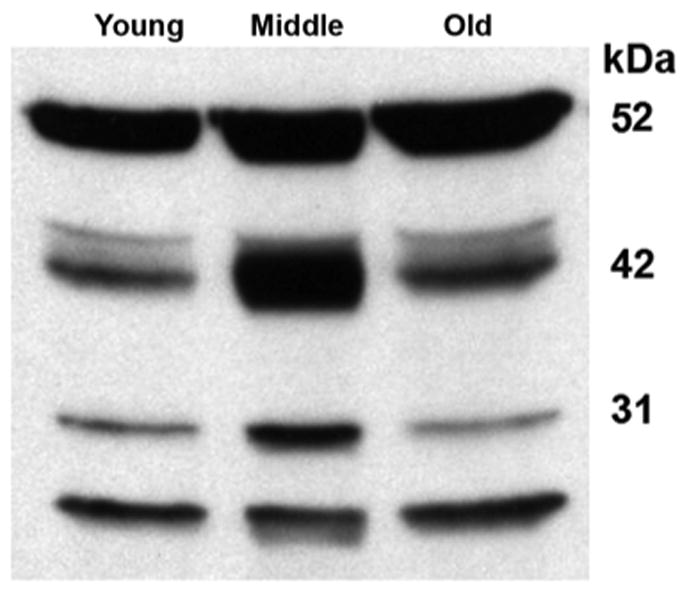
Western blotting analysis of cochlear extracts for cathepsin D showed that the inactive precursor (52 kDa) did not change significantly with aging, but the active form at molecular weights 42 and 31 kDa transiently increased in middle-aged mice. The figure is representative of 3 individual preparations at each age.
3.6 MAPK pathways increase in outer hair cells with aging
The activation of MAPK/JNK signaling is based on the phosphorylation of distinct proteins associated with the reaction chain and frequently associated with drug- and noise-induced hearting loss. To investigate whether MAPK stress-related pathways were also involved in outer hair cell death with aging, we examined the phosphorylation of p38 MAPK and SAPK/JNK as representative components. The staining intensity for phospho-p38 MAPK (fig. 6A) and SAPK/JNK (data not shown) increased with age in outer hair cells and spiral ganglion cells. Quantification of fluorescence intensity in surface preparations confirmed a significant increase of staining for phospho-p38 MAPK (Thr180/Tyr182) in the nuclei of outer hair cells with old age (fig. 6B).
Figure 6. Phospho-p38 MAP kinase increases in outer hair cells with aging.
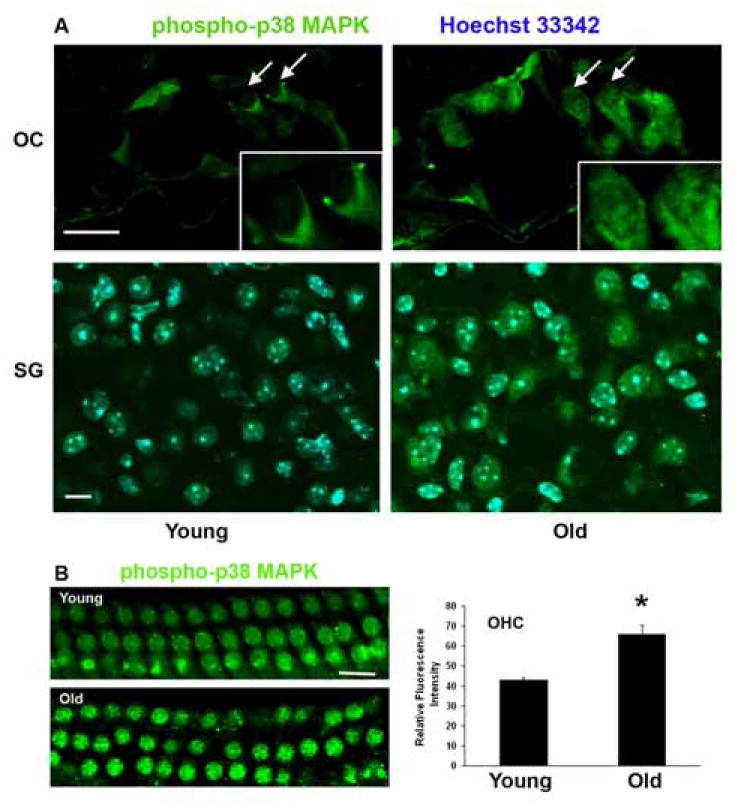
A: Fluorescent staining for phospho-p38 MAPK increased in the organ of Corti with aging. Staining was localized to the nuclei and cytoplasm of outer hair cells of the basal turn (arrows; boxed areas are high magnifications of outer hair cells) and to spiral ganglion cells. Green, phospho-p38 MAPK; blue (in the SG preparation), Hoechst 33342 for nuclei. The figure is representative of 4 individual preparations at each age. Scale bars = 10 μm.
