

Abstract
MicroRNAs (miRNAs) are short non-coding RNAs of cellular1 and viral origin2-7 that post-transcriptionally regulate gene expression through imperfect base pairing to their mRNA targets. Because the recognition sequences of miRNAs for their targets are short and may be discontinuous, bioinformatic prediction of targets is difficult. Here we present an approach to the experimental identification of the mRNA targets of miRNAs encoded by the Kaposi's sarcoma-associated herpesvirus (KSHV). KSHV encodes 17 miRNAs, derived from 12 pre-miRNAs expressed from a single locus during viral latency2,5-10. Our approach is based upon multiple screens that examine small changes in transcript abundance under different conditions of miRNA expression or inhibition, followed by searching the identified transcripts for seed sequence matches. This strategy led to the identification of the Bcl2-associated factor BCLAF1 as a target for miR-K5, and further analysis revealed that several other KSHV miRNAs also target this gene product. Our results support that this type of expression profiling provides a potentially general approach to the identification of miRNA targets.
To identify host RNA targets of KSHV miRNAs, we proceeded from the observation that RNAs targeted by miRNAs often display small reductions in their steady-state levels11-14. Accordingly, we performed mRNA expression profiling in B cells and endothelial cells, the main targets of KSHV, under four sets of conditions. First, we examined host mRNA profiles in BJAB B cells transfected with individual KSHV miRNAs vs control miRNAs. Second, we similarly profiled cells stably transduced with retroviruses expressing clusters of KSHV miRNAs. Third, we examined host mRNA expression in primary endothelial cells latently infected with KSHV; these cells express all KSHV miRNAs at their physiologic levels. In each of these cases, we looked for transcripts whose levels decreased upon viral miRNA expression. Fourth, we inhibited15,16 individual viral miRNAs in latently-infect BCBL-1 (B lymphoma) cells using antagomirs (Supplementary Fig. 5), and searched for transcripts whose levels rose in response to such inhibition (see Supplementary Materials). Transcripts whose levels were reduced in the presence of a KSHV miRNA and increased in the presence of the cognate antagomir represent candidate miRNA targets. We identified genes with this pattern across all experiments using t-test, fold-change, k-means clustering and rank sum analysis.
Typically, the array results revealed only small (< twofold) changes in mRNA levels, which is consistent with previous studies13,17-19. For each miRNA tested, we identified between 10-30 transcripts that passed all four expression filters. Here, we focus on the set of RNA target candidates identified for KSHV miR-K520. Fig. 1a shows the identities of the host RNAs that passed all the screens for targeting by this miR, along with their expression profiles. Next, we employed sequence analysis to look for genes with seed sequence matches for miR-K5 in their 3′ UTR. Though perfect complementarity may not always be found21-23, Grimson et al.17 observed that transcripts with complementary bases at positions 2-8 were more consistently and more strongly down regulated than RNAs with lesser degrees of seed homology. Accordingly, our initial analysis included this additional, very stringent filter for perfect seed complementarity on top of the four expression filters. Among the miR-K5-regulated transcripts, only one gene, that encoding the bcl-2 associated factor (BCLAF1; also called Btf, for bcl2-associated transcription factor), met all five criteria. BCLAF1 was initially identified24 in a yeast two-hybrid screen for factors binding to the adenovirus E1B-19K protein, an anti-apoptotic protein with sequence and functional homologies to the mammalian bcl-2 protein. That report suggested that overexpression of BCLAF1 induces apoptosis in HeLa cells.
Figure 1.
