

Abstract
Gerstmann–Sträussler–Scheinker syndrome (GSS) is a genetic prion disease typified clinically by the development of progressive ataxia and dementia, and histopathologically by the presence of prion protein (PrP) amyloid plaques in the CNS, especially within the cerebellum. Several mutations of the PrP gene (PRNP) are associated with GSS, but only the P102L mutation has been convincingly modeled in transgenic (Tg) mice. To determine whether other mutations carry specific GSS phenotypic information, we constructed Tg mice that express PrP carrying the mouse homolog of the GSS-associated A117V mutation. Tg(A116V) mice express approximately six times the endogenous levels of PrP, develop progressive ataxia by ∼140 d, and die by ∼170 d. Compared with a mouse model of transmissible Creutzfeldt–Jakob disease (CJD), the ataxia of Tg(A116V) mice is more prominent, and the course of disease is more protracted, paralleling that observed in human disease. Neuropathology includes mild scattered vacuolation and prominent, mainly cerebellar localized, thioflavin S-positive PrP plaques comprised of full-length PrPA116V. In some mice, more prominent vacuolation or a noncerebellar distribution of PrP plaques was evident, suggesting some variability in phenotype. The biophysical properties of PrP from Tg(A116V) mice and human GSS(A117V) revealed a similarly low fraction of insoluble PrP and a weakly protease-resistant ∼13 kDa midspan PrP fragment, not observed in CJD. Overall, Tg(A116V) mice recapitulate many clinicopathologic features of GSS(A117V) that are distinct from CJD, supporting PrPA116V to carry specific phenotypic information. The occasional variation in histopathology they exhibit may shed light on a similar observation in human GSS(A117V).
Introduction
Prion diseases are invariably fatal neurodegenerative disorders that result from accumulation of misfolded pathogenic isoforms (PrPSc) of the normal prion protein (PrPC) (Prusiner, 1998). Several mutations of the PrP gene (PRNP) are known to cosegregate with one of three major clinicopathologic phenotypes, including Creutzfeldt–Jakob disease (CJD), Gerstmann–Sträussler–Scheinker syndrome (GSS), and fatal insomnia. Although there is considerable overlap among them, these disease subtypes are distinguished on the basis of broadly defined clinical presentations, specific histopathological features, and a characteristic PrPSc conformation, indirectly defined by the Western blot pattern of proteinase K (PK)-resistant PrPSc (rPrPSc) (Collinge et al., 1996). Of the two most common phenotypes, the core clinicopathologic features of CJD include the onset of rapidly progressive dementia, subsequent development of ataxia, and pronounced spongiform degeneration of the brain, whereas GSS is typified by the onset of ataxia, a protracted course, the late development of dementia, and the presence of PrP amyloid plaque deposits in the brain (Mastrianni, 1998). The rPrPSc associated with CJD is generally easily detectable and comprised of three major fractions of rPrPSc, representing amino-truncated (PrP90–230) monoglycosylated, diglycosylated, and unglycosylated PrPSc, known as PrP27–30, whereas that found in GSS is a less prominent, unglycosylated fragment ranging in size from 7 to 14 kDa, depending on the mutation with which it is associated (Piccardo et al., 2001).
It is predicted that mutations within PRNP increase the propensity for PrPC to misfold into specific conformations of PrPSc that define the clinical, pathologic, and rPrPSc properties of each (Prusiner, 1998; Gambetti et al., 2003). To better understand the pathogenesis of familial prion diseases and to confirm linkage of specific mutations to specific phenotypes, several transgenic (Tg) mouse lines expressing mouse (Mo) or human (Hu) PrP with disease-associated mutations have been constructed (Weissmann and Flechsig, 2003). Although multiple mouse lines that express CJD-linked mutations, including T183A (DeArmond et al., 1997), E199K (Telling et al., 1996), a nine octarepeat segment insertion (Chiesa et al., 1998), and the D178N mutation (Dossena et al., 2008) have been generated, with varying success, only one GSS-linked mutation (P102L) has, thus far, been successfully modeled in mice (Hsiao et al., 1990, 1994).
To begin to confirm linkage of additional mutations to specific prion disease phenotypes, we constructed a Tg mouse line that expresses mouse PrP carrying an A116V mutation, the homolog of the GSS-associated A117V mutation. We show that these mice develop clinical, pathologic, and PrPSc characteristics that are distinct from human CJD and a mouse model of transmissible CJD, but overlap significantly with GSS(A117V), including the presence of a faint, but consistently present ∼13 kDa rPrPSc fragment that is not present in CJD and not reported in other Tg mouse models of prion disease. When viewed in the context of other mouse models, Tg(A116V) mice support the hypothesis that specific mutations of PRNP cosegregate with specific disease phenotypes and PrPSc conformations, and as such, they represent an important new transgenic model with which to study the underlying biology of prion disease and to test potential therapeutics.
Materials and Methods
Production of transgenic mice.
The substitution of valine (V) for methionine (M) at codon 128 of the mouse Prnp open reading frame was produced by PCR amplification with Pfu polymerase (Stratagene) using forward and reverse primers containing the M128V mutation. The following forward primer and its reverse complement (data not shown) were used: 5′-CTTGGTGGCTACGTGCTGGGGAGCGCCATG-3′. The parental DNA was digested with the DpnI restriction enzyme, and the remaining nonmethylated PCR product was used to transform XL-1 Blue supercompetent cells (Stratagene). The A116V mutation was introduced into the template carrying the M128V mutation by a similar process, using the following forward primer and its reverse complement (data not shown): 5′-CATGTGGCAGGGGCTGCGGTGGCTGGGGCAGTAGTGGGG-3′. The Prnp-A116V/128V construct was amplified from this plasmid using the forward primer (MoPrP-Xho-5′), 5′-CCCCCCTCGAGACCATGGCGAACCTTGGCTACTGGCTGC-3′, and the reverse primer (MoPrP-Xho-3′), 5′-GGGGGGCTCGAGTCATCCCACGATCAGGAAGATGAGG-3′. PCR conditions were as follows: 94°C (30 s), 55°C (30 s), and 72°C (60 s) for 30 cycles. The amplified PCR product was digested with XhoI and ligated into the XhoI cut MoPrP.Xho vector (ATCC JHU-2; American Type Culture Collection). Recombinant plasmids with inserts in the correct orientation were selected by restriction analysis and sequencing of the entire coding segment. The DNA construct was excised from the recombinant plasmids by digestion with NotI, purified by QIAquick gel extraction kit (QIAGEN), and injected into pronuclei of fertilized eggs from PrP knock-out [Tg(Prnpo/o)] parental mice at the University of Chicago Transgenic Mouse Facility.
