

Abstract
The serotonin 5-HT2A, 5-HT2B, and 5-HT2C G protein-coupled receptors signal primarily through Gαq to activate phospholipase C (PLC) and formation of inositol phosphates (IP) and diacylglycerol. The human 5-HT2C receptor, expressed exclusively in the central nervous system, is involved in several physiological and psychological processes. Development of 5-HT2C agonists that do not also activate 5-HT2A or 5-HT2B receptors is challenging because transmembrane domain identity is about 75% among 5-HT2 subtypes. This paper reports 5-HT2 receptor affinity and function of (1R,3S)-(−)-trans-1-phenyl-3-dimethylamino-1,2,3,4-tetrahydronaphthalene (PAT), a small molecule that produces anorexia and weight-loss after peripheral administration to mice. (−)-Trans-PAT is a stereoselective full-efficacy agonist at human 5-HT2C receptors, plus, it is a 5-HT2A/5-HT2B inverse agonist and competitive antagonist. The Ki of (−)-trans-PAT at 5-HT2A, 5-HT2B, and 5-HT2C receptors is 410, 1200, and 37 nM, respectively. Functional studies measured activation of PLC/[3H]-IP formation in clonal cells expressing human 5-HT2 receptors. At 5-HT2C receptors, (−)-trans-PAT is an agonist (EC50 = 20 nM) comparable to serotonin in potency and efficacy. At 5-HT2A and 5-HT2B receptors, (−)-trans-PAT is an inverse agonist (IC50 = 490 and 1,000 nM, respectively) and competitive antagonist (KB = 460 and 1400 nM, respectively) of serotonin. Experimental results are interpreted in light of molecular modeling studies indicating the (−)-trans-PAT protonated amine can form an ionic bond with D3.32 of 5-HT2A and 5-HT2C receptors, but, not with 5-HT2B receptors. In addition to probing 5-HT2 receptor structure and function, (−)-trans-PAT is a novel lead regarding 5-HT2C agonist/5-HT2A inverse agonist drug development for obesity and neuropsychiatric disorders.
Keywords: Drug addiction, Drug discovery, Obesity, Psychiatric disorder, Serotonin 5-HT2 receptor
1. Introduction
Serotonin (5-hydroxytryptamine, 5-HT) mediates its central and peripheral psychological and physiological effects through 14 mammalian serotonin receptor subtypes grouped into the 5-HT1–5-HT7 families. The 5-HT2 receptor family consists of the 5-HT2A, 5-HT2B, and 5-HT2C membrane-bound G protein-coupled receptors that signal primarily through Gαq to activate phospholipase C (PLC) and formation of inositol phosphates (IP) and diacylglycerol second messengers (Raymond et al., 2001). The human 5-HT2C receptor (Saltzman et al., 1991) is found exclusively in the central nervous system where it is widely expressed and putatively involved in several (patho)- physiological and psychological processes, including, ingestive behavior (Tecott et al., 1995), cocaine addiction (Fletcher et al., 2002), psychosis (Siuciak et al., 2007), anxiety (Heisler et al., 2007), depression (Rosenzweig-Lipson et al., 2007), and motor function (Fox and Brotchie, 2000a, 2000b). Thus, the importance of the 5-HT2C receptor as a pharmacotherapeutic target has been apparent for about 10 years, however, 5-HT2C receptor-selective drugs still are not available.
Drug discovery targeting the 5-HT2C receptor is challenging because it shares about 75% transmembrane sequence identity with 5-HT2A and 5-HT2B receptors. The highly conserved transmembrane domains and same second messenger signaling complicates development of selective 5-HT2C agonists, in particular. Meanwhile, there is compelling evidence that 5-HT2C receptor activation reduces food intake and leads to anti-obesity effects in lab animals and humans. For example, 5-HT2C knockout mice demonstrate increased feeding and obesity, and, they are resistant to the anorectic effects of the now banned weight-loss drug S-(+)-fenfluramine (Tecott et al., 1995; Vickers et al., 2001). S-(+)- and (±)-fenfluramine promote serotonin release (Rothman et al., 1999) and both were banned by the US Food and Drug Administration in 1997 because fenfluramine and its metabolite S-(+)-norfenfluramine cause activation of 5-HT2B receptors that can lead to valvular heart disease (Fitzgerald et al., 2000; Setola et al., 2005) and/or pulmonary hypertension (Launay et al., 2002)–fatalities have resulted.
There also is convincing preclinical evidence that activation of brain 5-HT2C receptors could be pharmacotherapeutic for cocaine and other psychostimulant addiction (Bubar and Cunningham, 2008). For example, the non-selective 5-HT2 agonist RO60175 reduces the rate of cocaine self-administration in rats and this effect is not blocked by M100907 (Fletcher et al., 2002), a 5-HT2A antagonist with 500-times and 15-times selectivity over 5-HT2B and 5-HT2C receptors, respectively (Knight et al., 2004). In addition to psychostimulant addiction, evidence for the pharmacotherapeutic relevance of 5-HT2C receptor activation in other neuropsychiatric disorders continues to come forth (e.g., Heisler et al., 2007; Siuciak et al., 2007; Rosenzweig-Lipson et al., 2007), and, there is intense interest by pharmaceutical companies to develop a genuinely selective 5-HT2C agonist—no 5-HT2C agonist devoid of 5-HT2A and/or 5-HT2B agonism has been reported previously (e.g., Siuciak et al., 2007; Rosenzweig-Lipson et al., 2007; Thomsen et al., 2008). Results reported in this paper, however, document that a novel compound synthesized in our laboratories, (1R,3S)-(−)-trans-1-phenyl-3-dimethylamino-1,2,3,4-tetrahydronaphthalene (PAT; Fig.1), is a full-efficacy agonist at human 5-HT2C receptors, plus, it is an inverse agonist at 5-HT2A and 5-HT2B receptors and competitively antagonizes 5-HT activation of these receptors. Recent studies (Rowland et al., 2008; Bivens et al., 2007) document that (−)-trans-PAT produces anorexia and weight-loss in mice after peripheral administration with potency and efficacy comparable to the commercially-available but nonselective 5-HT2C agonist WAY-161503 (Rosenzweig-Lipson et al., 2006). The PAT molecule exists as four stereoisomers (Fig. 1) and 5-HT2C receptor agonist activity resides only in the (1R,3S)-(−)-trans-PAT isomer. This striking PAT stereoselective functional activity is relevant not only for pharmacotherapeutic possibilities involving selective 5-HT2C activation, but, also, for delineating the 3-dimensional molecular determinants for ligand binding and function at 5-HT2 receptors. Accordingly, G protein-coupled receptor homology-based molecular models were constructed for the 5-HT2 receptor subtypes and (−)-trans-PAT docking experiments were undertaken to characterize molecular determinants for selective binding and inferences regarding agonism vs. inverse agonism functional activity.
Fig. 1.
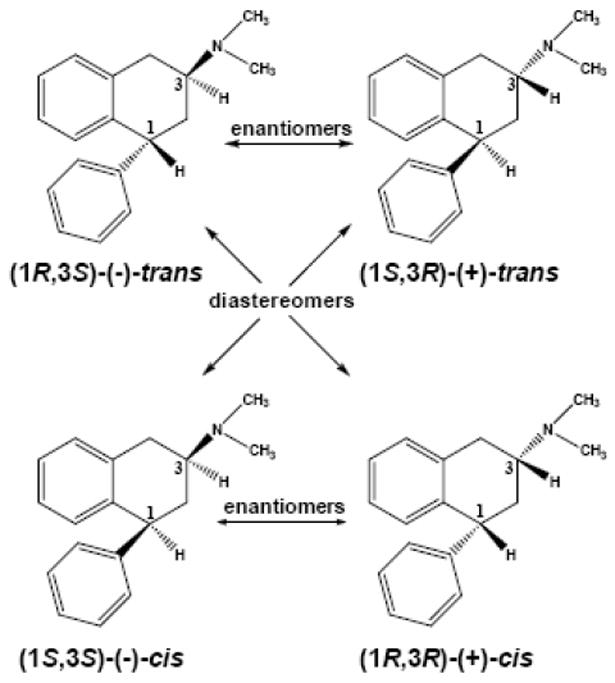
Structures of 1-phenyl-3-dimethylamino-1,2,3,4-tetrahydronaphthalene (PAT) stereoisomers.