B: Surface preparations of the cochlear epithelium of the basal turn were stained with phospho-p38 MAPK (green) and showed an increase in phospho-p38 MAPK in outer hair cells with age. Quantitative analysis of relative fluorescence intensity confirmed its elevation in outer hair cells. *p < 0.01, n = 3. Scale bar = 10 μm.
4. Discussion
Cell death pathways vary in a stress- and time-dependent manner. It is not surprising then that the cochlea displays a plethora of responses to aging at different stages of the deterioration of auditory function in CBA/J mice. Auditory deficits become apparent at 12 months of age and progress to a significant threshold shift and loss of hair cells by 22 to 24 months (Sha et al., 2008). However, the rate of pathological change varies between individuals so that near-normal thresholds and hair cell populations can be found among animals in older age groups. Therefore, the middle-aged and old animals in this study were selected for an elevated threshold at 24 kHz, reflecting damage to the basal turn of the cochlea, the area of this investigation.
Morphological evidence suggests the existence of both necrotic and apoptotic events in age-related hair cell death. The co-existence of several modes of death is not unusual, even in the same cell population (Unal-Cevik et al., 2004) and has similarly been observed in inner ear pathologies arising from drug damage (Nakagawa et al., 1998; Jiang et al., 2006) or noise trauma (Yang et al., 2004). Consistent with the occurrence of both apoptosis and necrosis, the molecular analyses show the involvement of caspases, Endo G and JNK/p38 MAP kinase, which are mostly associated with apoptotic pathways (Leist and Jäättelä 2001) and the activation of calpain/cathepsin which might relate to both apoptotic and necrotic cell death (Jiang et al., 2006).
A common denominator of the pathways active in the aging cochlea is their potential relation to oxidative stress. Mitochondrial cell death pathways reflect mitochondrial dysfunction, a condition that has been postulated for the aging inner ear based on observations of oxidative stress and mitochondrial lesions that correlate with loss of auditory sensitivity (Seidman 2000; Jiang et al., 2007). Mitochondrial pore opening, which leads to the release of pro-apoptotic proteins, is a critical step in initiating these pathways and the increase of cytochrome C levels in the cytosol is a hallmark of compromised mitochondrial integrity. Our data are consistent with the established sequence of events that cytochrome C, following its release from mitochondria, forms a complex with and activates caspase 9 which, in turn, will activate caspase 3, resulting in altered gene transcription promoting “caspase-dependent” apoptosis (Danial and Korsmeyer 2004). Another sequel of mitochondrial pore opening is the release of Endo G, also evident in the aging sensory cells. While sequestered within mitochondria, Endo G is involved in the replication of mitochondrial DNA. Following its translocation into nuclei, however, it is responsible for apoptotic oligonucleosomal DNA fragmentation, often observed under sustained oxidative stress (Li et al., 2001; Ishihara and Shimamoto 2006).
A further consequence of oxidative injury to mitochondria is a compromised calcium homeostasis (Green and Kroemer 2004; Ferri and Kroemer 2001) which has been associated with inner ear traumata, most notably those caused by noise overexposure (Fridberger et al., 1998) or aminoglycoside antibiotics (Dulon et al., 1989; Matsui et al., 2004). Calpains are calcium-sensitive neutral cysteine proteases present in all animal cells in which they can participate in the regulation of both apoptotic and necrotic pathways (Yamashima 2000; Leist and Jäättelä 2001). The activation of calpains in the cochlea supports such a scenario for the demise of aging hair cells, thereby establishing a parallel to other cochlear pathologies. Furthermore, cathepsin activation is another possible consequence of calcium imbalance and is observed transiently in middle-aged animals. Since cathepsin D can lead to either apoptosis or necrosis (Tsukuba et al., 2000; Yamashima 2004), its transient activation is in agreement with the fact that both forms of cell death are present at this age while necrotic nuclei seem absent from the outer hair cells of old animals when cathepsin D levels return to base line.