Transgenic founders were bred to Tg(Prnpo/o) mice, kindly provided by Stanley Prusiner (University of California, San Francisco, San Francisco, CA), which have been described in detail previously (Büeler et al., 1992). This line develops normally and has been repeatedly shown to be resistant to prion challenge. To screen for the transgene by PCR, genomic DNA was isolated from weanling tail clips, and 5′-AACCAAGTGTACTACAGGCCA-3′ (sense) and 5′-GTGGATACCCCCTCCCCCAGCCTAGACC-3′ (antisense) primers, which anneal to an internal coding segment of PrP and the 3′-untranslated region of the MoPrP.Xho vector are used with the following PCR conditions: 94°C (15 s), 62°C (15 s), and 72°C (60 s) for 42 cycles. The resultant 800 bp product is then subjected to restriction digestion with PvuII to identify the loss of a cleavage site at codon 116 introduced by the mutation.
Inoculation of mice.
Fresh frozen frontal cortex from a patient with CJD was used to prepare a 1% w/v brain homogenate in PBS, as previously described (Mastrianni et al., 1999). A total of 30 μl of brain homogenate was intracerebrally inoculated into Tg mice expressing human PrPC with Met at residue 129 on a mouse PrP knock-out background [Tg(HuPrP-129M)Prnpo/o], previously constructed and described (Telling et al., 1994). At the time of injection, the mouse was anesthetized with a xylazine/ketamine mixture, the head was fixed in a small animal stereotaxic instrument (David Kopf Instruments) fitted with mouse ear bars, the scalp was swabbed with alcohol, and a 25 gauge needle attached to a 1 cc syringe filled with inoculum was lowered 3 mm deep directly through the scalp into the brain.
Clinical observation of mice.
Mice were monitored daily for typical signs of prion disease in rodents, including ataxia, roughened fur, hunched posture, and righting reflex. Symptomatic mice were killed, and the brains harvested when death was imminent. Subtle signs of disease onset include widened stance of hindlimbs, and paws slipping through the wires when placed on the wired cage top.
Histological analysis.
Mice were asphyxiated with CO2 and perfused via cardiac puncture with 20 ml of PBS followed by 20 ml of 4% paraformaldehyde (Sigma-Aldrich). Brains were stored in 4% paraformaldehyde for 48 h, and then transferred to PBS containing 0.1% sodium azide (Sigma-Aldrich) until paraffin or Epon embedding. The blocks were cut with a microtome into 8 μm sections and stained with hematoxylin and eosin (H&E), periodic acid-Schiff (PAS), or with 0.05% thioflavin S (Sigma-Aldrich), for detection of amyloid deposits. Vacuolation scoring was performed by analysis of a single coronal section sampled from four brain regions, to include neocortex, hippocampus, thalamus, hypothalamus, caudate nucleus, pons, and cerebellum. The area occupied by vacuoles was scored, as previously described (Carlson et al., 1994), with slight modification. The following scale was used: 0, no vacuoles; 3, occasional vacuolation; 5, mild; 10, mild to moderate; 20, moderate; 30, moderate to severe; >40, severe.
For PrP immunohistochemistry, slides were baked and deparaffinized through xylenes and alcohols to water. Hydrolytic autoclaving was used as the method of antigen retrieval. Slides were then stained using recombinant F(ab) antibody Est123, HumP, D18, and R2 (InPro) against the N-terminal, central, and C-terminal residues of MoPrP. Visualization of these recombinant antibodies was accomplished using biotinylated anti-human IgG (Vector Laboratories).
For the evaluation of astrocytosis, 8 μm paraffin sections were stained with a rabbit antiserum against glial fibrillary acidic protein (GFAP) (Dako) using an automated tissue stainer (Ventana) after antigen retrieval in a citrate buffer at pH 6 for 40 min at 98°F.
Biochemical analyses of PrP.
Fresh frozen brain was used to prepare 10% (w/v) brain homogenates in lysis buffer (20 mm Tris-HCl, pH 7.4, 150 mm NaCl, 1 mm EDTA, 0.5% Triton X-100, 0.5% Na-deoxycholate). For deglycosylation, a 20 μl aliquot (20 μg) was incubated for 1 h at 37°C with PNGase F (New England Biolabs) according to the manufacturer's instructions. To detect protease-resistant PrP, brain homogenate was incubated with varying concentrations of PK for 1 h at 37°C and stopped with 2 mm phenylmethylsulfonyl fluoride. For the solubility assay, 10% brain homogenates were prepared in lysis buffer and centrifuged at 4°C for 1 h at 100,000 × g in a Sorvall RC M120EX ultramicrocentrifuge and T-100 rotor. The pellet was washed once with lysis buffer, centrifuged again, and then resuspended in the starting volume of lysis buffer. Equal fractions of supernatant and pellet were subjected to analysis by Western blotting. For all Western blots, 14% polyacrylamide gels were used for SDS-PAGE, and proteins were transferred to polyvinylidene difluoride membranes that were probed with anti-PrP monoclonal 3F4 antibody (gift from Richard Kascsak, Staten Island, NY) at 1:5000 dilution, followed by goat-anti-mouse IgG secondary antibody (1:5000; Santa Cruz Biotechnology) to detect human PrP, or anti-mouse PrP recombinant F(ab) D13 or D18 antibody (InPro) at 1:3000 dilution and goat anti-human IgG secondary antibody (1:5000; Santa Cruz Biotechnology). Signal was detected on a Chemi Doc XRS imager (Bio-Rad), using SuperSignal West Pico chemiluminescent substrate (Pierce). For dot blot analysis, equal volumes of 1% brain homogenates were serially diluted in lysis buffer and passed through a nitrocellulose membrane by vacuum before immunoblotting, using a Bio-Rad Bio-dot. Quantitation of dot blot was performed using Quantity One software (Bio-Rad) by plotting density measurements against the dilution factor in the linear range of signal.
Results
Construction of transgenic mice and analysis of PrP expression
To model GSS(A117V), we constructed MoPrP cDNA with a Val (GTG) substitution for Ala (GCA) at codon 116, the mouse Prnp gene equivalent of human PRNP A117V. We also introduced a Val at position 128, since Val coding at the polymorphic codon 129 is invariably allelic with the A117V mutation in all reported cases of GSS(A117V). Although its role in GSS is not known, the polymorphic codon 129 is well known to modify disease risk and/or phenotype of sporadic and some genetic forms of prion disease (Goldfarb et al., 1992; Gambetti et al., 2003). As such, we considered it to be important to accurately model GSS(A117V). The construct was ligated into the MoPrP.Xho vector and microinjected into the pronuclei of fertilized eggs from mice with an ablated PrP gene (Prnpo/o). The MoPrP.Xho vector includes the promoter and intron 1 region of the MoPrP gene, thereby mirroring expression of endogenous PrP. A founder was obtained and bred to Prnpo/o mice to establish the parental Tg(MoPrP-A116V,128V)Prnpo/o line, which we officially designated Tg1309, but refer to here as Tg(A116V) mice.
