

Abstract
Objective
To determine whether tumor necrosis factor (TNF) gene polymorphisms and/or the shared epitope are genetic predictors of response to adalimumab (ADA) in rheumatoid arthritis (RA).
Methods
This ancillary study to the Research in Active Rheumatoid Arthritis (ReAct) Phase IIIb study included a large cohort of Caucasian patients with RA from France (N=380) treated with ADA plus methotrexate (MTX) (n=182), ADA plus any other DMARD (N=96) or ADA alone (N=102). The primary outcome was ACR50 at 12 weeks. Patients underwent genotyping for HLA-DRB1 and 3 TNFα gene polymorphisms (-238A/G, -308A/G and -857C/T). Extended haplotypes involving HLA-DRB1 and TNFα loci were reconstructed by use of the PHASE program.
Results
A total of 152 patients (40%) had an ACR50 response at week 12. Neither the number of HLA-DRB1 SE copies nor presence of the 3 TNFα polymorphisms tested separately was significantly associated with ACR50 response at week 12. However, haplotype reconstruction of the TNFα locus revealed the GGC haplotype (-238G/-308G/–857C) in a homozygous form, present in more than half of the patients, significantly associated with a lower ACR50 response at 12 weeks (34% vs. 50% in patients without the haplotype) on treatment with ADA concomitant with MTX (P=0.0041; Pc=0.02). This effect was restricted to the subgroup of patients concomitantly treated with MTX.
Conclusion
This large pharmacogenetic study provides robust data indicating that a single TNFα locus haplotype (-238G/-308G/-857C), present on both chromosomes is associated with a lower response to ADA and MTX therapy in RA patients homozygous for this haplotype.
Keywords: Adult; Aged; Antibodies, Monoclonal; therapeutic use; Antirheumatic Agents; therapeutic use; Arthritis, Rheumatoid; drug therapy; genetics; Drug Therapy, Combination; Female; Genotype; HLA-DR Antigens; genetics; Haplotypes; Humans; Male; Methotrexate; therapeutic use; Middle Aged; Polymorphism, Single Nucleotide; Treatment Outcome; Tumor Necrosis Factor-alpha; genetics
Keywords: TNF-alpha, adalimumab, rheumatoid arthritis, genetic polymorphism, haplotype
The last ten years have seen a better understanding of the pathogenesis of rheumatoid arthritis (RA). Studies have led to the recognition of TNFα as one of the cornerstone cytokines involved in the synovial inflammatory process. Such results have provided the basis for the development of TNF blockers for treating RA. Three TNF blockers are currently used for RA treatment, one corresponding to a recombinant soluble form of TNF receptor, TNFRSF1B (etanercept), and two others corresponding an anti-TNFα monoclonal antibody (infliximab and adalimumab [ADA]). These TNF blockers act by inhibiting the binding of TNFs to TNF receptors on the cell surface and therefore interfere with TNF-driven signal transduction pathways. Etanercept binds to both TNFα and TNFβ (or lymphotoxine α [LTA]) and infliximab and adalimumab to TNFα only. TNF blockers are efficient in about 70% of patients, but 30% are resistant.
Taking into account the cost of these treatments in RA, the persisting questions about potential long-term adverse events (infections and cancers) and the availability of other efficient biotherapies, the identification of predictive factors of response to treatment is a key issue. In this field, pharmacogenetic approaches give promising hope. Nevertheless, few studies have been performed to date and some have led to contradictory results, especially those concerning the role of the shared epitope (SE) and/or TNF -308A/G polymorphism [1–12] (Table 1).
Table 1.
Pharmacogenetic studies on TNF blockers investigating TNF -308A/G polymorphism and HLA DRB1 shared epitope.