2. Materials and Methods
2.1 Chemicals
The four stereoisomers of 1-phenyl-3-N,N-dimethylamino-1,2,3,4-tetrahydronaphthalene (PAT) stereoisomers (Fig. 1) were synthesized by modification of our previously reported procedure (Wyrick et al., 1993) and the details will be reported elsewhere. Briefly, (E)-1,4-diphenylbut-1-en-3-one was cyclized to the corresponding β-tetralone intermediate using polyphosphoric acid in toluene under reflux conditions (18 h) and the product was purified by flash column chromatography. The tetralone was reduced with sodium borohydride to a mixture of (±)-cis- and (±)-trans-tetralols that could be separated by recrystallization. To obtain (±)-trans-PAT, the (±)-cis-tetralol was stirred with p-toluenesulfonyl chloride in pyridine for 2 days at room temperature and purified by flash column to obtain the corresponding tosylate intermediate. Stirring the tosylate with sodium azide in N,N-dimethylformamide for 2 days at room temperature yields the stereochemically-inverted (±)-trans-azido derivative, which was reduced by catalytic hydrogenation to the free amine. The (±)-trans-amine was converted to the diastereomeric salt using (1R)-(–)-camphor-10-sulfonic acid and the diastereomers were separated by fractional recrystallization. The (1R, 3S)-(+)-trans- and (1R, 3S)-(−)-trans-amine enantiomers isolated were dimethylated using formic acid/formaldehyde and purified by column chromatography to obtain the (1R, 3S)-(+)-trans- and (1R, 3S)-(−)-trans-PAT products. The (1R, 3R)-(+)-cis- and (1S, 3S)-(−)-cis-PAT enantiomers were obtained from the (±)-trans-tetralol using methods analogous to those used to synthesize the (+)- and (−)-trans isomers. All products were converted to the hydrochloride salt for use in pharmacological experiments. Purity and absolute configuration were determined by elemental analysis, 1H-NMR, mass spectrometry, melting point, X-ray crystallography, optical rotation, and HPLC using a polysaccaharide-based chiral column (Kromasil, Akzo Nobel, Brewster, NY).
[3H]-Ketanserin (specific activity 72.2 Ci/mmol) and myo-[2-3H(N)]-inositol (specific activity 18.5 Ci/mmol) were purchased from Perkin-Elmer Life Science (Boston, MA) and [N6-methyl-3H]-mesulergine (specific activity 72.0 Ci/mmol) was purchased from Amersham Biosciences (GE healthcare, Piscataway, NJ). Unless otherwise noted, all other compounds were obtained in highest purity from Sigma-Aldrich (St. Louis, MO).
2.2 Clonal cell culture and transfection
Chinese hamster ovary K1 cells (CHO, ATCC CCL-61) were maintained in Ham’s F-12 medium supplemented with 10% fetal bovine serum and 1% sodium bicarbonate (Mediatech 25-035-CI), 10 IU/ml penicillin and 10 ug/ml streptomycin. Human embryonic kidney 293 cells (HEK, ATCC CRL-1573) were maintained in Eagle minimum essential medium with 10% fetal bovine serum and 2 mM L-glutamine, 0.1 mM non-essential amino acids, 1.5 g/L sodium bicarbonate, 1.0 mM sodium pyruvate, 10 IU/ml penicillin, and 10 ug/ml streptomycin. Cells were grown in a humidified incubator at 37 °C with 5% carbon dioxide.
The cDNAs encoding the wild type human 5-HT2A, 5-HT2B, and 5-HT2C receptors were obtained from UMR (Rolla, MO). Serotonin 5-HT2A and 5-HT2B receptors were transiently expressed in HEK cells (Setola et al., 2005) and 5-HT2C receptors were transiently expressed in CHO cells (Porter et al., 1999). HEK cells were grown to 90-95% confluence in 100 mm dishes and transfected with 24 μg of plasmid DNA for the wild type 5-HT2A or 5-HT2B receptor sequences using 40 μl of Lipofectamine 2000 (Invitrogen, Carlsbad, CA) per dish. Transfection proceeded for 24 hrs, then, medium was replaced by fresh growth medium and cells were allowed to express 5-HT2A or 5-HT2B receptors for another 24 hrs. CHO cells were grown to 40% confluence in 100 mm dishes and transfected with 12 μg of plasmid DNA for the wild type 5-HT2C receptor sequence and 32 μl of Lipofectamine 2000, then, transfection and expression proceeded as above.
2.3 Radioreceptor assays
Radioreceptor saturation and competition binding assays were performed using membrane homogenates prepared from transfected HEK (5-HT2A, 5-HT2B) or CHO (5-HT2C) cells. [3H]-Ketanserin was used to radiolabel 5-HT2A receptors and [3H]-mesulergine was used to label 5-HT2B and 5-HT2C receptors (Knight et al., 2004). Forty-eight hours following transfection, cells were harvested and homogenized in 50 mM Tris-HCl containing 0.1 % ascorbic acid and 4.0 mM calcium chloride at pH 7.4 (assay buffer). The homogenate was centrifuged at 35,000 ×g for 25 min and the resulting membrane pellet was re-suspended in assay buffer. For saturation binding assays, membrane suspension containing 20 μg (for 5-HT2A receptor), 50 μg (for 5-HT2B receptor) or 100 μg (for 5-HT2C receptor) protein was incubated with 0.1 – 5.0 nM [3H]-ketanserin (5-HT2A receptors) or 0.1 – 20 nM [3H]-mesulergine (5-HT2B and 5-HT2C receptors) in a total assay buffer volume of 250 μl. Non-specific binding was determined in the presence of 10 μM methysergide (5-HT2A receptors) or 1.0 μM mianserin (5-HT2B and 5-HT2C receptors). Competition binding assays were conducted under the same conditions using 1.0 nM [3H]-ketanserin (5-HT2A receptors), 5.0 nM [3H]-mesulergine (5-HT2B receptors), or 1.0 nM [3H]-mesulergine (5-HT2C receptors) (~Kd concentration). Incubation of radioreceptor binding assay mixtures was for 1.0 h at 37°C, with terminat ion by rapid filtration through Whatman GF/B filters using a 96-well cell harvester (Tomtec, Hamden, CT). The membrane-bound [3H]-radioligand retained on the filter discs was quantified by liquid scintillation spectrometry. Data were analyzed by nonlinear regression using the sigmoidal curve-fitting algorithms in Prism 4.03 (GraphPad Software Inc., San Diego, CA), with the one-site model providing the best fit. Ligand affinity is expressed as an approximation of Ki values by conversion of the IC50 data to Ki values using the equation Ki = IC50/1 + L/Kd where L is the concentration of radioligand having affinity Kd. Each experimental condition was performed in triplicate and each experiment was performed a minimum of three times to determine mean Kd ± SEM.
2.4. Measurement of PLC Activation and [3H]-IP Formation
Functional activation of PLC was measured as [3H]-IP formation in HEK cells transiently expressing 5-HT2A or 5-HT2B receptors (Setola et al., 2005), or, CHO cells transiently expressing 5-HT2C receptors (Porter et al., 1999). HEK cells expressing 5-HT2A or 5-HT2B receptors, or, CHO cells expressing 5-HT2C receptors were seeded at 105 cells per well in 12-well plates in inositol-free Dulbecco’s modified Eagle’s medium (DMEM) for 12 – 24 hours with 1.0 μCi/ml myo-[2-3H]-inositol, the radiolabeled precursor of the PLC-β substrate phosphatidylinositol (with addition of 5% dialyzed fetal bovine serum to DMEM for HEK cells). Cells were subsequently washed and incubated in inositol-free DMEM containing 10 mM lithium chloride, 10 μM pargyline, and various concentrations of test ligand for 45 – 60 min at 37 °C and 5% CO2. After aspiration of media, cells were lysed by incubation with 50 mM formic acid (15 – 60 min). Formic acid was neutralized with ammonium hydroxide and contents from each well were added to individual AG1-X8 200-400 formate resin anion exchange columns. Ammonium formate/formic acid (1.2 M/0.1 M) was used to elute [3H]-IP directly into scintillation vials for counting of tritium by liquid scintillation spectrometry. Resulting data were analyzed using the nonlinear regression algorithms in Prism 4.03, with the one-site model providing the best fit. Data is expressed as mean percentage of basal control [3H]-IP formation, with potency expressed as concentration required to stimulate (EC50) or inhibit (IC50) maximal basal (constitutive) [3H]-IP formation by 50% ± SEM. (n ≥ 3).