Western blot of whole-brain homogenates prepared from Tg(A116V) mice show that PrPA116V is expressed as three major fractions ranging from ∼25 to 35 kDa, representing unglycosylated, monoglycosylated, and diglycosylated PrP (Fig. 1A). The pattern and predominance of PrPA116V glycoforms was similar to wild-type (WT) PrPC of non-Tg mice. The relative expression levels of the transgene in asymptomatic (30-d-old) and symptomatic (150-d-old) Tg(A116V) mice were compared withendogenous PrP expression of 150-d-old WT mice by semiquantitative dot blot analysis of serially diluted total-brain homogenate. PrP levels in young Tg(A116V) mice are approximately four to six times that of WT mice, but in older, symptomatic mice, the level of PrP is nearly eight times that of WT, suggesting accumulation of PrPA116V (Fig. 1B,C).
Figure 1.

PrPA116V in Tg(A116V) mice. A, Western blot of brain homogenates from Tg(A116V) and WT mice displays a similar pattern and proportion of PrP glycoforms. B, Serially diluted whole-brain homogenates from 30-d-old asymptomatic and 150-d-old symptomatic Tg(A116V) mice, compared with 150 d-old WT mice. C, Plot of densitometric quantification of dot blot. At a young age, Tg(A116V) mice express approximately six times the normal level of PrPC. When symptomatic, the total level is approximately eight times that of WT mice, suggesting accumulation of PrP. The image was processed on a Bio-Rad XRS document imager, and Quantity One software (Bio-Rad) was applied to calculate the relative density against the dilution factor. Anti-mouse PrP D13 F(ab) antibody was used in each case.
Spontaneous neurological symptoms in Tg(A116V) mice
Tg(A116V) mice display normal development and activity until ∼5 months of age (mean onset, ∼148 ± 4 d; n = 15), when they begin to manifest subtle but clear signs of ataxia, including widened stance and unsteady gait. These symptoms progress steadily over the course of ∼30 ± 3 d, leading to the development of severe ataxia with excessive falling, difficulty righting, and death at 176 ± 3 d (range, 150–196 d) (Fig. 2A). To determine whether this phenotype was distinct, we compared it directly to a mouse model of transmissible CJD. A group of six Tg mice that express HuPrP on an ablated mouse PrP background [i.e., Tg(HuPrP)Prnpo/o] were intracerebrally inoculated with a 1% w/v brain homogenate prepared from a case of sporadic Creutzfeldt–Jakob disease (sCJD) that was homozygous for Met at codon 129 (i.e., 129MM) and carried typical type 1 rPrPSc (i.e., ∼21 kDa migration rate of the unglycosylated fraction). Although the onset of disease may vary among sCJD types, we used sCJD(129MM1) since it produces disease after an incubation period that is relatively similar to the age at onset of Tg(A116V) mice, which allows us to make a reasonable comparison of the duration of disease with Tg(A116V). CJD-inoculated mice developed symptoms at 160 ± 5 d (n = 6) and a mean time to death of 170 ± 4 d, resulting in a much shorter disease duration of 9 ± 1 d (Fig. 2B–D). In addition, the clinical phenotype differed strikingly between the two; the ataxia of CJD-inoculated mice was preceded by other features, including decreased exploratory behavior and physical signs of rough coat, hunched back, and wasting, all of which developed long after ataxia had developed in Tg(A116V) mice. Finally, the ataxia in CJD-inoculated mice was notably less severe and best characterized as a teetering gait, compared with the exaggerated ataxic movements of Tg(A116V) mice (see video supplement 1, available at www.jneurosci.org as supplemental material).
Figure 2.

Characteristics of spontaneous disease in Tg(A116V) mice. A, Kaplan–Meier survival curve of the rate of death plotted as a function of age in 15 Tg(A116V) mice. B, Mean age at symptom onset (in days) of 15 Tg(A116V) mice (onset, 148 ± 4 d) compared with an incubation period of 160 ± 5 d in 6 Tg(HuPrP)Prnpo/o mice that were intracerebrally inoculated with CJD(129MM1) brain homogenate. C, Mean age at death (in days) of Tg(A116V) mice was 176 ± 3 d, compared with the time to death from inoculation of 170 ± 4 d in CJD-inoculated Tg(HuPrP)Prnpo/o mice. D, Duration of disease in Tg(A116V) mice was 30 ± 3 d, compared with a very rapid 9 ± 1 d, in CJD-inoculated Tg(HuPrP)Prnpo/o mice (p < 0.001, Student's t test).
Neuropathological changes in Tg(A116V) mice
The most characteristic feature was the presence of PrP-immunopositive plaques, which were easily identified by H&E, PAS, and PrP immunohistochemical (IHC) stains in all mice analyzed (Fig. 3). Most plaques had irregular round to oval shapes and measured 30–50 μm in greatest dimension and consisted of a single deposit, although smaller numbers of plaques displayed a central core surrounded by a halo of smaller PrP-positive deposits, similar to those that characterize human GSS (DeArmond et al., 2002). The plaques were stained with thioflavin S, confirming them to be true amyloid (Fig. 3E). To assess the overall regional distribution of PrP plaques, the mean number of total plaque counts from three tissue slices within each of four brain regions was analyzed in seven symptomatic Tg(A116V) mice. Overall, the cerebellum contained the highest numbers of plaques (mean ± SE, 26 ± 4), especially the granule cell layer, compared with all other regions combined (mean ± SE, 5.0 ± 0.32) (Fig. 3, Table 1). Interestingly, a variability in plaque deposition was notable in some mice, such as mouse E, in which plaques were not evident in the cerebellar sections analyzed, although they were present in high numbers in hippocampus, and mouse F, in which they were evenly distributed among the cerebellum, neocortex, and caudate. Because all mice develop profound and progressive ataxia, these findings suggest that the presence of plaques in the cerebellum is not a requisite for the ataxic phenotype, but their presence may hasten the onset of symptoms.
Figure 3.
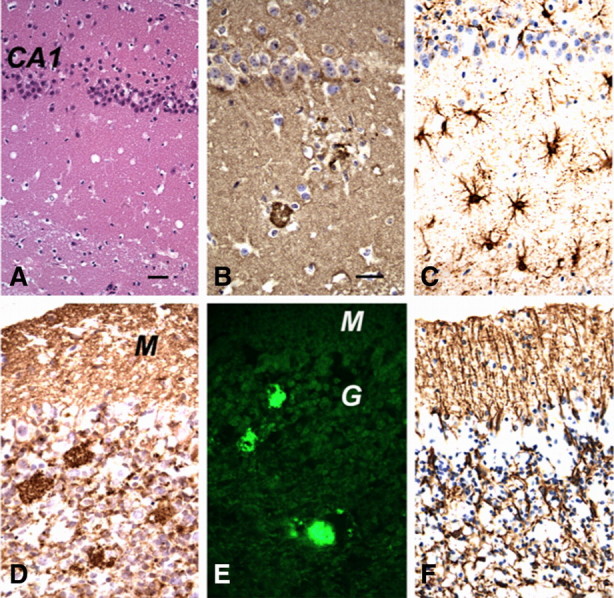
Neuropathological characteristics of Tg(A116V) mice. A, H&E stain shows sparse numbers of vacuoles in the hippocampus (CA1). B, PrP IHC with R2 antibody shows two PrP-positive amyloid plaques surrounded by vacuoles (florid plaques) in the hippocampus. C, GFAP IHC shows a moderate degree of reactive astrocytic gliosis in the hippocampus. D, PrP IHC (R2 antibody) shows PrP-positive amyloid plaques in the granule cell layer of the cerebellar cortex, as well as smaller punctate PrP-immunopositive deposits in both the molecular layer (M) and granular cell layer (G). E, Thioflavin S binds to the PrP plaques and displays fluorescence under UV light. F, GFAP IHC shows intense radial gliosis in the molecular layer and astrocytic gliosis in the granule cell layer. Scale bars: (in A) A, C, F, 50 μm; (in B) B, D, E, 50 μm.