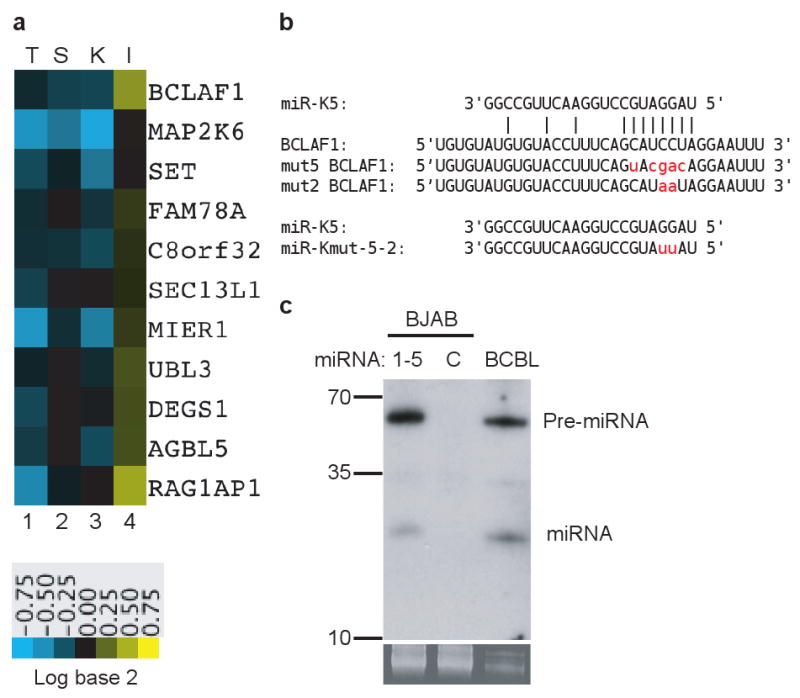
Candidate mRNA targets of KSHV miR-K5. a, Heatmap showing expression in the presence of transiently transfected miRNA mimics (T), stable cell lines transduced with retroviral vectors expressing miRNAs (S), HUVEC cells latently infected with KSHV (K) and BCBL-1 cells treat with specific miRNA inhibitor (I). Green denotes downregulation, red denotes upregulation; key below indicates magnitude of fold-changes in RNA abundance as a function of color intensity. b, miR-K5 sequence and seed matching site in BCLAF1 3′UTR. Mutant BCLAF1 3′UTR is shown as “mut5/2 BCLAF1”. Mutated bases in BCLAF1 3′UTR and miR-Kmut-5-2 are shown in lower case letters. c, Northern blot analysis of miR-K5 with RNA from stable BJAB cell line containing KSHV miR-K1, 2, 3, 4, 5; control line transduced with empty vector (C) or latently infected BCBL cells. Ethidium-stained low molecular weight RNA is shown as a loading control below blot.
To examine its targeting by KSHV miR-K5 in further detail, we first asked if the level of the endogenous BCLAF1 protein could be downregulated by miR-K5 expression, as judged by immunoblotting. Transient transfection of miR-K5 diminished BCLAF1 accumulation in 293 cells (Fig2a, left panel); the protein was similarly reduced in BJAB B cells stably transduced with a retrovirus coexpressing miRNAs K1, -2, -3, -4, and 5 (Supplementary Fig. 4a), compared with cells transduced with a retrovirus lacking the miRNA genes (center panel). Importantly, we also detected a strong decrease in BCLAF1 in HUVEC cells latently infected with authentic KSHV (right panel and Supplementary Fig. 1, 3, 4).
Figure 2.

BCLAF1 protein and reporter gene expression analysis. a, Western blot analysis of endogenous BCLAF1 and loading control, tubulin. 293 cells were transfected with negative control miRNA (C) or miR-K5 (5). BJAB stable lines transduced with empty vector or a cluster containing miR-K1, 2, 3, 4 and 5 (1-5). Primary HUVEC cells mock infected (-) or duplicate latent KSHV infections (+). b, 293 cells were transfected with firefly luciferase vector with various 3′ UTRs, internal TK-renilla control reporter and miRNAs miR-K5 (5) and mutant (m5-2). Reporters with BCLAF1 3′UTR (BC), a 5 or 2 base mutation in miR-K5 target site in the BCLAF1 3′UTR (mt5 or mt2 BC), and miR-K5 target site duplicated twice without remaining BCLAF 3′UTR (2XBC5) are shown. Mutant miR-Kmut-5-2 (m5-2) restores complementarity to the mutant BCLAF1 3′UTR (mt2 BC). Firefly:renilla ratios are normalized to same reporter transfected with the negative control miRNA (Ctl). Mean fold change values are from at least triplicate independent transfections. Error bars are 1 std. dev. P-values from t-tests less than 0.01 (**) and 0.001 (***) are shown. c, HUVEC cells were transfected with miR-K5 (5) or mutant miR-K5 (mut 5). MicroRNAs and reporters refer to sequences shown in Fig. 1b.