2.5 Molecular Modeling and Ligand Docking
Three-dimensional molecular models of the human 5-HT2A, 5-HT2B and 5-HT2C receptors were constructed by homology modeling according to the crystal structure of bovine rhodopsin (Protein Data Bank entry 1L9H) (Okada et al., 2002). The transmembrane (TM) portions were built using the Biopolymer module of Sybyl 7.3 by substituting the amino acid side chains in rhodopsin to the side chains of the amino acids in the respective 5-HT2 receptor. The rhodopsin helix 7 structure that expands parallel to the membrane also was retained in the 5-HT2 models. The resulting seven TM bundle was minimized with constrained C alpha atoms using Amber force field and atomic charges. The 5-HT2 receptor intracellular and extracellular loops were added, however, the loop connecting TM 5 and 6 was truncated to include ten residues leading out of helix 5 and ten residues leading into helix 6. The conserved disulfide bridge formed between the cysteine amino acids in TM 3 and extracellular loop 2 also was included in the 5-HT2 receptor models. Water molecules present in the rhodopsin crystal structure were not included. The crude 5-HT2 models were minimized using the Powell method implemented in Sybyl with Amber force field and atomic charges and all Cα atoms restrained. The resulting structure was inserted into a rectangular box containing a pre-equilibrated dipalmitoylphosphatidylcholine (DPPC) bilayer. The DPPC bilayer was constructed with the “genconf” tool implemented with Gromacs v3.3.1 software (Lindahl et al., 2001) using coordinates available from the website of Dr. Peter Tieleman at the University of Calgary (http://moose.bio.ucalgary.ca/index.php?page=Structures_and_Topologies). The starting receptor–lipid bilayer system (180 DPPC molecules and 14,774 water molecules) was neutralized by adding chlorine ions and then subjected to 500 steps of energy minimization, followed by an equilibration run (500 ps) with receptor Cα atoms restrained (1000 kJ × mol-1 × nm-1) and 2 – 6 ns unrestrained production run. Minimization and molecular dynamics simulations were performed with Gromacs v3.3.1 software using the optimized potentials for liquid simulations all-atom force field for protein (Jorgensen et al., 1996) and Berger parameters for DPPC molecules (Berger et al., 1997). The LINCS algorithm was used to restrain bond lengths (Hess et al., 1997). Lipids topology files were from Dr. Tieleman’s website and the water model was a simple rigid three-point charge model. All simulations are performed for an NPT ensemble (number of particles, pressure, temperature) using Berendsen’s barostat and thermostat (Berendsen et al., 1981). The system was simulated with semiisotropic coupling, with the pressure at 1.0 bar and 300K (protein, phospholipids, solvent, and ions were coupled separately). Electrostatic interactions were calculated using particle mesh Ewald (Darden et al., 1993) with a cutoff of 0.9 nm. Cutoff for van der Waals interactions was set at 1.4 nm and the time step for integration was 2 fs.
Flexible ligand docking was performed with the FlexiDock utility in Sybyl 7.3 that uses a genetic algorithm to probe conformational space defining possible interactions between the ligand and its putative binding site. Ligand structures were built as monocations using the Sketch Molecule module in Sybyl 7.3, applying Gastaiger-Huckel atomic charges and Tripos force fields while Amber charges and atom types were used for 5-HT2 receptors. The last 1 ns of dynamics trajectories was used to extract 5-HT2 receptor structures in 250 ps increments. Each ligand was pre-positioned in the putative 5-HT2 receptor binding pocket and all possible rotatable bonds in the ligand and side chains defining the putative binding site (120 rotatable bonds in total) were rotated during sampling of conformational space. Default FlexiDock parameters were set at 80,000-generation and the best docking solution, according to highest FlexiDock score, was minimized using an Amber force field (during minimization only backbone atoms of the receptor were constrained).
3. Results
3.1. Serotonin 5-HT2 receptor radioligand saturation binding analysis
There was no measurable specific radioligand binding using membranes prepared from null-transfected CHO and HEK cells. Using membranes prepared from cells transiently transfected with 5-HT2A, 5-HT2B, or 5-HT2C cDNA, however, saturable specific radioligand binding was observed. Fig. 2 shows representative binding data for [3H]-ketanserin labeled 5-HT2A receptors and [3H]-mesulergine labeled 5-HT2B receptors and 5-HT2C receptors. [3H]-Ketanserin binds to an apparent single population of 5-HT2A receptors (Bmax = 1.73 ± 0.11 pmol/mg protein) with high affinity (KD = 0.80 ± 0.03 nM). [3H]-Mesulergine labels a single population of 5-HT2B receptors with Bmax = 1.13 ± 0.39 pmol/mg protein and KD = 5.19 ± 0.36 nM. Analogously, [3H]-mesulergine also labels an apparent single population of 5-HT2C receptors (Bmax = 8.37 ± 0.15 pmol/mg prot) with high affinity (KD = 0.88 ± 0.03 nM).
Fig. 2.

- (A) [3H]-Ketanserin labeled 5-HT2A receptors; Bmax = 1.73 ± 0.11, Kd = 0.80 ± 0.03.
- (B) [3H]-Mesulergine labeled 5-HT2B receptors; Bmax = 1.13 ± 0.39, Kd = 5.19 ± 0.36.
- (C) [3H]-Mesulergine labeled 5-HT2C receptors; Bmax = 8.37 ± 0.15, Kd = 0.88 ± 0.03.
3.2. PAT binding to serotonin 5-HT2-type radiolabeled receptors
Fig. 3 shows representative 5-HT2A, 5-HT2B, and 5-HT2C receptor radioligand displacement curves for the PAT isomers. Curves are sigmoidal in shape and span 3 to 4 log ligand concentration units to achieve complete radioligand displacement, characteristic of competitive displacement of ~Kd radioligand concentration from a single population of G protein-coupled receptors. Affinity data for PAT isomers at each 5-HT2 receptor subtype is summarized in the Table.
Fig. 3.

Representative concentration–response curves for PAT isomer displacement of 5-HT2 receptor radioligands from 5-HT2A (A), 5-HT2B (B), and 5-HT2C (C) receptors. Ki and nH values are summarized in the Table.
Table 1.
| PAT Isomer | 5-HT2A Ki ± SEM (nM) | nHa | 5-HT2B Ki ± SEM (nM) | nHa | 5-HT2C Ki ± SEM (nM) | nHa |
|---|---|---|---|---|---|---|
| (−)-trans | 410 ± 38 | 1.1 ± 0.1 | 1,200 ± 6.8 | 1.0 ± 0.001 | 37.6 ± 3.02 | 0.9 ± 0.1 |
| (+)-trans | 520 ± 3.0 | 1.0 ± 0.2 | ~ 2,500 | ~1.0 | 1300 ± 80 | 1.0 ± 0.1 |
| (+)-cis | 780 ± 2.0 | 0.9 ± 0.2 | ~ 5,000 | ~1.0 | 980 ± 7.8 | 0.9 ± 0.1 |
| (−)-cis | 1500 ± 2.0 | 1.0 ± 0.1 | ~ 10,000 | ~1.0 | 430 ± 4.8 | 0.90 ± 0.04 |
None of the Hill slope values differs significantly (P>0.10) from unity.
The (−)-trans-PAT isomer has 10- to 30-times higher affinity (Ki ~ 38 nM) at 5-HT2C receptors compared to the other three PAT stereoisomers—in fact, (−)-trans-PAT is the isomer with highest affinity at all three 5-HT2 subtypes. For the other PAT isomers, rank order affinity potency is (+)-trans- > (+)-cis- > (−)-cis-PAT at 5-HT2A and 5-HT2B receptors, with the order reversed at 5-HT2C receptors—overall, however, affinity differences between these isomers at any particular receptor subtype is relatively small (between 2- and 4-times), and, affinity is low (Ki > 500nM) to nil (Ki ~ 10 μM) across 5-HT2 receptor subtypes. Thus, the (−)-trans-PAT isomer demonstrated highest 5-HT2C affinity and selectivity compared to the other isomers and detailed data for its 5-HT2 receptor functional activity is shown in the next section.
3.3. PAT functional activity at serotonin 5-HT2c receptors
The 5-HT2 G protein-coupled receptor family is constitutively active when expressed in CHO and HEK cells, coupling primarily to Gαq protein to activate PLC and IP formation (Raymond et al., 2001). In lysates of null-transfected CHO and HEK cells, no increase in PLC/[3H]-IP formation above basal activity was detected after incubation with up to 10 μM of the endogenous agonist serotonin for 45 min. In CHO cells expressing human 5-HT2C receptors, however, serotonin produces a concentration-dependent increase in basal activity of PLC/[3H]-IP formation, with EC50 = 7.30 ± 0.55 nM (nH = 1.04 ± 0.20) and Emax = 480 ± 12.4 % basal control activity (occurs at ~ 100 nM)–a representative graph of data is shown in Fig. 4. These results for serotonin activation of 5-HT2C receptors are consistent with results reported in the literature (e.g., Porter et al., 1999).
Fig. 4.

Representative concentration–response curves for serotonin (closed squares) and (−)-trans-PAT (closed circles) activation of PLC/[3H]-IP formation in clonal cells expressing 5-HT2C receptors. Single concentration effect of (−)-trans-PAT at 5-HT2A (open triangles) and 5-HT2B (open squares) receptors also is shown. EC50 and nH values are given in the text.