Table 1.
Plaque distribution of Tg(A116V) mice
| Mouse | Neocortex | Hippocampus | Caudate nucleus | Cerebellar cortex |
|---|---|---|---|---|
| A | 0 | 6 | 10 | 23 |
| B | 1 | 1 | 1 | 34 |
| C | 0 | 0 | 1 | 87 |
| D | 1 | 0 | 4 | 5 |
| E | 11 | 24 | 9 | 0 |
| F | 16 | 6 | 15 | 10 |
| G | 0 | 0 | 0 | 22 |
PrP plaque counts from each of four regions, determined as described in Materials and Methods.
Although the dominant neuropathological feature of CJD in humans and rodent models of CJD and scrapie, is vacuolation (i.e., spongiform degeneration) that occupies 20–90% of the cross-sectional area of cerebral gray matter, spongiform degeneration in GSS is typically mild. Consistent with this, the gray matter of Tg(A116V) mice contained sparse numbers of typical prion disease vacuoles (Fig. 3A,B,D), compared with CJD-inoculated mice that showed no plaque and prominent vacuolation (data not shown). A comparison of the regional vacuolation among eight brain regions within seven terminally ill Tg(A116V) mice suggested the thalamus to be the most affected area and the hypothalamus and molecular layer of the cerebellum to be the least affected (Fig. 4A). However, when individual mice were compared (Fig. 4B), it was clear that the majority had very low levels of spongiform degeneration, whereas mice B and F were exceptions that skewed the results of the distribution means. Mouse B had moderate-to-severe levels of vacuolation, especially within the thalamus and caudate, whereas mouse F had moderate levels, primarily within the thalamus. When compared with the plaque distribution of these “outlier” mice, no specific relationship was observed, although as a group, the thalamus had a higher level of spongiform degeneration but lacked plaque pathology. In contrast, within the cerebellum, vacuolation was more prominent within the granule cell layer, in association with plaque deposition.
Figure 4.

Regional vacuolation of Tg(A116V) mice. A, Mean vacuolation scores from eight brain regions of seven symptomatic Tg(A116V) mice. NC, Neocortex; Hp, hippocampus; Th, thalamus; Hy, hypothalamus; Cd, caudate nucleus; pons; cbm, cerebellum molecular layer; cbg, cerebellum granular layer. The scoring system is described in Materials and Methods. The dashed line represents the limit at which vacuolation is reliably detected (score, 3). Error bars indicate SEM. B, The individual vacuolation scores of each of seven mice are plotted. The majority of scores were similarly low among the group, with the exception of mouse B, which displayed much higher levels in thalamus and caudate. Mouse F also had more prominent vacuolation in the thalamus, compared with the remainder of the group.
IHC staining with anti-GFAP antibody revealed gliosis that was severe in the cerebellar cortex and moderate in the hippocampus (Fig. 3C,F) and brainstem, but patchy in the thalamus (data not shown). The neocortex, caudate, and septum lacked obvious gliosis.
Plaques are composed of full-length PrP
IHC staining of PrP plaques in human GSS has been previously described (Ghetti et al., 1996). The central core appears to be composed of truncated PrP peptides, whereas the periphery of the plaques is stained by N- and C-terminal-specific PrP antibodies, suggesting that N- and C-terminal cleavage occurs subsequent to plaque formation. To determine whether plaques in Tg(A116V) mice are composed of full-length PrP or truncated polypeptide fragments, we immunostained Tg(A116V) brain sections with four antibodies that recognize the N and C termini, as well as the central amyloidogenic region, of mature PrP (Fig. 5). All plaques in all seven mice were intensely and uniformly immunostained by each of the four antibodies, suggesting, in contrast to human GSS plaques, they are composed of full-length PrP.
Figure 5.

PrP amyloid plaques in Tg(A116V) mice are composed of full-length PrPA116V. IHC was performed using four PrP antibodies that recognize four distinct regions of mature PrP23-230, as indicated. All plaques are from the granule cell layer of the cerebellar cortex. “No Primary Ab” signifies all of the IHC steps were performed except incubation of the slide with a primary anti-PrP antibody.
PrPA116V develops properties of PrPSc
The two principal biophysical properties that distinguish PrPSc from PrPC are detergent insolubility and a relative resistance to PK. We questioned whether PrPA116V expressed in the brain of Tg(A116V) mice acquires either or both of these properties. Ten percent (w/v) brain homogenates from a symptomatic Tg(A116V) mouse, a symptomatic scrapie-inoculated (RML isolate) WT mouse, and an uninoculated WT mouse, were prepared in lysis buffer and centrifuged at 100,000 × g for 1 h at 4°C, to separate insoluble PrP. At 30 d of age, Tg(A116V) mice displayed a low level of insoluble PrPA116V, which was similar in level to that recovered from 150-d-old WT mice. However, in 160-d-old symptomatic Tg(A116V) mice, the insoluble fraction of PrPA116V is much greater, yet still significantly lower than that recovered from a symptomatic scrapie-infected WT mouse, in which it constitutes >90% of total brain-derived PrP (Fig. 6A). To determine whether the relatively small fraction of insoluble PrP in Tg(A116V) mice is a characteristic of human GSS, or a feature specific to these mice, we prepared similar fractions of human brain homogenates from a normal brain, one affected with GSS(A117V), and one with sCJD(129MM1). The insoluble fraction of PrP in GSS(A117V) is evident but significantly lower than that from a representative case of sCJD(129MM1) (Fig. 6B). Thus, the small fraction of insoluble PrP in Tg(A116V) mice compares extremely well with GSS(A117V) and contrasts with the large fraction of insoluble PrP in humans with CJD.
Figure 6.