To determine the cis-acting sequence necessary for down-regulation, we cloned the BCLAF1 3′UTR25,26 downstream of a luciferase (LUC) reporter gene, and examined LUC expression following cotransfection of this construct with miR-K5 or control microRNA (Fig. 2b). While expression of miR-K5 had no effect on the control LUC vector lacking the BCLAF1 3′UTR (lane 7), we observed a modest but highly reproducible down-regulation of the luciferase reporter containing the BCLAF1 3′ UTR (lane 2). This region contains a perfect 8-mer target site for the miR-K5 seed25, and multimerization of sequences spanning this site strongly enhanced inhibition of luciferase expression by miR-K5 (Fig. 2b, lane 12). When the seed-complementary region in the 3′ UTR was mutated, downregulation by miR-K5 was abolished. (Fig. 2b, lanes 5 and 9). Cotransfection of the mutant 3′ UTR plasmid with a mutant of miR-K5 in which complementarity to the UTR was restored resulted in renewed repression of the reporter by the mutant of miR-K5 compared to wild type miR-K5 (lanes 9-10). Similarly, mutating two bases in the seed of miR-K5 itself also ablated its ability to inhibit (i) expression of the wild-type BCLAF1 UTR-containing luciferase reporter construct (Fig. 2b, lane 3), and (ii) the accumulation of endogenous BCLAF1 itself (Fig. 2c). These data unequivocally establish that the BCLAF1 3′UTR is directly targeted by miR-K5.
Viruses often encode redundant functions when inactivation of a key host target is critical – e.g. the deployment of multiple viral proteins to inactivate interferon induction or signaling27. For this reason, we wondered if any other KSHV miRNAs might also target BCLAF1. Consistent with this possibility, we noted that the downregulation of BCLAF1 in latently infected cells, which express all 17 viral miRNAs, is substantially stronger than that observed following expression of miR-K5 alone (Fig. 2a). Accordingly, we examined the levels of BCLAF1 protein in HUVEC cells transfected with each of the 17 KSHV miRs. Fig. 3a shows that, in addition to miR-K5, several other KSHV miRs down-regulated this protein reproducibly – notably, miRs K9, K10a and K10b. Each of these miRNAs was tested for the ability to downregulate the LUC-BCLAF1 3′UTR reporter construct (Fig. 3b). While miRs K9 and K10b repressed the chimera as expected, miR K10a did not, suggesting that either (i) its targets lie elsewhere in the BCLAF1 transcript, or (ii) it modulates BCLAF1 levels indirectly.
Figure 3.

BCLAF1 protein and reporter gene expression analysis with various KSHV miRNAs. a, Western blot analysis of endogenous BCLAF1 and loading control, actin, in HUVEC cells transfected with individual KSHV miRs, harvested 48 hr. after transfection. MiRNA “1” refers to miR-K1 and so on. “-” refers to cells treated with transfection reagent without any miRNA. “No txn” refers to untransfected cells. Note: the apparent downregulation of BCLAF1 by miR-K4-3 was not reproducible in replicate experiments (data not shown). b, Luciferase assays as in Fig 2b, except fold change values were calculated using the normalized ratio of firefly luciferase activity from the BCLAF1 UTR reporter to the activity of the control vector lacking the BCLAF1 UTR due to effects on the control vector by specific miRNAs. Mean fold change values are from at least triplicate independent transfections. Error bars are 1 std. dev. c, Western blot analysis of endogenous BCLAF1 and loading control, actin. BJAB stable lines transduced with empty vector (C) or a region containing miR-K9. d, HUVEC cells transfected with wild type or mutant (m) miRNAs. e, Western blot analysis of BCBL-1 cells were treated with the indicated miRNA inhibitors and harvested five days after treatment.
To further validate this downregulation, we examined the levels of BCLAF1 protein in cells stably expressing miR-K9 from an integrated retroviral vector (Supplementary Fig 3a, 4b). As shown in Fig. 3c., miR-K9 expression in this context also downregulated BCLAF1 protein accumulation. Moreover, transient transfection with mutant versions of miRs -K9, and –K10 bearing lesions in their seed sequences impaired the downregulation of BCLAF protein accumulation (Fig. 3d). Finally, cotransfection of latently infected BCBL-1 cells with antagomirs to miR-K5 or the combination of miRs-K5,9, 10a and 10b raised the level of endogenous BCLAF1 (Fig. 3e). Together, these results affirm that downregulation of BCLAF1 by these miRs is authentic and specific.
What is the function of BCLAF1 modulation by the viral miRNAs? The reported ability of BCLAF1 to promote apoptosis24 suggested to us that its downregulation by KSHV miRNAs might be associated with reduced susceptibility to apoptosis. To investigate this, we tested miR–K5 for the ability to impair etoposide-induced caspase activation, as measured by PARP cleavage28 As shown in Fig. 4a, miR–K5 inhibited etoposide-induced PARP-cleavage, while the seed-disrupting mutant of miR-K5 did not (Fig. 4b). Similarly, miRs– K-9, K-10a and K-10b, which also downregulate BCLAF1, also impair etoposide-induced caspase activation (Fig. 4c). Conversely, antagomir inhibition of these miRs in latently infected BCBL-1 cells increases PARP cleavage in the absence of etoposide (Fig. 5c).