Relative to serotonin, (−)-trans-PAT is a full-efficacy 5-HT2C receptor agonist that produces a concentration-dependent increase in basal activity of PLC/[3H]-IP formation, spanning about 4 log concentration units to achieve maximal functional efficacy, characteristic of agonist activation of a single population of GPCRs, according to the ternary complex model (Fig. 4). For (−)-trans-PAT activation of 5-HT2C receptors, EC50 = 19.9 ± 2.22 nM (nH = 0.75 ± 0.13; not significantly different from unity, P>0.10). The potency (EC50) of (−)-trans-PAT to activate 5-HT2C receptors appears to be about 2-times greater than its 5-HT2C affinity (Ki), however, it is noted that the 5-HT2C radioligand used to determine affinity, [3H]-mesulergine, is an antagonist that likely does not selectively label the agonist-preferring conformation of the receptor (Knight et al., 2004). Regarding (−)-trans-PAT efficacy as a 5-HT2C agonist, Emax = 464 ± 10.8% basal control activity (occurs at ~ 10 μM), and, there is no significant difference (P = 0.2) between the Emax values for (−)-trans-PAT and serotonin. None of the other three PAT isomers stimulated 5-HT2C basal PLC/[3H]-IP formation at concentrations up to 10 μM (data not shown).
Having identified (−)-trans-PAT as an agonist for 5-HT2C receptors, further studies were conducted with this isomer to determine its functional activity at 5-HT2A and 5-HT2B receptors. It was observed that (−)-trans-PAT does not stimulate PLC/[3H]-IP formation in cells expressing 5-HT2A or 5-HT2B receptors—in fact, beginning at concentrations of about 1.0 μM, (−)-trans-PAT appeared to inhibit 5-HT2A and 5-HT2B receptor basal (constitutive) activity (Fig. 4). Accordingly, experiments were undertaken to evaluate antagonist, as well as, inverse agonist functional activity of (−)-trans-PAT at 5-HT2A and 5-HT2B receptors. In view of the very low affinity (Ki ~ 0.5 – 10 μM, Table) of the other PAT isomers at 5-HT2A and 5-HT2B receptors, further functional experiments were not conducted with these isomers.
3.4. Assessment of (−)-trans-PAT antagonist activity at serotonin 5-HT2A and 5-HT2B receptors
In these experiments, the ability of (−)-trans-PAT to antagonize serotonin stimulation of 5-HT2A and 5-HT2B receptor-mediated activation of PLC/[3H]-IP formation was assessed in HEK cells expressing 5-HT2A or 5-HT2B receptors. Cells were incubated with 10-10 to 10-5 M serotonin alone, or, with 0.1, 1.0 and 10 μM (−)-trans-PAT. Fig. 5 shows that serotonin stimulation of 5-HT2A and 5-HT2B receptor-mediated activation of PLC/[3H]-IP formation is competitively antagonized by (−)-trans-PAT, as indicated by a serotonin EC50 value that increases as concentration of (−)-trans-PAT increases (shift to the right in concentration–response curve). In contrast, the serotonin Emax values (625.7 ± 13.80% and 549.9 ± 15.36% for 5-HT2A and 5-HT2B receptors, respectively) are not affected significantly (P>0.05) by 0.1, 1.0 or 10 μM of (−)-trans-PAT.
Fig. 5.
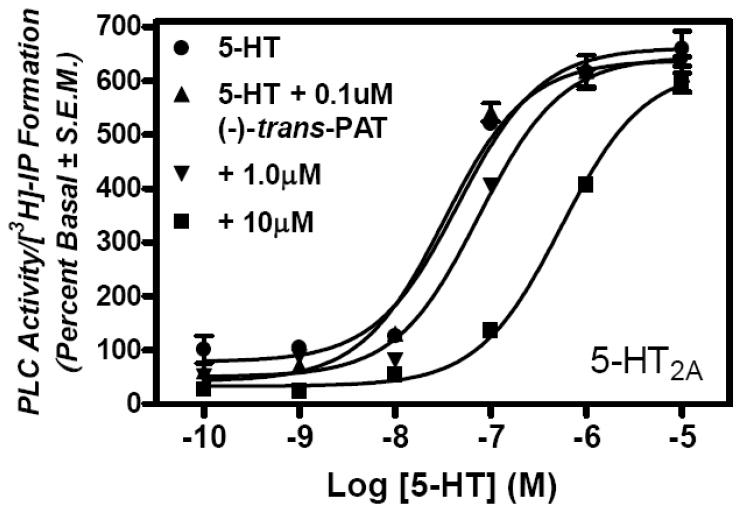
Representative graphed data for (−)-trans-PAT competitive antagonism of serotonin activation of 5-HT2A (A) and 5-HT2B (B) receptors.
At 5-HT2A receptors (Fig. 5A), the serotonin EC50 is 30.0 ± 7.88 nM in absence of (−)-trans-PAT, but, serotonin EC50 significantly increases to 77.8 ± 0.20 nM (P=0.0182) and 570 ± 70 nM (P=0.0020) in the presence of 1.0 and 10 μM of (−)-trans-PAT, respectively; 0.1 μM (−)-trans-PAT produced no significant change in the serotonin EC50 value (37.4 ± 7.15 nM; P=0.5677), as could be expected given its relatively low 5-HT2A receptor affinity (Ki ~ 0.4 μM, Table). The data in Fig. 5A yield an apparent KB = 0.46 ± 0.11 μM (pA2 = 6.3) for (−)-trans-PAT competitive antagonism of serotonin activation of 5-HT2A receptors—the (−)-trans-PAT KB and Ki values at 5- HT2A receptors are in close agreement.
At 5-HT2B receptors (Fig. 5B), the serotonin EC50 is 20.6 ± 1.65 nM in absence of (−)-trans-PAT, but, the serotonin EC50 significantly increases to 36.9 ± 2.15 nM (P=0.0172) and 110 ± 5.5nM (P=0.0001) in the presence of 1.0 and 10 μM of (−)-trans-PAT, respectively; 0.1 μM (−)-trans-PAT produced no significant change in the serotonin EC50 value (21.9 ± 2.75 nM; P=0.6973), as could be expected given its very low 5-HT2B receptor affinity (Ki ~ 1.2 μM, Table). The data in Fig. 5B yield an apparent KB = 1.4 ± 0.8 μM (pA2 = 5.8) for (−)-trans-PAT competitive antagonism of serotonin activation of 5-HT2B receptors—the (−)-trans-PAT KB and Ki values for 5-HT2B receptors are in close agreement.
3.5. Assessment of (−)-trans-PAT inverse agonist activity at serotonin 5-HT2A and 5-HT2B receptors
The functional data in Figs. 4 and 5 indicate that at higher concentrations (i.e., 1.0 and 10 μM), (−)-trans-PAT inhibits 5-HT2A and 5-HT2B receptor constitutive (basal) activity, suggesting, inverse agonism at these receptors. To assess (−)-trans-PAT inverse agonist activity, HEK cells expressing 5-HT2A or 5-HT2B receptors were incubated with 0.01 – 10 μM of (−)-trans-PAT and activity of PLC/[3H]-IP formation was measured. Results shown in Fig. 6 indicate (−)-trans-PAT is an inverse agonist at 5-HT2A and 5-HT2B receptors, with IC50 values of 490 ± 96 nM (nH= 0.85 ± 0.01) and 1000 ± 0.5 nM (nH = 0.87 ± 0.23), respectively. The inverse agonist potency values (IC50) for (−)-trans-PAT at 5-HT2A and 5-HT2B receptors are consistent with its affinity potency (Ki ~ 0.4 and 1.2 μM, respectively, Table) and competitive antagonist potency (KB ~ 0.5 and 1.4, respectively, Fig. 5) at these receptors. (−)-Trans-PAT solubility limited the maximal test concentration to 10 μM, at which, (maximal) inhibition of constitutive signaling is 57.6 ± 5.66 and 35.3 ± 2.00 % at 5-HT2A and 5-HT2B receptors, respectively.
Fig. 6.