Oxidative stress in the cochlea can also activate non-mitochondrial pathways of cell death, exemplified by the MAPK cascade. SAPK/JNK and p38 MAPK are crucial downstream elements in responses to a variety of stress stimuli, including physical, chemical and biological stresses. Both SAPK/JNK and p38 MAPK are translated into the nucleus after activation and regulate gene expression through their effects on transcription factors (Kyriakis and Avruch 2001; Leppa and Bohmann 1999). The age-related elevated phosphorylation of these key elements of the signaling cascade is again reminiscent of events in cochlear noise and drug trauma. C-Jun, as a component of the AP-1 transcription factor, is up-regulated following noise exposure (Ogita et al., 2000) and aminoglycoside antibiotics as well as cisplatin trigger MAPK pathways (Matsui et al., 2004; Rybak et al., 2008).
The focus of the analyses presented is on the outer hair cells. In contrast to human presbycusis (Schuknecht et al., 1974) and other animal models (Mills et al., 1990), loss of supporting cells or atrophy of stria vascularis is not observed in the CBA/J strain during the life span covered in this study (Sha et al., 2008). Spiral ganglion cells, however, degenerate. The appearance of various cell death markers in these cells with advancing age is consistent with the fact that ganglion cell density in CBA/J mice is reduced by about 30% in old animals (Sha et al., 2008).
It is interesting to compare our results of changes at the protein level to the changes of gene expression (Tadros et al., 2008) not only for consistencies and potential discrepancies but also for the complementary nature of the two types of analysis. Both analytical approaches show a diversity of pathways affected by age and an upregulation or activation of caspases as a potentially major factor in apoptosis. The interpretation of the data, however, appears to diverge on calpain and the MAP kinase pathway. Gene expression suggested a down regulation of calpain II and an upregulation of DUSP9, a p38 MAP kinase modulator that could possibly blunt MAP kinase signaling. In contrast, we see activation of calpain II and of MAP kinase. In both cases, however, the activation is based on a post-translational modification: a proteolytic cleavage in the case of calpain, and a phosphorylation in the case of p-38 MAP kinase. Post-translational modifications are important regulatory mechanisms that are independent of gene expression and whose detection remains in the realm of proteomics.
Pathway analyses of an extended process such as age-related hearing loss can only rely on selected snapshots in time. Such snapshots can delineate potential but not definitive mechanisms of cell death because of extensive “cross-talk” between signaling pathways. Different cell death pathways might be coordinated or follow in a time-dependent fashion but they can also be antagonized by cell survival responses, and it is the balance of the two that determines cell fate. For example, some cell types lack “competence to die” even after cytochrome c release because of low expression of pro-apoptotic regulators (Deshmukh and Johnson 1998) while other cells might quickly progress to their demise. Likewise, apoptosis is not an inevitable outcome of a presumed death signal such as JNK activation (Liu et al., 1996) when other anti-apoptotic or homeostatic pathways intervene (for an account of cell death and survival relevant to the inner ear, see Green et al. 2008). This is the context in which we must view the early appearance of apoptotic signals in cell populations that do not die until much later or not at all as illustrated by calpain expression in surviving pillar cells.
In conclusion, our study demonstrates the complexity of cell death pathways in age-related hearing loss (fig. 7) with apoptosis as a major type of outer hair cell death. Post-translational modifications of signaling cascades contribute in a fashion overlapping with but not necessarily identical to cochlear pathologies induced by noise and drugs. Understanding cell death in the aging cochlea will shed light not only on the phenomenon of hair cell loss in sensori-neural presbycusis but might also aid in designing protective or palliative treatments.
Figure 7. Schematic diagram of potential cell death pathways in the aging auditory system.

For clarity, the proposed scheme only depicts components of pathways actually analyzed in this study and omits putative intermediates and cross-talk.