Figure 4.
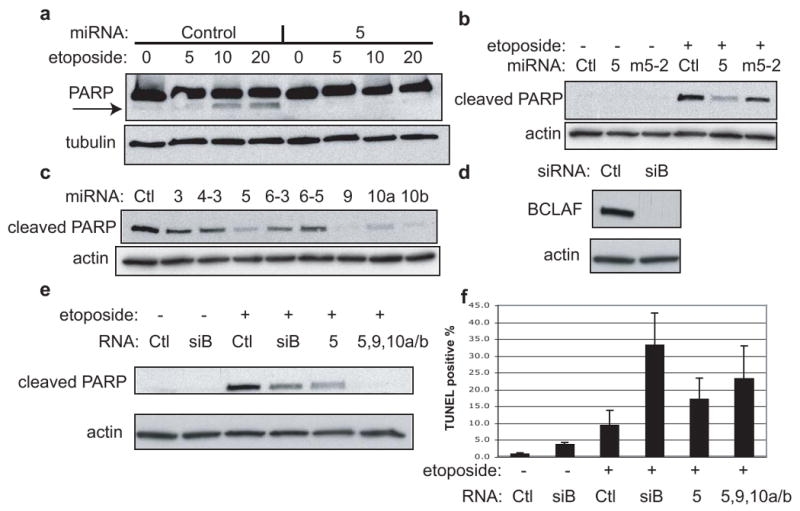
Apoptosis assays in HUVEC with various miRNAs. a, Cells were transfected with miRNAs (control or miR-K5) for 48 hr., followed by 24 hr. treatment with etoposide (μM conc.). Samples were probed with PARP antibody (recognizes full length and cleaved form) and the loading control antibody, tubulin. Arrow indicates cleaved PARP. b, Similar to (a) except cells were transfected with miRNAs (negative control “Ctl”, miR-K5 “5”, or mutant miR-Km5-2 “m5-2”) for 24 hr., followed by 24 hr. treatment with 50 μM etoposide. In this case, samples were probed with antibodies specific for the cleaved PARP chain, and for the loading control, actin. c, All cells shown were transfected with miRNAs for 48 hr., followed by 24 hr. treatment with 20 μM etoposide. Samples were probed with antibodies specific for cleaved PARP and for the loading control, actin. d, Western blot analysis of endogenous BCLAF1 and loading control, actin, in HUVEC cells transfected with siRNAs directed to BCLAF1 (siB) or control (ctl). e and f, HUVEC cells were transfected with the indicated miRNAs or siRNAs, split to low density and treated with 50 μM etoposide followed by western blotting (e) TUNEL staining and flow cytometry (f). Values reflect average from at least three experiments with errors bars showing 1 std. dev.
Figure 5.
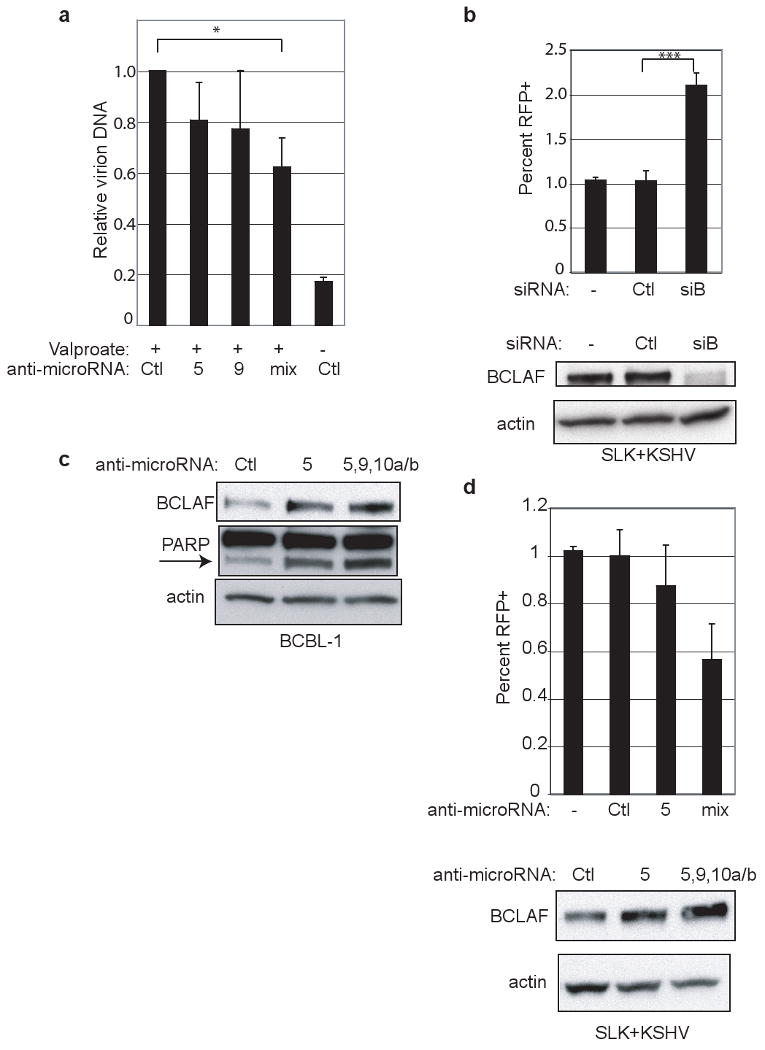
Inhibition of miRNAs and BCLAF1 expression. a, BCBL-1 cells were treated with miRNA inhibitors (“mix” is inhibitors for miR-K5, -9, -10a, -10b) followed by lytic induction by valproate treatment. Virions were purified from filtered supernatants. DNA content was measured by qPCR and normalized to a spike-in control. Values reflect average from three experiments with errors bars showing 1 std. dev. “*” refers to p-value less than 0.05. b, SLK endothelial cells expressing a recombinant virus with RFP under the control of a lytic promoter were transfected with no (-), control siRNA (Ctl), or BCLAF1 siRNA (siB). Cells were analyzed by flow for expression of RFP as a reporter of lytic induction. Values reflect average from at least six experiments with errors bars showing 1 std. dev. “***” refers to p-value less than 0.0001. Bottom panel shows western blot analysis from samples used for flow cytometry. c, Western blot analysis of BCBL-1 cells were treated with miRNA inhibitors and harvested five days after treatment. This is from the same experiment in Fig 3e with additional probing for PARP. Arrow indicates PARP cleavage product. d, Same cell line as in b were treated with miRNA inhibitors as in a and analyzed by flow cytometry. Western blot analysis shows endogenous BCLAF1 expression.