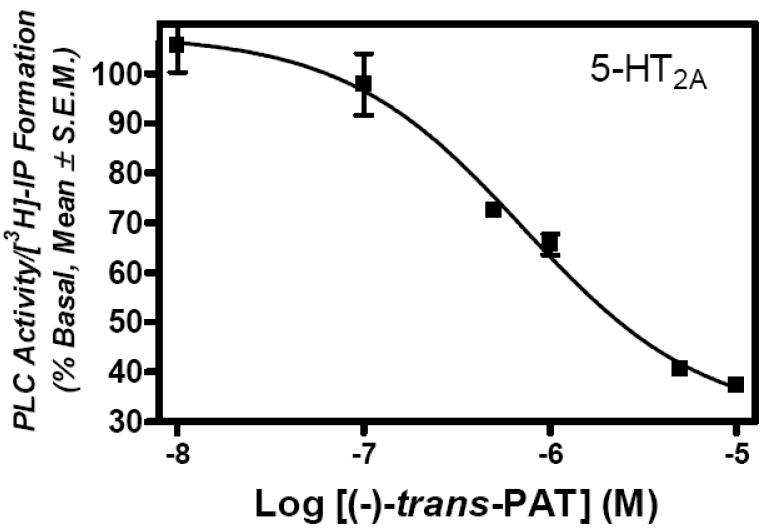
Representative graphed data for (−)-trans-PAT inverse agonist activity at serotonin 5-HT2A (A)and 5-HT2B (B) receptors.
3.6. (−)-Trans-PAT molecular interaction with the 5-HT2A receptor (Fig 7A)
Fig. 7.
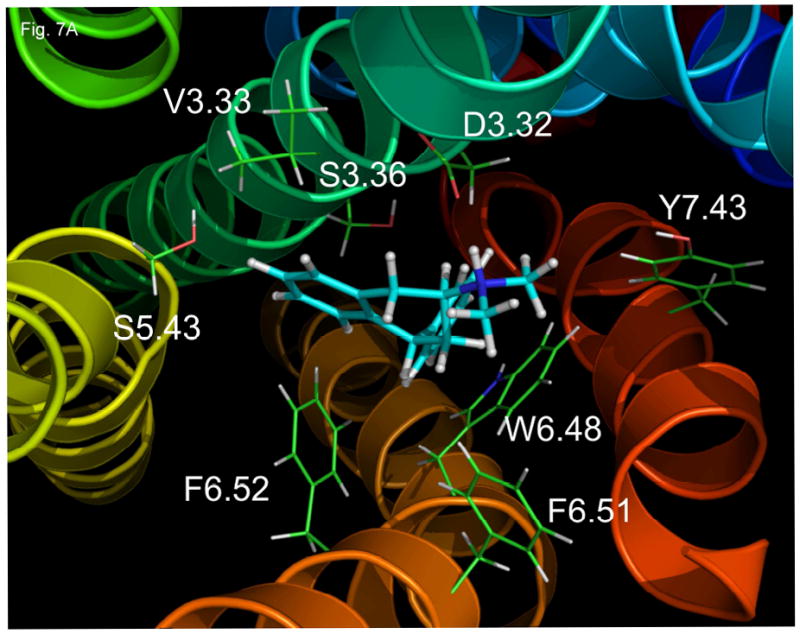


Molecular models of the 5-HT2A (A), 5-HT2B (B), and 5-HT2C (C) receptors with (−)-trans-PAT docked at the putative binding pocket.
When the energy minimized structure of (−)-trans-PAT is docked to the 5-HT2A molecular model, the PAT dimethylamine group (pKa ~ 9), protonated at physiological pH, can form an ionic bond with the 5-HT2A residue D3.32 (distance between PAT proton and aspartate carboxylic acid side chain is 1.7 Å)–amino acid residues are numbered according to Ballesteros et al. (2001). The aromatic portion of the PAT tetrahydronaphthalene moiety is in close proximity to the 5-HT2A residues V.3.33 (2.3 Å), S3.36 (3.2 Å) S5.43 (3.2 Å), and F6.52 (2.3 Å). The aliphatic portion of the PAT tetrahydronaphthalene and one of the methyl amine groups interact with 5-HT2A F6.51 (distance is 2.3 and 3.4 Å, respectively) and the other PAT methyl group orients towards Y7.43 (4.4 Å). The PAT pendant (C1) phenyl moiety interacts with 5-HT2A W6.48 (distance between centroids of PAT phenyl and tryptophan indole is 3.8 Å).
3.7. (−)-Trans-PAT molecular interaction with the 5-HT2B receptor (Fig. 7B)
There are significant differences in binding mode of (−)-trans-PAT to the 5-HT2B receptor in comparison to 5-HT2A (Fig. 7A) and 5-HT2C (Fig. 7C, below). Most importantly, there is no interaction between the PAT protonated dimethylamine moiety and the 5-HT2B residue D3.32 (the PAT amine proton orients away from D3.32 at a distance of 5.5 Å). Instead, the PAT dimethylamine group is much closer to residue F6.51 (2.3 Å). The two aromatic moieties of (−)-trans-PAT sandwich residue V3.33 midway between them (2.4 Å). The PAT tetrahydronaphthalene phenyl is close (2.7 Å) to 5-HT2B residue S3.36, as is the case for binding at 5-HT2A, however, no interaction of (−)-trans-PAT with 5-HT2B residue S5.43 is observed (distance between closest atoms is 6.9 Å). The PAT tetrahydronaphthalene phenyl interacts with the 5-HT2B W6.48 residue, likely, via п–п electron T-stacking interaction (distance 2.9 Å)—note that at the 5-HT2A receptor the PAT pendant (C1) phenyl interacts with W6.48. At 5-HT2B, the PAT pendant (C1) phenyl interacts with F6.52 (4.4 Å)—recall that at 5-HT2A (above), it is the PAT tetrahydronaphyl phenyl that interacts with F6.52. Meanwhile, there is the same interaction between the methyl group of (−)-trans-PAT and 5-HT2B Y7.43 (4.9 Å) that is apparent for the 5-HT2A receptor.
3.8 (−)-Trans-PAT interaction with the 5-HT2C receptor (Fig. 7C)
The PAT basic nitrogen of the dimethylamine moiety apparently forms an ionic bond with 5-HT2C residue D3.32 (distance 1.7 Å), similar to the result for 5-HT2A. At the 5-HT2C receptor, the PAT methyl groups orient differently than at 5-HT2A, interacting with 5-HT2C residues V.3.33 (distance 2.5 Å) and F6.51 (2.2 Å). The PAT tetrahydronaphthyl system is situated between the indole ring of 5-HT2C residue W6.48 (2.7 Å) and S3.36 (3.5 Å), which, is quite different than its orientation at 5-HT2A. In addition, S5.43 of the 5-HT2C receptor is further away (4.9 Å) from the PAT tetrahydronaphthalene moiety as compared to (−)-trans-PAT binding at the 5-HT2A receptor (3.2 Å). Meanwhile, There is no interaction between (−)-trans-PAT and 5-HT2C residue F6.52 (the distance between closest atoms is 6.2 Å), whereas, the PAT tetrahydronaphthyl system interacts relatively closely (2.3 Å) with this residue at the 5-HT2A receptor. Another important difference in (−)-trans-PAT at the 5-HT2C vs. 5-HT2A receptors is with regard to the pendant phenyl (C1) moiety—it is in close proximity to 5-HT2C residues S3.36 (2.5 Å) and Y7.43 (3.3 Å), but, it interacts mainly with W6.48 at the 5-HT2A receptor.
4. Discussion
The results presented establish (−)-trans-PAT as a stereoselective agonist at human 5-HT2C receptors with full-efficacy relative to the endogenous agonist serotonin. The selectivity of (−)-trans-PAT for activation of 5-HT2C versus 5-HT2A and 5-HT2B receptors is unequivocal in light of its inverse agonist activity and competitive antagonism of serotonin at 5-HT2A and 5-HT2B receptors. The results for docking of (−)-trans-PAT to molecular models of the 5-HT2 receptor subtypes help to rationalize (−)-trans-PAT selectivity for receptor binding, albeit, only clues can be gleaned to explain its unique 5-HT2-type multi-functional activity.
Regarding molecular determinants for 5-HT2 receptor binding, an obvious correlation is that (−)-trans-PAT has very low affinity (Ki ~ 1.2 μM) at 5-HT2B receptors and ligand docking results indicate (−)-trans-PAT does not interact with the 5-HT2B D3.32 residue. The corresponding D3.32 is conserved among aminergic GPCRs and mutagenesis studies indicate D3.32 interacts with positively charged amines of endogenous agonists and other ligands, including, for all three 5HT2 receptor subtypes (Muntasir et al., 2000a,b; 2007; Wang et al., 2003). In fact, for the human histamine H1 GPCR, phylogenetically closely related to the 5-HT2 family, D3.32 is required for [3H]-(−)-trans-PAT binding (Bakker et al., 2004). At 5-HT2A and 5-HT2C receptors, differences in the binding mode of (−)-trans-PAT are subtle, and, correspondingly, the difference in (−)-trans-PAT affinity at these receptors is modest (10-times). The major difference in (−)-trans-PAT binding at 5-HT2A and 5-HT2C receptors involves the S5.43 and F6.52 residues—(−)-trans-PAT interacts with these residues at the 5-HT2A but not at the 5-HT2C receptor. Regarding the change in (−)-trans-PAT functional activity from inverse agonism at 5-HT2A/5-HT2B receptors to agonism at 5-HT2C receptors, the modeling results are not very informative. Currently, there is not 5-HT2 G protein-coupled receptor structure–function information available to link PAT–5HT2 differential binding to stabilization of receptor conformations that lead to activation (5-HT2C) versus inactivation (5-HT2A and 5-HT2B), however, we have recently initiated mutagenesis studies to this end.