Starting with an increase in reactive oxygen species in the inner ear with aging (Jiang et al., 2007) redox-sensitive cell death pathways are being triggered. The observation of necrotic nuclei of outer hair cells coincides with the activation of cathepsin D in middle-aged animals. Release of cytochrome C from the mitochondria and its complexation with and activation of caspase 9 points to the involvement of caspase-dependent apoptosis. Endonuclease G tanslocating from the mitochondria to the nuclei of outer hair cells is a potential trigger of caspase-independent apoptosis. In addition, the activation of p38MAPK and JNK in the organ of Corti, especially in the outer hair cells, implies that MAPK cascades combine with other cell death pathways in age-related hearing loss.
Acknowledgments
This study was supported by program project grant AG-025164 from the National Institute on Aging and core grant P30 DC-05188 from the National Institute on Deafness and Other Communication Disorders, NIH.
Abbreviations
- ABR
auditory brain stem evoked response
- Endo G
endonuclease G
- JNK
c-Jun-N-terminal kinase
- MAPK
mitogen-activated protein kinases
- PBS
phosphate-buffered saline
- ROS
reactive oxygen species
- SAPK
stress-activated protein kinase
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Brunk UT, Dalen H, Roberg K, Hellquist HB. Photo-oxidative disruption of lysosomal membranes causes apoptosis of cultured human fibroblasts. Free Radic Biol Med. 1997;23:616–626. doi: 10.1016/s0891-5849(97)00007-5. [DOI] [PubMed] [Google Scholar]
- Danial NN, Korsmeyer SJ. Cell death: critical control points. Cell. 2004;116:205–219. doi: 10.1016/s0092-8674(04)00046-7. [DOI] [PubMed] [Google Scholar]
- Deshmukh M, Johnson EM., Jr Evidence of a novel event during neuronal death: development of competence-to-die in response to cytoplasmic cytochrome c. Neuron. 1998;21:695–705. doi: 10.1016/s0896-6273(00)80587-5. [DOI] [PubMed] [Google Scholar]
- Dulon D, Zajic G, Aran JM, Schacht J. Aminoglycoside antibiotics impair calcium entry but not viability and motility in isolated cochlear outer hair cells. J Neurosci Res. 1989;24:338–346. doi: 10.1002/jnr.490240226. [DOI] [PubMed] [Google Scholar]
- Ferri KF, Kroemer G. Organelle-specific initiation of cell death pathways. Nat Cell Biol. 2001;3:E255–263. doi: 10.1038/ncb1101-e255. [DOI] [PubMed] [Google Scholar]
- Fridberger A, Flock A, Ulfendahl M, Flock B. Acoustic overstimulation increases outer hair cell Ca2+ concentrations and causes dynamic contractions of the hearing organ. Proc Natl Acad Sci USA. 1998;95:7127–7132. doi: 10.1073/pnas.95.12.7127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green DR, Kroemer G. The pathophysiology of mitochondrial cell death. Science. 2004;305:626–629. doi: 10.1126/science.1099320. [DOI] [PubMed] [Google Scholar]
- Green SH, Altschuler RA, Miller JM. Cell death and cochlear protection. In: Schacht J, Popper AN, Fay RR, editors. Handbook of Auditory Research. Vol. 31. Springer; New York: 2008. pp. 275–319. [Google Scholar]
- Guicciardi ME, Deussing J, Miyoshi H, Bronk SF, Svingen PA, Peters C, Kaufmann SH, Gores GJ. Cathespin B contributes to TNF-α-mediated hepatocyte apoptosis by promoting mitochrondrial release of cytochrome c. J Clin Invest. 2000;106:1127–1137. doi: 10.1172/JCI9914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson D, Hu B, Bielefeld E. Patterns and mechanisms of noise-induced cochlear pathology. In: Schacht J, Popper AN, Fay RR, editors. Handbook of Auditory Research. Vol. 31. Springer; New York: 2008. pp. 195–217. [Google Scholar]