PrPA116V insolubility compares with human GSS(A117V). A, Supernatant (S) and pellet (P) fractions prepared from brain homogenates of 150-d-old WT mice, asymptomatic 30-d-old and symptomatic 160-d-old Tg(A116V) mice, and a terminally ill scrapie-infected WT mouse (RML isolate). B, The solubility profiles of PrP from homogenates prepared from normal human brain (CTL), GSS(A117V), and sporadic CJD, are compared. Brain homogenates prepared as 10% w/v in lysis buffer and centrifuged at 100,000 × g at 4°C for 1 h to separate insoluble PrP, as described in Materials and Methods.
rPrPSc is relatively low and often difficult to detect in GSS, however; when present, it is characterized by the presence of small PrP fragments, ranging from 6 to 14 kDa, that represent the central segment of PrP that has been N- and C-terminally cleaved (Piccardo et al., 2001; Tagliavini et al., 2001). To assess the presence of rPrPSc in Tg(A116V) mice, we subjected 5% w/v brain homogenates from 160-d-old symptomatic mice and human GSS(A117V) to a range of 1–50 μg/ml PK for 1 h at 37°C, and assayed by Western blot. Using D13 antibody (residues 96-106) to detect mouse PrP and 3F4 (residues 109-112) to detect human PrP, we identified a faint ∼13 kDa PrP fragment in 160-d-old symptomatic Tg(A116V) mice that corresponded to a similarly sized fragment in human GSS(A117V). The relative resistance to PK of this band is modest in both mice and humans, as it could not be readily detected beyond a concentration of 10 μg/ml PK, although the relative proportion of this fragment from human GSS was greater than from Tg(A116V) mice (Fig. 7B,C). This fragment appears to be disease-associated, since it was not detected in 60-d-old asymptomatic Tg(A116V) mice (Fig. 7A).
Figure 7.

A ∼13 kDa rPrPSc fragment is detected in symptomatic Tg(A116V) mice and in GSS(A117V). PK titration of brain homogenates from a 60-d-old asymptomatic Tg(A116V) mouse (A), a 160-d-old symptomatic Tg(A116V) mouse (B), and human GSS(A117V) (C). Brain homogenates were digested with indicated concentrations of PK for 1 h at 37°C. Membranes were probed with D13 for MoPrP detection, and 3F4 to detect human PrP, as labeled. The open arrowheads indicate the ∼13 kDa fragments. A single fragment is detected in Tg(A116V) mice, whereas GSS(A117V) displayed a very faint ∼15 kDa fragment and a smaller doublet of ∼13 kDa.
Endogenous cleavage of PrP in Tg(A116V) differs from CJD-inoculated mice
PrP undergoes endocytic recycling and processing to yield two principal C-terminal fragments, C1 and C2, that differ in size and relative proportions in normal and prion-affected brain (Chen et al., 1995). Because they are glycosylated, these endogenously cleaved fragments are better visualized after deglycosylation. C1 is an ∼18 kDa C-terminal fragment that results from the cleavage of PrP at residue ∼111, by metalloproteinases, whereas C2 is a ∼21 kDa C-terminal fragment resulting from cleavage at residue ∼90 that is attributed to calpain activity on the conformationally altered PrP (Vincent et al., 2001; Yadavalli et al., 2004). To determine whether endogenous processing of PrPA116V in affected Tg(A116V) mice differs from rPrPSc typically associated with scrapie or CJD, we compared the Western blot pattern of PNGase F-treated PrP from Tg(A116V) mice with RML-infected, and uninfected, WT mice (Fig. 8). Samples were probed with D13 (epitope 96-106) and D18 (epitope 132-158) antibodies to detect C1 and C2. In each case, the full-length ∼25 kDa PrP (i.e., PrP23-231) and the ∼21 kDa C2 fragment were detected by D13. As expected, the C2 fraction was very low in noninfected WT mice and very high in RML-infected WT mice. In Tg(A116V) mice, it was intermediate in level between the two, but much less than in RML. When probed with D18, in addition to C2, the ∼18 kDa C1 fragment that is not recognized by D13 was visualized. Since D18 recognizes both C1 and C2, the relative levels of each fragment within the same sample can be compared and, because the protein loads were similar, supported by the similar intensities of the 25 kDa band, a relative comparison of C1 and C2 among the samples can be made. In WT, C1 was only slightly more prominent than C2, although both are low, relative to full-length PrP. In RML-infected mice, C2 is severalfold higher than C1, the latter of which is lower than in WT mice, supporting a predominance of misfolded PrP that is alternatively cleaved to produce PrP90–231. In contrast, the C1 fragment was much more prominent in Tg(A116V) mice than in WT or RML-infected mice, and although C2 is present in Tg(A116V) mice, it is significantly lower than in RML-infected mice and only slightly greater than in WT mice. Thus, PrPA116V from affected mice displays a cleavage pattern that differs significantly from prion disease associated with a high level of rPrPSc. The low level of the ∼13 kDa rPrPSc fragment in symptomatic Tg(A116V) mice is likely to correspond to the low level of C2, and may be derived from C2, as both are recognized by D13.
Figure 8.

Mapping endogenous cleavage products of Tg(A116V). Brain homogenates from an uninfected WT mouse (WT), a clinically sick WT mouse inoculated with mouse-adapted RML scrapie prions (RML), and a symptomatic (160-d-old) Tg(A116V) mouse (Tg), before (−) and after (+) deglycosylation with PNGase F, separated by SDS-PAGE on a 16% gel and probed separately with D13 or D18 antibodies. C1 and C2 are endogenous degradation products of PrPA116V that differ from typical scrapie.
Discussion
The first and only currently available transgenic mouse shown to specifically model GSS was produced in 1990 (Hsiao et al., 1990, 1994). These mice express mouse PrP with a P101L mutation to model human GSS(P102L). With the Tg(A116V) mouse line, we expand the available resources with which to study genetic prion disease. In addition, this mouse line further underscores the tenet that specific mutations of PrP induce specific conformations of PrPSc that are linked to the clinicopathologic phenotype of disease.
Tg(A116V) mice display all the characteristic features of GSS, which are easily distinguished from CJD and a mouse model of CJD, including the onset of ataxia, a protracted yet progressive course to death, and the histological appearance of PrP amyloid plaques in association with a low level of spongiform degeneration (Farlow et al., 1989; Ghetti et al., 1989). We further show that the biochemical properties of PrPA116V expressed in Tg(A116V) mice are remarkably similar to those of human PrPA117V in GSS(A117V). Whereas CJD and scrapie-infected mice carry a large fraction of insoluble PrP and rPrPSc with a migration rate of ∼19–21 kDa, human GSS(A117V) and Tg(A116V) mice harbor comparably low levels of insoluble PrP and a small fraction of a ∼13 kDa rPrPSc fragment. Several lines of evidence suggest the conformation of PrPSc, which is indirectly assessed by the electrophoretic mobility of rPrPSc, is linked to the phenotype of disease (Bessen and Marsh, 1992), and the presence of small ∼7–14 kDa rPrPSc fragments are associated with human GSS(A117V) (Piccardo et al., 2001). Thus, the ∼13 kDa fragment we detect in Tg(A116V) mice and human GSS(A117V), suggests they are specific to this mutation and likely the same as the ∼14 kDa fragment reported by others. Although the level of this fragment is low, its presence in symptomatic, but not asymptomatic, Tg(A116V) mice argue that it is disease-associated.