In addition to characterization of 5-HT2 G protein-coupled receptor structure and function, the unique 5-HT2 multi-functional activity of (−)-trans-PAT is promising with regard to development of novel 5-HT2 receptor-based pharmacotherapy. For example, activation of brain 5-HT2C receptors is mainly responsible for the pharmacotherapeutic effects of the anti-obesity drug S-(+)-fenfluramine (Tecott et al., 1995; Vickers et al., 2001). As mentioned, however, fenfluramine also indirectly leads to activation of 5-HT2B receptors that can produce heart valve damage (Fitzgerald et al., 2000; Setola et al., 2005) and/or pulmonary hypertension (Launay et al., 2002) and resulting fatality. In light of the unfortunate but informative experience with fenfluramine, evidence for lack of 5-HT2B receptor activation should be unequivocal regarding development of a weight-loss drug that is intended to act via 5-HT2C receptors. In this regard, (−)-trans-PAT shows no evidence of 5-HT2B receptor activation, and, in fact, it is a 5-HT2B inverse agonist as well as antagonist.
Although toxicity associated with 5-HT2B receptor activation has (deservedly) received much attention with regard to development of 5-HT2 receptor-based obesity pharmacotherapy, indiscriminant 5-HT2A receptor activation also is problematic. For example, 5-HT2A receptor activation is associated with psychomimetic (hallucinogenic) effects (Nichols, 2004) that obviously should be avoided, especially, given the potentially large patient population that may use a weight-loss drug, and, the sometimes subtle and complex nature of psychiatric disturbances. In this regard, the unequivocal 5-HT2A inverse agonist/antagonist activity demonstrated by (−)-trans-PAT suggests psychiatric side-effects linked to 5-HT2A receptor activation would not be an issue for this compound in a clinical setting.
In contrast, the only 5-HT2C agonist to undergo clinical trials, lorcaserin (APD356), also activates 5-HT2A receptors (75% efficacy) and 5-HT2B receptors (100% efficacy) (Jensen, 2006; Smith et al., 2006; Thomsen et al., 2008). Affinity of lorcaserin for 5-HT2C receptors (Ki ~ 15 nM) is only about 7.5-times higher than its affinity at 5-HT2A receptors (Ki ~ 112 nM), thus, activation of both receptors is likely in vivo, with possible 5-HT2A receptor-mediated psychiatric effects. In a 12-week clinical trial, about one-third of patients taking lorcaserin showed approximately 5% weight loss, however, except for dizziness, incidence of other central nervous system or psychiatric side-effects (e.g., 5-HT2A-mediated) was not specified. Affinity of lorcaserin for human 5-HT2C over 5-HT2B receptors (Ki ~ 174 nM) also is modest (about 12-times), however, echocardiograms performed at baseline and at the end of the trial suggested no drug effect on heart valves or pulmonary artery pressure. Nevertheless, the 12-week time-frame of the lorcaserin clinical trial is far short of the 5-month time period used to demonstrate in vivo valvular heart disease by serotonin in rats (Droogmans et al., 2007).
The 5-HT2C functional potency of lorcaserin (EC50 ~ 10 nM) (Thomsen et al., 2008) and (−)-trans-PAT (EC50 ~ 20 nM) is similar regarding activation of PLC/IP formation in cells transiently expressing the cloned human 5-HT2C receptor, and, both ligands are full efficacy 5-HT2C agonists compared to serotonin. Preclinical in vivo studies with lorcaserin and (−)-trans-PAT are not directly comparable because they used different paradigms and species—oral single-dose administration of lorcaserin to rats reduced food intake maximally by about 60% at 24 mg/kg, with ED50 = 18 mg/kg (Thomsen et al., 2008), and, intraperitoneal single-dose administration of (−)-trans-PAT to mice reduced food intake maximally by about 75% at 10 mg/kg, with ED50 = 4.2 mg/kg (Rowland et al., 2008). For both cases, the in vivo results unequivocally demonstrate human 5-HT2C receptor-mediated activation of PLC/IP signaling in clonal cells can predict anti-obesity activity, and, development of 5-HT2C agonists can lead to efficacious anti-obesity drugs—safety of such drugs will be a paramount concern, and, in this regard, the 5-HT2A/2B inverse agonist/antagonist activity of (−)-trans-PAT suggests it would not carry the potential psychiatric and cardiopulmonary toxicity liability that could result from activation of these receptors in vivo.
In addition to their role in treatment of obesity, 5-HT2C agonists also show efficacy in rodent models of schizophrenia (Siuciak et al., 2007). In this regard, the multi-functional activity of (−)-trans-PAT at multiple 5-HT2 receptor subtypes (5-HT2C agonism and 5-HT2A/2B inverse agonism/antagonism) is in line with new dogma that proposes drugs with a multi-faceted pharmacological profile may be more effective than single-target drugs in treatment of neuropsychiatric disorders–the qualified effectiveness of atypical antipsychotic drugs, such as, olanzapine supports this new treatment paradigm. Atypical antipsychotics generally have clinically-relevant affinity at several neurotransmitter receptor types, including, adrenergic (α1 and α2), dopamine (D1, D3 and D4), histamine (H1), muscarinic, and serotonin (5-HT1A, 5-HT2A, 5-HT2C, 5-HT6 and 5-HT7), and, their functional activity includes partial-agonism, as well as, inverse agonism and antagonism at these receptors. While 5-HT2A receptor inverse agonism/antagonism is thought to contribute to the efficacy of atypical antipsychotic drugs, most, such as olanzapine, also are about equi-potent at blocking 5-HT2C receptors, which could contribute to their propensity to cause weight gain. Antagonist activity at the histamine H1 G protein-coupled receptor also correlates with atypical antipsychotic-induced weight gain (Kroeze et al., 2003). In fact, histamine H1 receptors and the serotonin 5-HT2 receptor family are phylogenetically closely related and propensity of atypical antipsychotics to cause weight gain is most highly correlated with affinity for both H1 and 5-HT2C receptors, as well as, adrenergic α1 receptors (Kroeze et al., 2003).
Receptor profiling studies indicate (−)-trans-PAT has very low (Ki> 1 μM) or virtually no (Ki > 5 μM) affinity for 35 tested CNS receptor systems, including neurotransmitter (adrenergic; cholinergic; GABAA; histamine H2, H3, H4; serotonin 5-HT1A, 5-HT1B, 5-HT5A, 5-HT6, 5-HT7), neuromodulator (adenosine, benzodiazepine, opiate, NMDA/PCP, sigma), neurotransporter (dopamine, norepinephrine, serotonin), ion channel (Ca++, Cl−, K+), and second messenger (adenylyl cyclase, PLC) (PDSP, 2005). In contrast, (−)-trans-PAT has high affinity (Ki ~ 2 nM) at human H1 receptors, where it is an agonist that activates H1-linked adenylyl cyclase signaling and catecholamine (dopamine, norepinephrine) synthesis in mammalian brain and adrenal tissues (Moniri et al., 2004; Moniri and Booth, 2006). (−)-Trans-PAT activation of brain catecholamine synthesis via H1 receptor activation may result in effects similar to norepinephrine—when injected into rat brain, norepinephrine inhibits feeding, putatively, by activation of hypothalmus α1 receptors (Wellman and Davies, 1991). Selective brain-penetrating norepinephrine uptake inhibitors that inhibit feeding behavior in rats have been considered as potential drugs for obesity (Gehlert et al., 1998).
Regarding cocaine addiction, there is compelling evidence that 5-HT2C receptor agonism can attenuate neurobehavioral effects of cocaine, as can 5-HT2A receptor antagonists (Bubar and Cunningham, 2008). For example, the reinforcing effects of cocaine are reduced by 5-HT2C agonists, whereas, discriminative stimulus and reinstating effects are sensitive to attenuation by both 5-HT2C agonists and 5-HT2A antagonists (Bubar and Cunningham, 2008). Both 5-HT2C agonism and 5-HT2A antagonism/inverse agonism are demonstrated by (−)-trans-PAT, suggesting, (−)-trans-PAT may be a lead for psychostimulant addiction pharmacotherapy.