- Hu BH, Henderson D, Nicotera TM. Involvement of apoptosis in progression of cochlear lesion following exposure to intense noise. Hear Res. 2002;166:62–71. doi: 10.1016/s0378-5955(02)00286-1. [DOI] [PubMed] [Google Scholar]
- Ishihara Y, Shimamoto N. Involvement of endonuclease G in nucleosomal DNA fragmentation under sustained endogenous oxidative stress. J Biol Chem. 2006;281:6726–6733. doi: 10.1074/jbc.M510382200. [DOI] [PubMed] [Google Scholar]
- Jiang H, Sha SH, Forge A, Schacht J. Caspase-independent pathways of hair cell death induced by kanamycin in vivo. Cell Death Differ. 2006;13:20–30. doi: 10.1038/sj.cdd.4401706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang H, Talaska AE, Schacht J, Sha SH. Oxidative imbalance in the aging inner ear. Neurobiol Aging. 2007;28:1605–1612. doi: 10.1016/j.neurobiolaging.2006.06.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelekar A. Autophagy. Ann NY Acad Sci. 2006;1066:259–271. doi: 10.1196/annals.1363.015. [DOI] [PubMed] [Google Scholar]
- Kyriakis JM, Avruch J. Mammalian mitogen-activated protein kinase signal transduction pathways activated by stress and inflammation. Physiol Rev. 2001;81:807–869. doi: 10.1152/physrev.2001.81.2.807. [DOI] [PubMed] [Google Scholar]
- Lee CK, Klopp RG, Weindruch R, Prolla TA. Gene expression profile of aging and its retardation by caloric restriction. Science. 1999;285:1390–1393. doi: 10.1126/science.285.5432.1390. [DOI] [PubMed] [Google Scholar]
- Leist M, Jäättelä M. Triggering of apoptosis by cathepsins. Cell Death Differ. 2001;8:324–326. doi: 10.1038/sj.cdd.4400859. [DOI] [PubMed] [Google Scholar]
- Leppa S, Bohmann D. Diverse functions of JNK signaling and c-Jun in stress response and apoptosis. Oncogene. 1999;18:6158–6162. doi: 10.1038/sj.onc.1203173. [DOI] [PubMed] [Google Scholar]
- Li LY, Luo X, Wang X. Endonuclease G is an apoptotic DNase when released from mitochondria. Nature. 2001;412:95–99. doi: 10.1038/35083620. [DOI] [PubMed] [Google Scholar]
- Liu ZG, Hsu H, Goeddel DV, Karin M. Dissection of TNF receptor 1 effector functions: JNK activation is not linked to apoptosis while NF-kB activation prevents cell death. Cell. 1996;87:565–576. doi: 10.1016/s0092-8674(00)81375-6. [DOI] [PubMed] [Google Scholar]
- Matsui JI, Gale JE, Warchol ME. Critical signaling events during the aminoglycoside-induced death of sensory hair cells in vitro. J Neurobiol. 2004;61:250–266. doi: 10.1002/neu.20054. [DOI] [PubMed] [Google Scholar]
- McFadden SL, Ding D, Burkard RF, Jiang H, Reaume AG, Flood DG, Salvi RJ. Cu/Zn SOD deficiency potentiates hearing loss and cochlear pathology in aged 129, CD-1 mice. J Comp Neurol. 1999;413:101–112. [PubMed] [Google Scholar]
- McFadden SL, Ding D, Reaume AG, Flood DG, Salvi RJ. Age-related cochlear hair cell loss is enhanced in mice lacking copper/zinc superoxide dismutase. Neurobiol Aging. 1999;20:1–8. doi: 10.1016/s0197-4580(99)00018-4. [DOI] [PubMed] [Google Scholar]
- McFadden SL, Ding D, Salvi R. Anatomical, metabolic and genetic aspects of age-related hearing loss in mice. Audiology. 2001;40:313–321. [PubMed] [Google Scholar]
- Mills JH, Schmiedt RA, Kulish LF. Age-related changes in auditory potentials of Mongolian gerbil. Hear Res. 1990;46:201–210. doi: 10.1016/0378-5955(90)90002-7. [DOI] [PubMed] [Google Scholar]
- Nakagawa T, Yamane H, Takayama M, Sunami K, Nakai Y. Apoptosis of guinea pig cochlear hair cells following chronic aminoglycoside treatment. Eur Arch Otorhinolaryngol. 1998;255:127–131. doi: 10.1007/s004050050027. [DOI] [PubMed] [Google Scholar]
- Ogita K, Matsunobu T, Schacht J. Acoustic trauma enhances DNA binding of transcription factor AP-1 in the guinea pig inner ear. Neuroreport. 2000;11:859–862. doi: 10.1097/00001756-200003200-00040. [DOI] [PubMed] [Google Scholar]