Although it cannot be determined with certainty, it is suspected that this fragment results from the C-terminal cleavage of the small pool of C2, since both are detected at their N termini by the D13 antibody, in contrast to the C1 fragment that is cleaved around residue 111 and, as such, eliminates the D13 epitope. However, previous work by others suggests the core of GSS(A117V) plaques are composed of N- and C-terminally truncated PrP polypeptides, which may constitute the 7–14 kDa rPrPSc fragments observed by Western blot (Tagliavini et al., 2001). Immunohistochemical staining of the plaques suggests the central core of the plaque is composed, at least to some extent, of PrP polypeptides with either or both ends trimmed, whereas the periphery of the plaques contain the trimmed ends (Farlow et al., 1989; Ghetti et al., 1989). This suggests that the plaque may be formed from full-length PrP that is processed in situ, to generate the midspan cleavage fragments detected by Western blot. In support of this, we find the plaques in Tg(A116V) mice to be composed, at least predominantly, of full-length PrP, based on the IHC studies. Although it may be possible that a small level of trimming is present and undetected by IHC, plaque purification and sequencing will be necessary to confirm this. This finding may also explain the relatively low levels of the ∼13 kDa rPrPSc fragment in Tg(A116V) mice, compared with human GSS(A117V). In addition, close examination of Figure 7 suggests that, in GSS(A117V), but not in Tg(A116V) mice, the ∼13 kDa band is already present before PK digestion, supporting the idea that N- and C-terminal trimming is greater in human GSS(A117V). The lack of detectable in situ processing of PrP amyloid in mice could reflect a difference in processing between mouse and human, or simply reflect the fact that plaque deposits in human brains are present for years rather than weeks and are therefore exposed to potential processing events for a much longer period of time. In any event, our Tg(A116V) mice confirm that full-length, rather than truncated, PrPA116V is the substrate for amyloid production.
It is important to note that GSS(A117V) in humans has been reported to present with either ataxia (Mastrianni et al., 1995) or cognitive impairment (Hsiao et al., 1991). Whether these two major presentations result from differing genetic backgrounds of affected individuals or is inherent with the expression of PrPA117V, is not known. Since Tg(A116V) mice have a homogeneous genetic background and all Tg(A116V) mice develop progressive and profound ataxia, the A117V mutation appears to predispose primarily to an ataxic phenotype and other genes may be responsible for the telencephalic phenotype. However, we did note that two of seven mice displayed a higher proportion of plaques in hippocampus and neocortex than the cerebellum, compared with the rest of the group. In addition, compared with the group, mice B and F had higher levels of spongiform degeneration in the thalamus and caudate. Thus, despite the same genetic background, these variations in histopathology suggest an inherent variability in the phenotype resulting from PrPA116V expression. Although formal cognitive assessments have not yet been performed on these mice, it is intriguing to speculate that some degree of cognitive impairment might precede ataxia in the minority of mice that display predominant hippocampal and/or neocortical pathology, compared with the more common cerebellar phenotype.
The Tg(P101L) mice (Hsiao et al., 1990, 1994) were constructed with the CosTet vector to drive expression of MoPrP by a hamster promoter segment. They express approximately eight times the normal level of PrP and develop ataxia at ∼190 d of age, similar to our mice that express approximately four to six times endogenous PrP. Nazor et al. (2005) subsequently reproduced several lines of mice expressing the same transgene at levels varying from 0.5 to 12 times normal WT levels, with markedly delayed onset times. For instance, their line expressing approximately six times endogenous PrP, comparable with Tg(A116V) mice, developed disease at a mean age of ∼400 d. The reason for this dramatic difference in sensitivity to PrPP101L levels in those mice is difficult to resolve. However, all mice overexpressing mouse PrPP101L develop pathologic features in their brains similar to Tg(A116V) mice, including mild spongiform degeneration, astrocytic gliosis, and PrP amyloid plaques, although the regional distribution and the composition of plaques have not been detailed in Tg(P101L) mice. GSS neuropathology was not observed in Prnp gene targeted mice expressing a single MoPrP-P101L gene (Manson et al., 2000), arguing that the disease of Tg(P101L) mice represents an artificial proteinopathy resulting from overexpression of mutant PrP. However, that the development of disease is specific to the mutant transgene and not simply a result of PrP overexpression is underscored by the lack of spontaneous disease or rPrPSc in Tg mice that express five to seven times the normal level of PrP (Fischer et al., 1996; Chiesa et al., 1998).
PrP plaque pathology has also been observed in Tg mice that express PrP lacking the glycosylphosphatidylinositol (GPI) anchor, suggesting full-length PrP is secreted to generate plaques. In contrast to our mice, those expressing PrP(GPI−) did not develop significant clinical features of disease unless PrP(GPI−) was coexpressed with full-length PrPC, suggesting the presence of membrane-localized PrP is necessary for the phenotypic expression of disease. Because PrPA116V is expressed with the GPI anchor, it provides its own source of membrane-bound PrP that might be necessary for the development of progressive neurodegeneration. In addition, the A117V mutation of human PrP has been shown to favor a transmembrane (CtmPrP) topology that might be linked directly to the underlying disease pathogenesis (Hegde et al., 1998, 1999).
In contrast to the GSS phenotype of Tg(P101L) and Tg(A116V) mice, transgenic mouse models of CJD have been less clearcut. An important new transgenic model of CJD linked to the D178N mutation has been recently described (Dossena et al., 2008). These mice display cognitive dysfunction but not the profound ataxia observed in Tg(A116V) mice, and their histopathology includes punctate PrP deposits, and gliosis, but no amyloid plaques (Dossena et al., 2008). PK-resistant PrP is also reported at low concentrations of PK, but no fragments smaller than 20 kDa were reported. Thus, although this CJD model has some limitations, the clinical and histopathologic phenotype, in addition to the molecular phenotype of rPrPSc, clearly differ from the profile of Tg(A116V), providing additional support that specific PrP mutations determine the distinct clinical subtypes of familial prion disease.
Is PrPA116V transmissible? The potential for transmissibility of disease from Tg(A116V) mice is currently under study. It should be noted that, in our hands (our unpublished observations) and others (Brown et al., 1994; Tateishi and Kitamoto, 1995), human GSS is less transmissible than CJD, possibly because of its inherently different strain or because the mutations associated with GSS fall within regions of PrP that require homology between PrPSc and PrPC to efficiently transfer the pathogenic conformation. In support of the latter hypothesis, transmission of GSS(P102L) to a rodent model was successful only when the receptive mouse expressed low levels of the equivalent mutated PrP (Telling et al., 1996). The A117V mutation may pose an even greater barrier to transmission, as it lies within the palindromic segment (AGAAAAGA) that extends from residue 113 to 120 and is necessary for the generation of PrPSc and propagation of prions (Norstrom and Mastrianni, 2005). In addition, previous work has suggested that the PrP amyloid deposits of GSS are composed of only mutant PrP (Tagliavini et al., 1994), suggesting that mutant PrP that forms amyloid either does not, or very inefficiently, interacts with WT PrP. Ongoing studies that cross Tg(A116V) mice with WT mice will allow us to explore these questions.