Curiously, we noted that under these experimental conditions (nondividing cells plated at confluence) etoposide did not induce visible cell death in HUVEC cells. When the cells were plated at lower density (conditions under which etoposide exposure leads to prominent cell death), we were surprised to observe no protection from apoptosis by KSHV miRNA expression (Fig. 4f). These unexpected findings led us to re-examine the function of BCLAF1, utilizing BCLAF-specific siRNAs to dramatically and specifically inhibit its expression (Fig. 4d). Dividing HUVEC cells were pretreated with siRNAs to BCLAF1, then exposed to etoposide and assayed for apoptosis by caspase activity assays and TUNEL staining (Fig. 4e-f). This showed that, contrary to expectation, BCLAF1 actually impairs apoptosis - its inhibition sensitizes cells to etoposide-induced cell death (Fig. 4f). This effect was phenocopied by co-expression of miRs-K5,9,10a and 10b, and to a lesser extent by miR-K5 alone (Fig. 4e-f) – further affirming their ability to target BCLAF1. It therefore appears that the original inference, derived from overexpression studies, that BCLAF1 is pro-apoptotic24 is either highly context-dependent or a result of supra-physiological expression. Clearly, further experiments will be required to understand the role of BCLAF1 in both uninfected and infected cells.
Nonetheless, the functional consequence of this downregulation for KSHV latency can be assessed by examining the effects of BCLAF1 modulation on viral replication. When latently infected cells are chemically induced to lytic KSHV growth, antagonism of miRs K5,9, 10a and 10b is associated with decreased virion production (Fig. 5a) and increased BCLAF1 expression (Fig. 5c). This suggests that BCLAF1 action acts to impair lytic viral replication. If so, then its inhibition by exogenous siRNA should sensitize cells to lytic reactivation. To test this notion, we employed SLK endothelial cells latently infected with a recombinant KSHV (rKSHV.214) in which a red fluorescent protein reporter gene has been placed under the control of a strong delayed-early lytic promoter29. In the ground state, most cells are latently infected, but approximately 1% spontaneously reactivate, as judged by RFP expression. When ambient levels of BCLAF1 are further reduced by specific RNAi to its mRNA, spontaneous lytic reactivation is doubled (Fig. 5b). Furthermore, in these same endothelial cells we observe decreased spontaneous lytic reactivation and increased BCLAF1 expression as a result of antagonism of miRs K5,9, 10a and 10b (Fig 5d).