| Authors (Ref) | Evaluation criteria | Patients, N (Ethnicity) | TNF blocker (Disease) | TNF -308A/G | Shared Epitope |
|---|---|---|---|---|---|
| Padyukof et al. (1) | ACR20 and/or DAS28/M3 | 123 (Cau) | ETN (RA) | No association | ND |
| Criswell et al. (2) | ACR50/M12 | 151 (Cau) | ETN (RA) | No association | Associated with better response |
| Mascheretti et al. (3) | CD activity score/W4 | 90 + 444 (Cau) | IFX (CD) | No association | ND |
| Mugnier et al. (4) | DAS28/W22 | 59 (Cau) | IFX (RA) | GG better responders | ND |
| Fonseca et al. (8) | DAS28/M24 | 22 (Cau) | ETN (RA) | GG better responders | ND |
| Kang et al. (9) | ACR70 vs non ACR20/W12 | 70 (Asi) | ETN (RA) | No association | No association |
| Marotte et al. (11) | ACR20/W30 | 198 (Cau) | IFX (RA) | No association | No association |
| Seitz et al. (12) | DAS28/W24 | 54 (Cau) | ETN, IFX, ADA (RA, PsA, AS) | GG better responders | ND |
| Our study | ACR50/W12 | 380 (Cau) | ADA (RA) | No association | No association |
Ref: references
M: month
W: week
Cau: Caucasian
Asi: Asian
CD: Crohn disease
PsA: Psoriatic arthritis
AS: ankylosing spondylitis
ETN: etanercept
IFX: infliximab
ADA: adalimumab
ND: not done
DAS: disease activity status
ACR20, ACR50, ACR70: 20%, 50%, 70% improvement according to the American College of Rheumatology (ACR) criteria
RA: rheumatoid arthritis
The aim of this study was to determine whether the SE and/or TNFα gene polymorphisms, analyzed separately or after haplotype reconstruction, are genetic predictors of response to ADA in patients with RA.
METHODS
Patients
This pharmacogenetic study was ancillary to the Research in Active Rheumatoid Arthritis (ReAct) protocol performed at various sites in Europe and Australia. In the parent ReAct study, 6610 patients were included to assess the safety and effectiveness of ADA, a fully human IgG1 anti-TNF monoclonal antibody. The objectives of the ReAct study were to evaluate patients for efficacy and tolerance of ADA in combination with a variety of disease-modifying antirheumatic drugs (DMARDs), including those previously treated with etanercept or infliximab. Briefly, patients enrolled in the ReAct study were men and women ≥ 18 years of age with active, adult-onset RA as defined by the 1987 revised criteria of the American College of Rheumatology (ACR) [13]. Inclusion criteria required a disease duration ≥ 3 months; a disease activity score based on erythrocyte sedimentation rate and an evaluation of 28 joints (DAS28) [14] ≥3.2, indicating at least moderate disease activity; and failure of treatment with at least 1 traditional DMARD. Previous therapy with biologic response modifiers including other TNF antagonists was allowed if the medication was discontinued at least 2 months before enrolment.
The pharmacogenetic study we describe here concerns a large cohort of 398 French patients included in the ReAct study. All the patients provided written informed consent. The study was approved by the local ethics committee. Eight patients were excluded owing to their Asian or African descent (Figure 1). Ten other patients were excluded from the main statistical analysis because of missing ACR50 response data at 12 weeks. Thus, 380 patients from the original population were eligible for the study and underwent treatment with ADA plus MTX (n=182), ADA plus any other DMARD (n=96) or ADA alone (n=102).
Figure 1.
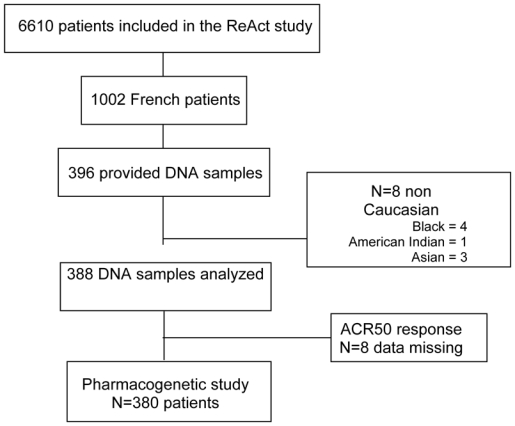
Flow chart of patients through the study.
Collection of clinical and biological data and outcome measures
The clinical and biological data were from the original ReAct protocol. At baseline and weeks 2, 6, and 12, scores for variables necessary to assess DAS 28 and ACR response were recorded, as were scores on the Health Assessment Questionnaire-Disability Index (HAQ). Outcomes were measured after 12 weeks of treatment. The primary outcome for the genetic study was ACR50 response, secondary outcomes were ACR20 and ACR70 responses.