An intriguing possibility for (−)-trans-PAT-type multi-functional drugs involves new treatment paradigms based on classification of over-eating and drug addiction as compulsive behavioral disorders directed toward different objects, food and drugs (Simansky, 2005). Although clinical efficacy of (−)-trans-PAT 5-HT2 in obesity and/or neuropsychiatric disorders is highly speculative at this point, recent studies using rodents give cause for optimism. In addition to the in vivo anorexic and weight-loss effects of (−)-trans-PAT in mice (Rowland et al., 2008; Bivens et al, 2007), preliminary results indicate (−)-trans-PAT also reduces amphetamine-induced locomotion in mice after intraperitoneal administration (Bivens et al., 2007), indicative of antipsychotic neurobehavioral activity. Taken together, the in vitro and in vivo activity of (−)-trans-PAT suggests it may show activity as an antipsychotic drug without propensity for weight gain.
Acknowledgments
This research was supported by United States Public Health Service (NIH) Grants MH068655, MH081193, DA023928, and the University of Florida Opportunity Fund. Receptor binding profile data for (−)-trans-PAT was generously provided by the National Institute of Mental Health’s Psychoactive Drug Screening Program, Contract # NO1MH32004 (NIMH PDSP). The NIMH PDSP is Directed by Bryan L. Roth MD, PhD at the University of North Carolina at Chapel Hill and Project Officer Jamie Driscol at NIMH, Bethesda MD, USA.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bakker RA, Dees G, Carrillo JJ, Booth RG, Lopez-Gimenez JF, Milligan G, Strange PG, Leurs R. Domain swapping in the human histamine H1 receptor. J Pharmacol Exp Ther. 2004;311:131–138. doi: 10.1124/jpet.104.067041. [DOI] [PubMed] [Google Scholar]
- Ballesteros JA, Shi L, Javitch JA. Structural mimicry in G protein-coupled receptors: implications of the high-resolution structure of rhodopsin for structure-function analysis of rhodopsin-like receptors. Mol Pharmacol. 2001;60:1–19. [PubMed] [Google Scholar]
- Berendsen HJC, Postma JPM, van Gunsteren WF, Hermans J. Intermolecular Forces. D. Reidel Publishing Company; Dordrecht, the Netherlands: 1981. Interaction models for water in relation to protein hydration; pp. 331–342. [Google Scholar]
- Berger O, Edholm O, Jähnig F. Molecular dynamics simulations of a fluid bilayer of dipalmitoylphosphatidylcholine at full hydration, constant pressure, and constant temperature. Biophys J. 1997;72:2002–2013. doi: 10.1016/S0006-3495(97)78845-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bivens NM, Weisstaub NV, Bradley-Moore M, Kevin K, Booth RG, Gingrich JA. 5HT2A and 5HT2C receptors in mouse models of psychosis and obesity Impact of a novel compound. American College of Neuropsychopharmacology Abstracts Annual Meeting; Boca Raton, Florida. December 9-13.2007. [Google Scholar]
- Bubar MJ, Cunningham KA. Prospects for serotonin 5-HT2R receptor pharmacology in psychostimulant abuse. Prog Brain Res. 2008;172:319–346. doi: 10.1016/S0079-6123(08)00916-3. [DOI] [PubMed] [Google Scholar]
- Droogmans S, Franken PR, Garbar C, Weytjens C, Cosyns B, Lahoutte T, Caveliers V, Pipeleers-Marichal M, Bossuyt A, Schoors D, van Camp G. In vivo model of drug-induced valvular heart disease in rats: pergolide-induced valvular heart disease demonstrated with echocardiography and correlation with pathology. Eur Heart J. 2007;28:2156–2162. doi: 10.1093/eurheartj/ehm263. [DOI] [PubMed] [Google Scholar]
- Fitzgerald LW, Burn TC, Brown BS, Patterson JP, Corjay MH, Valentine PA, Sun JH, Link JR, Abbaszade I, Hollis JM, Largent BL, Hartig PR, Hollis GF, Meunier PC, Robichaud AJ, Robertson DW. Possible role of valvular serotonin 5-HT(2B) receptors in the cardiopathy associated with fenfluramine. Mol Pharmacol. 2000;57:75–81. [PubMed] [Google Scholar]
- Fletcher PJ, Grottick AJ, Higgins GA. Differential effects of the 5-HT(2A) receptor antagonist M100907 and the 5-HT(2C) receptor antagonist SB242084 on cocaine-induced locomotor activity, cocaine self-administration and cocaine-induced reinstatement of responding. Neuropsychopharmacology. 2002;27:576–586. doi: 10.1016/S0893-133X(02)00342-1. [DOI] [PubMed] [Google Scholar]
- Fox SH, Brotchie JM. 5-HT(2C) receptor antagonists enhance the behavioural response to dopamine D(1) receptor agonists in the 6-hydroxydopamine-lesioned rat. Eur J Pharmacol. 2000a;398:59–64. doi: 10.1016/s0014-2999(00)00238-7. [DOI] [PubMed] [Google Scholar]
- Fox SH, Brotchie JM. 5-HT2C receptor binding is increased in the substantia nigra pars reticulata in Parkinson’s disease. Mov Disord. 2000b;15:1064–1069. doi: 10.1002/1531-8257(200011)15:6<1064::aid-mds1002>3.0.co;2-c. [DOI] [PubMed] [Google Scholar]
- Gehlert DR, Dreshfield L, Tinsley F, Benvenga MJ, Gleason S, Fuller RW, Wong DT, Hemrick-Luecke SK. The selective norepinephrine reuptake inhibitor, LY368975, reduces food consumption in animal models of feeding. J Pharmacol Exp Ther. 1998;287:122–127. [PubMed] [Google Scholar]
- Heisler LK, Tecott LH. A paradoxical locomotor response in serotonin 5-HT(2C) receptor mutant mice. J Neurosci. 2000;20:RC71. doi: 10.1523/JNEUROSCI.20-08-j0003.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heisler LK, Zhou L, Bajwa P, Hsu J, Tecott LH. Serotonin 5-HT(2C) receptors regulate anxiety-like behavior. Genes Brain Behav. 2007;6:491–496. doi: 10.1111/j.1601-183X.2007.00316.x. [DOI] [PubMed] [Google Scholar]
- Hess B, Bekker H, Fraaije JGEM. LINCS: A Linear Constraint Solver for Molecular Simulations. J Comput Chem. 1997;18:1463–1472. [Google Scholar]
- Jensen MD. Potential role of new therapies in modifying cardiovascular risk in overweight patients with metabolic risk factors. Obesity. 2006;14:143S–149S. doi: 10.1038/oby.2006.294. [DOI] [PubMed] [Google Scholar]
- Jorgensen WL, Maxwell DS, Tirado-Rives J. Development and Testing of the OPLS All-Atom Force Field on Conformational Energetics and Properties of Organic Liquids. J Am Chem Soc. 1996;118:11225–11236. [Google Scholar]
- Knight AR, Misra A, Quirk K, Benwell K, Revell D, Kennett G, Bickerdike M. Pharmacological characterisation of the agonist radioligand binding site of 5-HT(2A), 5-HT(2B) and 5-HT(2C) receptors. Naunyn Schmiedeberg’s Arch Pharmacol. 2004;370:114–123. doi: 10.1007/s00210-004-0951-4. [DOI] [PubMed] [Google Scholar]
- Kroeze WK, Hufeisen SJ, Popadak BA, Renock SM, Steinberg S, Ernsberger P, Jayathilake K, Meltzer HY, Roth BL. H1-histamine receptor affinity predicts short-term weight gain for typical and atypical antipsychotic drugs. Neuropsychopharmacology. 2003;28:519–526. doi: 10.1038/sj.npp.1300027. [DOI] [PubMed] [Google Scholar]
- Launay JM, Herve P, Peoc’h K, Tournois C, Callebert J, Nebigil CG, Etienne N, Drouet L, Humbert M, Simonneau G, Maroteaux L. Function of the serotonin 5-hydroxytryptamine 2B receptor in pulmonary hypertension. Nat Med. 2002;8:1129–1135. doi: 10.1038/nm764. [DOI] [PubMed] [Google Scholar]
- Lindahl E, Hess B, van der Spoel D. GROMACS 3.0: a package for molecular simulation and trajectory analysis. J Mol Model. 2001;7:306–317. [Google Scholar]
- Maffuid P, Smith B, Thomsen B, Behan D, Prosser W, Anderson C, Shanahan W. 5-HT2C Agonists in Obesity Drug Discovery and Development: Success and Challenges. San Diego Bo-Pharma Conference. 2006 Abstract and presentation available at http://www.sabpa.org/web/bio-conf-program.php.