Latency is an essential genetic program for herpesviruses, and recent data suggest that some latent viral miRNAs act to stabilize latency by opposing lytic reactivation30. The KSHV miRNAs affecting BCLAF1 appear to function differently: they sensitize latently infected cells to stimuli that will induce lytic reactivation. Why might this be useful? A key feature of latency is reversibility – without this feature, latency would be a dead-end pathway. By removing one safety lock on the trigger mechanism governing reactivation, we propose that these viral miRs help ensure that KSHV can exit latency when environmental conditions warrant.
Our multiscreen approach to miRNA target identification has both attractive features and limitations. On the positive side, it is very rigorous, and the approach is potentially applicable to host as well as to viral miRNAs. However, it is labor-intensive, and the number and stringency of the tests makes it nearly inescapable that some bona fide targets will be overlooked. For example, BCLAF1 passed only 2 of the 3 expression screens as a target for miRNAs-K9 and -K10b (Supplementary Fig 2). Beyond these technical factors, not all miRNA targets will display the modestly diminished transcript accumulation upon which the method is based; in addition, the fact that our screen used both B cells and endothelial cells means that cell type-specific targets will also fail to pass all screens. But by far the most restrictive screen is the bioinformatic one – requiring a perfect 8mer seed match is surely too stringent for routine use25, since many bona fide targets will fail this test (as BCLAF1 did for miR-K9, -K10a, -K10b, Supplementary Fig. 2c). Of course, the stringency of seed homology as well as the number of positive expression screens required by the experimenter can be relaxed. We anticipate that as our understanding of the nature of hybridization between miRNAs and targets progresses, more refined criteria will replace our stringent but insensitive initial settings.
Methods
miRNA expression
Synthetic miRNA mimics and inhibitors were from Ambion. BJAB and BCBL-1 cells were electroporated with siPORT system (Ambion) at 30 nM for mimics and 100 nM for inhibitors (RNA harvested 48 and 72 hr. after transfection, respectively). 293 and HUVEC cells were transfected with Lipofectamine 2000 (Invitrogen) and Dharmacon Dharmafect 1 at 15 and 10 nM, respectively. For stable cell lines, approximately 250 bp of flanking sequence surrounding the miRNA gene(s) were cloned into a gateway vector (Invitrogen) and then transferred into pMSCV-puro. BJAB cells were transduced and selected with puromycin. Northern blots were as described7.
Microarrays
Total RNA was harvested with RNAbee, quality controlled with Bioanalyzer 2100 (Agilent), and labeled with Low RNA Input Linear Amp kit plus (Agilent). Experimental samples were co-hybridized with labeled Universal Reference RNA (Stratagene) to Agilent Whole Human Genome arrays. Arrays were scanned on GenePix 4000B (Axon), extracted with Agilent Feature Extraction Software and analyzed with Genespring GX. Transient and stable BJAB arrays were performed in duplicate. Transient BJAB datasets were analyzed for genes that met either t-test p-value 0.05 cutoff or a combination of fold change and K-means clustering filters. Genes from other datasets were ranked in the rank sum analysis if the ratio was greater or less than 1.0 relative to the negative control miRNA.
Sequence Analysis
Datasets containing 3′UTR sequences were obtained from PACdb (http://harlequin.jax.org/) and TargetScan servers. TargetScan 4.0, miRANDA v1.9 and an ad-hoc perl script were used to search transcript sequences for seed matches without conservation analysis.
Luciferase assays
A Gateway cloning cassette (Invitrogen) was cloned into pMIR-Report (Ambion). PCR products containing the BCLAF1 3′UTR were cloned into a gateway entry vector and then transferred into the luciferase vector. The mutant was made using the QuikChange™ mutagenesis system (Stratagene). 293 cells were transfected with firefly reporter, TK renilla reporter and miRNA mimics using Lipofectamine 2000 (Invitrogen). Firefly activity was normalized to TK renilla internal control. Each transfection was assayed in triplicate and at least three independent transfections were performed.