In summary, Tg(A116V) mice recapitulate the clinicopathologic phenotype of GSS, including early onset of a protracted and progressive course of ataxia, PrP amyloid deposition in the brain, and the biochemical characteristics of rPrPSc that distinguish GSS from CJD. Specific features that relate to the histopathologic distribution of plaques, the endogenous cleavage of PrPA116V, and the PK-resistant molecular phenotype, suggest this model may provide important clues to the underlying pathogenesis of prion disease, and serve as an important model with which to test therapeutic agents against prion disease.
Footnotes
This work was supported by National Institutes of Health Grants R01NS046037 and R01NS051480, The Brain Research Foundation, and The Pioneer Fund. We thank Dr. Stanley Prusiner (University of California, San Francisco, San Francisco, CA) for his kind gift of Tg(Prnpo/o) mice, Dr. Richard Kascsak (Staten Island, NY) for the 3F4 antibody, and Dr. Thomas Bird (University of Washington, Seattle, WA) for a GSS(A117V) sample.
References
- Bessen RA, Marsh RF. Biochemical and physical properties of the prion protein from two strains of the transmissible mink encephalopathy agent. J Virol. 1992;66:2096–2101. doi: 10.1128/jvi.66.4.2096-2101.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown P, Gibbs CJ, Jr, Rodgers-Johnson P, Asher DM, Sulima MP, Bacote A, Goldfarb LG, Gajdusek DC. Human spongiform encephalopathy: the National Institutes of Health series of 300 cases of experimentally transmitted disease. Ann Neurol. 1994;35:513–529. doi: 10.1002/ana.410350504. [DOI] [PubMed] [Google Scholar]
- Büeler H, Fischer M, Lang Y, Bluethmann H, Lipp HP, DeArmond SJ, Prusiner SB, Aguet M, Weissmann C. Normal development and behaviour of mice lacking the neuronal cell-surface PrP protein. Nature. 1992;356:577–582. doi: 10.1038/356577a0. [DOI] [PubMed] [Google Scholar]
- Carlson GA, Ebeling C, Yang SL, Telling G, Torchia M, Groth D, Westaway D, DeArmond SJ, Prusiner SB. Prion isolate specified allotypic interactions between the cellular and scrapie prion proteins in congenic and transgenic mice. Proc Natl Acad Sci U S A. 1994;91:5690–5694. doi: 10.1073/pnas.91.12.5690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen SG, Teplow DB, Parchi P, Teller JK, Gambetti P, Autilio-Gambetti L. Truncated forms of the human prion protein in normal brain and in prion diseases. J Biol Chem. 1995;270:19173–19180. doi: 10.1074/jbc.270.32.19173. [DOI] [PubMed] [Google Scholar]
- Chiesa R, Piccardo P, Ghetti B, Harris DA. Neurological illness in transgenic mice expressing a prion protein with an insertional mutation. Neuron. 1998;21:1339–1351. doi: 10.1016/s0896-6273(00)80653-4. [DOI] [PubMed] [Google Scholar]
- Collinge J, Sidle KC, Meads J, Ironside J, Hill AF. Molecular analysis of prion strain variation and the aetiology of “new variant” CJD. Nature. 1996;383:685–690. doi: 10.1038/383685a0. [DOI] [PubMed] [Google Scholar]
- DeArmond SJ, Sánchez H, Yehiely F, Qiu Y, Ninchak-Casey A, Daggett V, Camerino AP, Cayetano J, Rogers M, Groth D, Torchia M, Tremblay P, Scott MR, Cohen FE, Prusiner SB. Selective neuronal targeting in prion disease. Neuron. 1997;19:1337–1348. doi: 10.1016/s0896-6273(00)80424-9. [DOI] [PubMed] [Google Scholar]
- DeArmond SJ, Kretzschmar HA, Prusiner SB. Prion diseases. In: Graham DI, Lantos PL, editors. Greenfield's neuropathology. Ed 7. London: Arnold; 2002. pp. 273–323. [Google Scholar]
- Dossena S, Imeri L, Mangieri M, Garofoli A, Ferrari L, Senatore A, Restelli E, Balducci C, Fiordaliso F, Salio M, Bianchi S, Fioriti L, Morbin M, Pincherle A, Marcon G, Villani F, Carli M, Tagliavini F, Forloni G, Chiesa R. Mutant prion protein expression causes motor and memory deficits and abnormal sleep patterns in a transgenic mouse model. Neuron. 2008;60:598–609. doi: 10.1016/j.neuron.2008.09.008. [DOI] [PubMed] [Google Scholar]
- Farlow MR, Yee RD, Dlouhy SR, Conneally PM, Azzarelli B, Ghetti B. Gerstmann-Straussler-Scheinker disease. I. Extending the clinical spectrum. Neurology. 1989;39:1446–1452. doi: 10.1212/wnl.39.11.1446. [DOI] [PubMed] [Google Scholar]
- Fischer M, R̈ulicke T, Raeber A, Sailer A, Moser M, Oesch B, Brandner S, Aguzzi A, Weissmann C. Prion protein (PrP) with amino-proximal deletions restoring susceptibility of PrP knockout mice to scrapie. EMBO J. 1996;15:1255–1264. [PMC free article] [PubMed] [Google Scholar]
- Gambetti P, Kong Q, Zou W, Parchi P, Chen SG. Sporadic and familial CJD: classification and characterisation. Br Med Bull. 2003;66:213–239. doi: 10.1093/bmb/66.1.213. [DOI] [PubMed] [Google Scholar]
- Ghetti B, Tagliavini F, Masters CL, Beyreuther K, Giaccone G, Verga L, Farlow MR, Conneally PM, Dlouhy SR, Azzarelli B, Bugiani O. Gerstmann-Straussler-Scheinker disease. II. Neurofibrillary tangles and plaques with PrP-amyloid coexist in an affected family. Neurology. 1989;39:1453–1461. doi: 10.1212/wnl.39.11.1453. [DOI] [PubMed] [Google Scholar]
- Ghetti B, Piccardo P, Frangione B, Bugiani O, Giaccone G, Young K, Prelli F, Farlow MR, Dlouhy SR, Tagliavini F. Prion protein amyloidosis. Brain Pathol. 1996;6:127–145. doi: 10.1111/j.1750-3639.1996.tb00796.x. [DOI] [PubMed] [Google Scholar]
- Goldfarb LG, Petersen RB, Tabaton M, Brown P, LeBlanc AC, Montagna P, Cortelli P, Julien J, Vital C, Pendelbury WW, Haltia M, Wills PR, Hauw JJ, Mckeever PE, Monari L, Schrank B, Swergold GD, Autiliogambetti L, Gajdusek DC, Lugaresi E. Fatal familial insomnia and familial Creutzfeldt-Jakob disease: disease phenotype determined by a DNA polymorphism. Science. 1992;258:806–808. doi: 10.1126/science.1439789. [DOI] [PubMed] [Google Scholar]