- Moniri NH, Covington-Strachan D, Booth RG. Ligand-directed functional heterogeneity of histamine H1 receptors: novel dual-function ligands selectively activate and block H1-mediated phospholipase C and adenylyl cyclase signaling. J Pharmacol Exp Ther. 2004;311:274–281. doi: 10.1124/jpet.104.070086. [DOI] [PubMed] [Google Scholar]
- Moniri NH, Booth RG. Role of PKA and PKC in histamine H1 receptor-mediated activation of catecholamine neurotransmitter synthesis. Neurosci Lett. 2006;407:249–253. doi: 10.1016/j.neulet.2006.08.051. [DOI] [PubMed] [Google Scholar]
- Muntasir HA, Takahashi J, Rashid M, Ahmed M, Komiyama T, Hossain M, Kawakami J, Nashimoto M, Nagatomo T. Site-directed mutagenesis of the serotonin 5-Hydroxytryptamine2c receptor: identification of amino acids responsible for sarpogrelate binding. Biol Pharm Bull. 2006;29:1645–1650. doi: 10.1248/bpb.29.1645. [DOI] [PubMed] [Google Scholar]
- Muntasir HA, Rashid M, Komiyama T, Kawakami J, Nagatomo T. Identification of amino acid residues important for sarpogrelate binding to the human 5-hydroxytryptamine2A serotonin receptor. J Pharmacol Sci. 2006;102:55–63. doi: 10.1254/jphs.fp0060171. [DOI] [PubMed] [Google Scholar]
- Muntasir HA, Hossain M, Bhuiyan MA, Komiyama T, Nakamura T, Ozaki M, Nagatomo T. Identification of a key amino acid of the human 5-HT(2B) serotonin receptor important for sarpogrelate binding. J Pharmacol Sci. 2007;104:274–277. doi: 10.1254/jphs.sc0060241. [DOI] [PubMed] [Google Scholar]
- Nichols DE. Hallucinogens. Pharmacol Ther. 2004;101:131–181. doi: 10.1016/j.pharmthera.2003.11.002. [DOI] [PubMed] [Google Scholar]
- Okada T, Fujiyoshi Y, Silow M, Navarro J, Landau EM, Shichida Y. Functional role of internal water molecules in rhodopsin revealed by X-ray crystallography. Proc Natl Acad Sci USA. 2002;99:5982–5987. doi: 10.1073/pnas.082666399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PDSP. Psychoactive Drug Screening Program, BL Roth, Director. NIMH Contract NO2MH80002, University of North Carolina, Chapel Hill, NC Novascreen (1996) NIMH Psychotherapeutic Drug Discovery and Development Program, Oceanix Biosciences Corporation 2005 [Google Scholar]
- Porter RH, Benwell KR, Lamb H, Malcolm CS, Allen NH, Revell DF, Adams DR, Sheardown MJ. Functional characterization of agonists at recombinant human 5-HT2A, 5-HT2B and 5-HT2C receptors in CHO-K1 cells. Br J Pharmacol. 1999;128:13–20. doi: 10.1038/sj.bjp.0702751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raymond JR, Mukhin YV, Gelasco A, Turner J, Collinsworth G, Gettys TW, Grewal JS, Garnovskaya MN. Multiplicity of mechanisms of serotonin receptor signal transduction. Pharmacol Ther. 2001;92:179–212. doi: 10.1016/s0163-7258(01)00169-3. [DOI] [PubMed] [Google Scholar]
- Rosenzweig-Lipson S, Sabb A, Stack G, Mitchell P, Lucki I, Malberg JE, Grauer S, Brennan J, Cryan JF, Sukoff Rizzo SJ, Dunlop J, Barrett JE, Marquis KL. Antidepressant-like effects of the novel, selective, 5-HT(2C) receptor agonist WAY-163909 in rodents. Psychopharmacology (Berl) 2007;192:159–170. doi: 10.1007/s00213-007-0710-6. [DOI] [PubMed] [Google Scholar]
- Rosenzweig-Lipson S, Zhang J, Mazandarani H, Harrison BL, Sabb A, Sabalski J, Stack G, Welmaker G, Barrett JE, Dunlop J. Antiobesity-like effects of the 5-HT2C receptor agonist WAY-161503. Brain Res. 2006:1073–1074. 240–251. doi: 10.1016/j.brainres.2005.12.052. [DOI] [PubMed] [Google Scholar]
- Rothman RB, Ayestas MA, Dersch CM, Baumann MH. Aminorex, fenfluramine and chlorphentermine are serotonin transporter substrates: implications for primary pulmonary hypertension. Circulation. 1999;100:869–875. doi: 10.1161/01.cir.100.8.869. [DOI] [PubMed] [Google Scholar]
- Rowland NE, Crump EM, Nguyen N, Robertson K, Sun Z, Booth RG. Effect of (-)-trans-PAT, a novel 5-HT2C receptor agonist, on intake of palatable food in mice. Pharmacol Biochem Behav. 2008;91:176–180. doi: 10.1016/j.pbb.2008.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saltzman AG, Morse B, Whitman MM, Ivanshchenko Y, Jaye M, Felderm S. Cloning of the human serotonin 5-HT2 and 5-HT1C receptor subtypes. Biochem Biophys Res Commun. 1991;181:1469–7148. doi: 10.1016/0006-291x(91)92105-s. [DOI] [PubMed] [Google Scholar]
- Setola V, Dukat M, Glennon RA, Roth BL. Molecular determinants for the interaction of the valvulopathic anorexigen norfenfluramine with the 5-HT2B receptor. Mol Pharmacol. 2005;68:20–33. doi: 10.1124/mol.104.009266. [DOI] [PubMed] [Google Scholar]
- Simansky KJ. NIH symposium series: ingestive mechanisms in obesity, substance abuse and mental disorders. Physiol Behav. 2005;86:1–4. doi: 10.1016/j.physbeh.2005.06.016. [DOI] [PubMed] [Google Scholar]
- Siuciak JA, Chapin DS, McCarthy SA, Guanowsky V, Brown J, Chiang P, Marala R, Patterson T, Seymour PA, Swick A, Iredale PA. CP-809,101, a selective 5-HT2C agonist, shows activity in animal models of antipsychotic activity. Neuropharmacology. 2007;52:279–290. doi: 10.1016/j.neuropharm.2006.07.024. [DOI] [PubMed] [Google Scholar]
- Smith SR, Prosser W, Donahue D, Anderson C, Shanahan W. Lorcaserin Phase 2b Clinical Study. American Diabetes Association 2006 [Google Scholar]
- Tecott LH, Sun LM, Akana SF, Strack AM, Lowenstein DH, Dallman MF, Julius D. Eating disorder and epilepsy in mice lacking 5-HT2c serotonin receptors. Nature. 1995;374:542–546. doi: 10.1038/374542a0. [DOI] [PubMed] [Google Scholar]
- Thomsen WJ, Grottick AJ, Menzaghi F, Reyes-Saldana H, Espitia S, Yuskin D, Whelan K, Martin M, Morgan M, Chen W, Al-Shamma H, Smith B, Chalmers D, Behan D. Lorcaserin, a novel selective human 5-hydroxytryptamine2C agonist: in vitro and in vivo pharmacological characterization. J Pharmacol Exp Ther. 2008;325:577–587. doi: 10.1124/jpet.107.133348. [DOI] [PubMed] [Google Scholar]
- Vickers SP, Dourish CT, Kennett GA. Evidence that hypophagia induced by D-fenfluramine and D-norfenfluramine in the rat is mediated by 5-HT2C receptors. Neuropharmacology. 2001;41:200–209. doi: 10.1016/s0028-3908(01)00063-6. [DOI] [PubMed] [Google Scholar]
- Wang CD, Gallaher TK, Shih JC. Site-directed mutagenesis of the serotonin 5-hydroxtryptamine 2 receptor: identification of amino acids necessary for ligand binding and receptor activation. Mol Pharmacol. 1993;43:931–940. [PubMed] [Google Scholar]
- Wellman PJ, Davies BT. Reversal of phenylpropanolamine anoxexia in rats by the alpha-1 receptor antagonist benoxathian. Pharmacol Biochem Behav. 1991;38:905–908. doi: 10.1016/0091-3057(91)90261-y. [DOI] [PubMed] [Google Scholar]
- Wyrick SD, Booth RG, Myers AM, Owens CE, Kula NS, Baldessarini RJ, Mailman RB. Synthesis and pharmacological evaluation of 1-phenyl-3-amino-1,2,3,4-tetrahydronaphthalenes as ligands for a novel receptor with sigma-like neuromodulatory activity. J Med Chem. 1993;36:2542–2551. doi: 10.1021/jm00069a013. [DOI] [PubMed] [Google Scholar]
- York DM, Darden TA, Pedersen LG, Anderson MW. Molecular dynamics simulation of HIV-1 protease in a crystalline environment and in solution. Biochemistry. 1993;32:1443–1453. doi: 10.1021/bi00057a007. [DOI] [PubMed] [Google Scholar]