Immunoblotting and apoptosis
After 48 or 72 hr. of transfection (and 24 hr. etoposide treatment for apoptosis assays), cells were lysed in RIPA buffer and equal total protein amounts were loaded on gel. Antibodies against BCLAF1 (Bethyl), PARP, cleaved PARP (Cell signaling), actin and tubulin (Sigma) were detected with HRP-conjugated secondary antibodies using ECL (GE). TUNEL staining data used the In Situ Cell Death Detection Kit (Roche).
Microarray data
MIAME-compliant HUVEC array data is available at http://puma.princeton.edu/. The GEO accession number for the BJAB and BCBL-1 array data is GSE12967.
Supplementary Material
Acknowledgments
We are grateful to S. Chandriani for sharing the HUVEC expression data, expertise and advice. We thank A. Goga and P. Lengyel for sharing a custom gateway cloning vector. The recombinant KSHV virus was a gift from J. Vieira. The infected SLK cell line was generated by J. Myoung. PUMAdb is supported by NIH grant P50 GM071508. J.M.Z. is a Damon Runyon Fellow supported by the Damon Runyon Cancer Research Foundation (DRG-1793). D.G. is an investigator of the Howard Hughes Medical Institute.
Footnotes
Author Contributions D.G, C.S.S. and J.M.Z. designed the experiments. C.S.S. generated the retroviruses, stable cell lines and performed northern blots. Sanjay Chandriani generated the HUVEC microarray data. J.M.Z. conducted the remaining experiments shown in the figures. D.G. directed and supervised the experimental progress. J.M.Z and D.G. wrote the manuscript.
Author Information The authors declare no competing financial interests.
Contributor Information
Joseph M. Ziegelbauer, Howard Hughes Medical Institute, G. W. Hooper Foundation and Departments of Medicine and Microbiology, University of California, San Francisco, CA 94143-0552
Christopher S. Sullivan, The University of Texas at Austin, Molecular Genetics & Microbiology, NMS 3.212, 2506 Speedway, Austin, TX 78712-1095
Don Ganem, Howard Hughes Medical Institute, G. W. Hooper Foundation and Departments of Medicine and Microbiology, University of California, San Francisco, CA 94143-0552.
References
- 1.Landgraf P, et al. A Mammalian microRNA Expression Atlas Based on Small RNA Library Sequencing. Cell. 2007;129:1401–14. doi: 10.1016/j.cell.2007.04.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pfeffer S, et al. Identification of microRNAs of the herpesvirus family. Nat Methods. 2005;2:269–76. doi: 10.1038/nmeth746. [DOI] [PubMed] [Google Scholar]
- 3.Sullivan C, Ganem D. MicroRNAs and viral infection. Mol Cell. 2005;20:3–7. doi: 10.1016/j.molcel.2005.09.012. [DOI] [PubMed] [Google Scholar]
- 4.Stern-Ginossar N, et al. Host immune system gene targeting by a viral miRNA. Science. 2007;317:376–81. doi: 10.1126/science.1140956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cai X, et al. Kaposi's sarcoma-associated herpesvirus expresses an array of viral microRNAs in latently infected cells. Proc Natl Acad Sci USA. 2005;102:5570–5. doi: 10.1073/pnas.0408192102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Samols MA, Hu J, Skalsky RL, Renne R. Cloning and identification of a microRNA cluster within the latency-associated region of Kaposi's sarcoma-associated herpesvirus. J Virol. 2005;79:9301–5. doi: 10.1128/JVI.79.14.9301-9305.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Grundhoff A, Sullivan C, Ganem D. A combined computational and microarray-based approach identifies novel microRNAs encoded by human gamma-herpesviruses. RNA. 2006;12:733–50. doi: 10.1261/rna.2326106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Samols M, et al. Identification of cellular genes targeted by KSHV-encoded microRNAs. PLoS Pathog. 2007;3:e65. doi: 10.1371/journal.ppat.0030065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Skalsky RL, et al. Kaposi's sarcoma-associated herpesvirus encodes an ortholog of miR-155. J Virol. 2007;81:12836–45. doi: 10.1128/JVI.01804-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gottwein E, et al. A viral microRNA functions as an orthologue of cellular miR-155. Nature. 2007;450:1096–9. doi: 10.1038/nature05992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Krützfeldt J, et al. Silencing of microRNAs in vivo with ‘antagomirs’. Nature. 2005;438:685–9. doi: 10.1038/nature04303. [DOI] [PubMed] [Google Scholar]