- Roberg K, Kagedal K, Ollinger K. Microinjection of cathepsin d induces caspase-dependent apoptosis in fibroblasts. AM J Pathol. 2002;161:89–96. doi: 10.1016/S0002-9440(10)64160-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rybak LP, Talaska AE, Schacht J. Drug-induced hearing loss. In: Schacht J, Popper AN, Fay RR, editors. Handbook of Auditory Research. Vol. 31. Springer; New York: 2008. pp. 219–256. [Google Scholar]
- Ryter SW, Kim HP, Hoetzel A, Park JW, Nakahira K, Wang X, Choi AM. Mechanisms of cell death in oxidative stress. Antioxid Redox Signal. 2007;9:49–89. doi: 10.1089/ars.2007.9.49. [DOI] [PubMed] [Google Scholar]
- Schuknecht HF. Further observations on the pathology of presbycusis. Arch Otolaryngol. 1964;80:369–382. doi: 10.1001/archotol.1964.00750040381003. [DOI] [PubMed] [Google Scholar]
- Schuknecht HF, Watanuki K, Takahashi T, Belal AA, Jr, Kimura RS, Jones DD, Ota CY. Atrophy of the stria vascularis, a common cause for hearing loss. Laryngoscope. 1974;84:1777–1821. doi: 10.1288/00005537-197410000-00012. [DOI] [PubMed] [Google Scholar]
- Seidman MD. Effects of dietary restriction and antioxidants on presbyacusis. Laryngoscope. 2000;110:727–738. doi: 10.1097/00005537-200005000-00003. [DOI] [PubMed] [Google Scholar]
- Sha SH, Kanicki A, Dootz G, Talaska AE, Halsey K, Dolan D, Altschuler R, Schacht J. Age-related auditory pathology in the CBA/J mouse. Hear Res. 2008;243:87–94. doi: 10.1016/j.heares.2008.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sohal RS, Dubey A. Mitochondrial oxidative damage, hydrogen peroxide release, and aging. Free Radic Biol Med. 1994;16:621–626. doi: 10.1016/0891-5849(94)90062-0. [DOI] [PubMed] [Google Scholar]
- Sohal RS, Weindruch R. Oxidative stress, caloric restriction, and aging. Science. 1996;273:59–63. doi: 10.1126/science.273.5271.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tadros SF, D'Souza M, Zhu X, Frisina RD. Apoptosis-related genes change their expression with age and hearing loss in the mouse cochlea. Apoptosis. 2008;13:1303–1321. doi: 10.1007/s10495-008-0266-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trachootham D, Lu W, Ogasawara MA, Nilsa RD, Huang P. Redox regulation of cell survival. Antioxid Redox Signal. 2008;10:1343–1374. doi: 10.1089/ars.2007.1957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsukuba T, Okamoto K, Yasuda Y, Morikawa W, Nakanishi H, Yamamoto K. New functional aspects of cathepsin D and cathepsin E. Mol Cells. 2000;10:601–611. doi: 10.1007/s10059-000-0601-8. [DOI] [PubMed] [Google Scholar]
- Unal-Cevik I, Kilinc M, Can A, Gursoy-Ozdemir Y, Dalkara T. Apoptotic and necrotic death mechanisms are concomitantly activated in the same cell after cerebral ischemia. Stroke. 2004;35:2189–2194. doi: 10.1161/01.STR.0000136149.81831.c5. [DOI] [PubMed] [Google Scholar]
- Yamashima T. Implication of cysteine proteases calpain, cathespin and caspase in ischemic neuronal death of primates. Prog Neurobiol. 2000;62:273–395. doi: 10.1016/s0301-0082(00)00006-x. [DOI] [PubMed] [Google Scholar]
- Yamashima T. Ca2+-dependent proteases in ischemic neuronal death: a conserved ‘calpain-cathepsin cascade’ from nematodes to primates. Cell Calcium. 2004;36:285–293. doi: 10.1016/j.ceca.2004.03.001. [DOI] [PubMed] [Google Scholar]
- Yamashita D, Miller JM, Jiang HY, Minami SB, Schacht J. AIF and Endo G in noise-induced hearing loss. Neuroreport. 2004;15:2719–2722. [PubMed] [Google Scholar]
- Yang WP, Henderson D, Hu BH, Nicotera TM. Quantitative analysis of apoptotic and necrotic outer hair cells after exposure to different levels of continuous noise. Hear Res. 2004;196:69–76. doi: 10.1016/j.heares.2004.04.015. [DOI] [PubMed] [Google Scholar]