- Hegde RS, Mastrianni JA, Scott MR, DeFea KA, Tremblay P, Torchia M, DeArmond SJ, Prusiner SB, Lingappa VR. A transmembrane form of the prion protein in neurodegenerative disease. Science. 1998;279:827–834. doi: 10.1126/science.279.5352.827. [DOI] [PubMed] [Google Scholar]
- Hegde RS, Tremblay P, Groth D, DeArmond SJ, Prusiner SB, Lingappa VR. Transmissible and genetic prion diseases share a common pathway of neurodegeneration. Nature. 1999;402:822–826. doi: 10.1038/45574. [DOI] [PubMed] [Google Scholar]
- Hsiao KK, Scott M, Foster D, Groth DF, DeArmond SJ, Prusiner SB. Spontaneous neurodegeneration in transgenic mice with mutant prion protein. Science. 1990;250:1587–1590. doi: 10.1126/science.1980379. [DOI] [PubMed] [Google Scholar]
- Hsiao KK, Cass C, Schellenberg GD, Bird T, Devine-Gage E, Wisniewski H, Prusiner SB. A prion protein variant in a family with the telencephalic form of Gerstmann-Straussler-Scheinker syndrome. Neurology. 1991;41:681–684. doi: 10.1212/wnl.41.5.681. [DOI] [PubMed] [Google Scholar]
- Hsiao KK, Groth D, Scott M, Yang SL, Serban H, Rapp D, Foster D, Torchia M, Dearmond SJ, Prusiner SB. Serial transmission in rodents of neurodegeneration from transgenic mice expressing mutant prion protein. Proc Natl Acad Sci U S A. 1994;91:9126–9130. doi: 10.1073/pnas.91.19.9126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manson JC, Barron R, Jamieson E, Baybutt H, Tuzi N, McConnell I, Melton D, Hope J, Bostock C. A single amino acid alteration in murine PrP dramatically alters TSE incubation time. Arch Virol. 2000;(Suppl):95–102. doi: 10.1007/978-3-7091-6308-5_8. [DOI] [PubMed] [Google Scholar]
- Mastrianni JA. The prion diseases: Creutzfeldt-Jakob, Gerstmann-Straussler-Scheinker, and related disorders. J Geriatr Psychiatry Neurol. 1998;11:78–97. doi: 10.1177/089198879801100206. [DOI] [PubMed] [Google Scholar]
- Mastrianni JA, Curtis MT, Oberholtzer JC, Da Costa MM, DeArmond S, Prusiner SB, Garbern JY. Prion disease (PrP-A117V) presenting with ataxia instead of dementia. Neurology. 1995;45:2042–2050. doi: 10.1212/wnl.45.11.2042. [DOI] [PubMed] [Google Scholar]
- Mastrianni JA, Nixon R, Layzer R, Telling GC, Han D, DeArmond SJ, Prusiner SB. Prion protein conformation in a patient with sporadic fatal insomnia. N Engl J Med. 1999;340:1630–1638. doi: 10.1056/NEJM199905273402104. [DOI] [PubMed] [Google Scholar]
- Nazor KE, Kuhn F, Seward T, Green M, Zwald D, Pürro M, Schmid J, Biffiger K, Power AM, Oesch B, Raeber AJ, Telling GC. Immunodetection of disease-associated mutant PrP, which accelerates disease in GSS transgenic mice. EMBO J. 2005;24:2472–2480. doi: 10.1038/sj.emboj.7600717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norstrom EM, Mastrianni JA. The AGAAAAGA palindrome in PrP is required to generate a productive PrPSc-PrPC complex that leads to prion propagation. J Biol Chem. 2005;280:27236–27243. doi: 10.1074/jbc.M413441200. [DOI] [PubMed] [Google Scholar]
- Piccardo P, Liepnieks JJ, William A, Dlouhy SR, Farlow MR, Young K, Nochlin D, Bird TD, Nixon RR, Ball MJ, DeCarli C, Bugiani O, Tagliavini F, Benson MD, Ghetti B. Prion proteins with different conformations accumulate in Gerstmann-Straussler-Scheinker disease caused by A117V and F198S mutations. Am J Pathol. 2001;158:2201–2207. doi: 10.1016/S0002-9440(10)64692-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prusiner SB. Prions. Proc Natl Acad Sci U S A. 1998;95:13363–13383. doi: 10.1073/pnas.95.23.13363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tagliavini F, Prelli F, Porro M, Rossi G, Giaccone G, Farlow MR, Dlouhy SR, Ghetti B, Bugiani O, Frangione B. Amyloid fibrils in Gerstmann-Straussler-Scheinker disease (Indiana and Swedish kindreds) express only PrP peptides encoded by the mutant allele. Cell. 1994;79:695–703. doi: 10.1016/0092-8674(94)90554-1. [DOI] [PubMed] [Google Scholar]
- Tagliavini F, Lievens PM, Tranchant C, Warter JM, Mohr M, Giaccone G, Perini F, Rossi G, Salmona M, Piccardo P, Ghetti B, Beavis RC, Bugiani O, Frangione B, Prelli F. A 7-kDa prion protein (PrP) fragment, an integral component of the PrP region required for infectivity, is the major amyloid protein in Gerstmann-Straussler-Scheinker disease A117V. J Biol Chem. 2001;276:6009–6015. doi: 10.1074/jbc.M007062200. [DOI] [PubMed] [Google Scholar]
- Tateishi J, Kitamoto T. Inherited prion diseases and transmission to rodents. Brain Pathol. 1995;5:53–59. doi: 10.1111/j.1750-3639.1995.tb00577.x. [DOI] [PubMed] [Google Scholar]
- Telling GC, Scott M, Hsiao KK, Foster D, Yang SL, Torchia M, Sidle KC, Collinge J, DeArmond SJ, Prusiner SB. Transmission of Creutzfeldt-Jakob disease from humans to transgenic mice expressing chimeric human-mouse prion protein. Proc Natl Acad Sci U S A. 1994;91:9936–9940. doi: 10.1073/pnas.91.21.9936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Telling GC, Haga T, Torchia M, Tremblay P, DeArmond SJ, Prusiner SB. Interactions between wild-type and mutant prion proteins modulate neurodegeneration in transgenic mice. Genes Dev. 1996;10:1736–1750. doi: 10.1101/gad.10.14.1736. [DOI] [PubMed] [Google Scholar]
- Vincent B, Paitel E, Saftig P, Frobert Y, Hartmann D, De Strooper B, Grassi J, Lopez-Perez E, Checler F. The disintegrins ADAM10 and TACE contribute to the constitutive and phorbol ester-regulated normal cleavage of the cellular prion protein. J Biol Chem. 2001;276:37743–37746. doi: 10.1074/jbc.M105677200. [DOI] [PubMed] [Google Scholar]
- Weissmann C, Flechsig E. PrP knock-out and PrP transgenic mice in prion research. Br Med Bull. 2003;66:43–60. doi: 10.1093/bmb/66.1.43. [DOI] [PubMed] [Google Scholar]
- Yadavalli R, Guttmann RP, Seward T, Centers AP, Williamson RA, Telling GC. Calpain-dependent endoproteolytic cleavage of PrPSc modulates scrapie prion propagation. J Biol Chem. 2004;279:21948–21956. doi: 10.1074/jbc.M400793200. [DOI] [PubMed] [Google Scholar]