- 12.Giraldez AJ, et al. Zebrafish MiR-430 promotes deadenylation and clearance of maternal mRNAs. Science. 2006;312:75–9. doi: 10.1126/science.1122689. [DOI] [PubMed] [Google Scholar]
- 13.Lim L, et al. Microarray analysis shows that some microRNAs downregulate large numbers of target mRNAs. Nature. 2005;433:769–73. doi: 10.1038/nature03315. [DOI] [PubMed] [Google Scholar]
- 14.Rodriguez A, et al. Requirement of bic/microRNA-155 for normal immune function. Science. 2007;316:608–11. doi: 10.1126/science.1139253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Meister G, Landthaler M, Dorsett Y, Tuschl T. Sequence-specific inhibition of microRNA- and siRNA-induced RNA silencing. Rna. 2004;10:544–50. doi: 10.1261/rna.5235104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hutvagner G, Simard MJ, Mello CC, Zamore PD. Sequence-specific inhibition of small RNA function. PLoS Biol. 2004;2:E98. doi: 10.1371/journal.pbio.0020098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Grimson A, et al. MicroRNA Targeting Specificity in Mammals: Determinants beyond Seed Pairing. Mol Cell. 2007;27:91–105. doi: 10.1016/j.molcel.2007.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Selbach M, et al. Widespread changes in protein synthesis induced by microRNAs. Nature. 2008;455:58–63. doi: 10.1038/nature07228. [DOI] [PubMed] [Google Scholar]
- 19.Baek D, et al. The impact of microRNAs on protein output. Nature. 2008;455:64–71. doi: 10.1038/nature07242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gottwein E, Cai X, Cullen BR. A novel assay for viral microRNA function identifies a single nucleotide polymorphism that affects Drosha processing. J Virol. 2006;80:5321–6. doi: 10.1128/JVI.02734-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Didiano D, Hobert O. Perfect seed pairing is not a generally reliable predictor for miRNA-target interactions. Nat Struct Mol Biol. 2006;13:849–51. doi: 10.1038/nsmb1138. [DOI] [PubMed] [Google Scholar]
- 22.Reinhart BJ, et al. The 21-nucleotide let-7 RNA regulates developmental timing in Caenorhabditis elegans. Nature. 2000;403:901–6. doi: 10.1038/35002607. [DOI] [PubMed] [Google Scholar]
- 23.Yekta S, Shih IH, Bartel DP. MicroRNA-directed cleavage of HOXB8 mRNA. Science. 2004;304:594–6. doi: 10.1126/science.1097434. [DOI] [PubMed] [Google Scholar]
- 24.Kasof G, Goyal L, White E. Btf, a novel death-promoting transcriptional repressor that interacts with Bcl-2-related proteins. Mol Cell Biol. 1999;19:4390–404. doi: 10.1128/mcb.19.6.4390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lewis B, Burge C, Bartel D. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell. 2005;120:15–20. doi: 10.1016/j.cell.2004.12.035. [DOI] [PubMed] [Google Scholar]
- 26.Jopling C, Yi M, Lancaster A, Lemon S, Sarnow P. Modulation of hepatitis C virus RNA abundance by a liver-specific MicroRNA. Science. 2005;309:1577–81. doi: 10.1126/science.1113329. [DOI] [PubMed] [Google Scholar]
- 27.Offermann MK. Kaposi sarcoma herpesvirus-encoded interferon regulator factors. Curr Top Microbiol Immunol. 2007;312:185–209. doi: 10.1007/978-3-540-34344-8_7. [DOI] [PubMed] [Google Scholar]
- 28.Oliver FJ, et al. Importance of poly(ADP-ribose) polymerase and its cleavage in apoptosis. Lesson from an uncleavable mutant. J Biol Chem. 1998;273:33533–9. doi: 10.1074/jbc.273.50.33533. [DOI] [PubMed] [Google Scholar]
- 29.Vieira J, O'Hearn PM. Use of the red fluorescent protein as a marker of Kaposi's sarcoma-associated herpesvirus lytic gene expression. Virology. 2004;325:225–40. doi: 10.1016/j.virol.2004.03.049. [DOI] [PubMed] [Google Scholar]
- 30.Murphy E, Vanicek J, Robins H, Shenk T, Levine AJ. Suppression of immediate-early viral gene expression by herpesvirus-coded microRNAs: implications for latency. Proc Natl Acad Sci U S A. 2008;105:5453–8. doi: 10.1073/pnas.0711910105. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.