Genetic polymorphisms in TNF and HLA genes
We chose to analyze the TNF gene polymorphisms on the basis of a previous report showing four main haplotypes consisting of TNF +488, −238 and −308 single-nucleotide polymorphisms (SNPs) among a Caucasian population [15]. However, TNF+488 has been reported to have strong linkage disequilibrium (LD) with TNF-857 (LD value, Somer’s D=0.92 in a Caucasian population [16]). Moreover, TNF -857 was recently reported to influence clinical response to etanercept [9], so we chose to genotype TNF-857 instead of TNF+488. The three studied SNPs encompasse the entire TNF promoter locus, leading hence, as expected, to obtain the four main haplotypes cited below in our Caucasian population of RA patients.
Because definite HLA-DRB1 alleles have been previously reported to play an important role in RA susceptibility -- the SE hypothesis [17] -- and severity [18], we genotyped our RA patients for HLA-DRB1 alleles by direct sequencing. The alleles considered to have the SE were HLA-DRB1*0101, *0102, *0401, *0404, *0405, *0408, *0413, *1001, and *1402 [17]. To analyze the SE contribution in response to ADA treatment, patients were classified as having 0, 1 or 2 copies of the SE, or as being SE carriers or not. Extended haplotypes comprising HLA-DRB1 alleles and TNF SNPs were reconstructed as well.
Genotyping methods
Patients were genotyped for HLA-DRB1 and 3 TNFα gene polymorphisms (-857C/T, -308A/G and -238A/G). HLA-DRB1 alleles were determined by polymerase chain reaction (PCR) amplification and DNA sequencing by use of an ABI 3700 sequencer (PE Applied Biosystems, Courtaboeuf, France).
TNF –238A/G PCR gene polymorphisms was genotyped by mismatch polymerase chain reaction (PCR) - restriction fragment full length polymorphism (RFLP) using the MspI restriction enzyme. Primers used for PCR amplification were: forward 5′ATCTGGAGGAAGCGGTAGTG3′ and reverse 5′AGAAGACCCCCCTCGGAACC3′. Forward primer contained a purposeful 3′ mismatch, so that when incorporated into the PCR products they create a MspI restriction site with the G allele but not with the A allele.
TNF -308A/G polymorphism was genotyped by allelic discriminating TaqMan PCR by use of the PreDeveloped TaqMan assay kit C_7514879 (PE Applied Biosystems). Amplifications involved use of a 7900HT Applied Biosystems real-time thermal cycler (PE Applied Biosystems).
TNFα-857C/T genotyping was performed by a TaqMan® 5′-nuclease assay (PE Applied Biosystems) with allele-specific fluorogenic oligonucleotide probes (5′CCCTGTCTTCATTAAG and 5′CCCTGTCTTCGTTAAG) using an ABI 7000 sequence detection system (PE Applied Biosysytem).
Statistical analysis
All quantitative data are expressed as the mean +/− SD and all qualitative data as frequencies and percentages. All genotyped SNPs were in Hardy-Weinberg equilibrium. Within the TNF gene, a measure of the LD between the different SNPs was estimated using Somer’s D′. Since the LD was significantly different among all TNF SNPs and between TNF SNPs and HLA-DRB1 alleles, we also considered the haplotypes for TNF and extended haplotypes comprising HLA DRB1 alleles and TNF. We used the software PHASE (version 2.1) to perform haplotype reconstructions. This Bayesian algorithm provides the most-likely pairs of haplotypes carried by each subject [19, 20]. The average probability of PHASE certainty in haplotype inference was 99% for TNF haplotypes and 83% for SE-TNF extended haplotypes.
For each gene, genotypes and haplotypes were tested for association with ACR50 response to ADA at week 12. Differences in genotype distribution for efficacy were tested using contingency tables (3 × 2 crosstabs for each genotype and 2 × 2 crosstabs for each possible combination of homozygote and heterozygote genotypes) with the 2-sided chi-square test. A Bonferroni correction was applied for multiple comparisons. Both adjusted and unadjusted P values are presented. P values less than 0.05 were considered significant.
The factors influencing the probability of achieving an ACR50 response at week 12 were studied through multivariate logistic regression. In addition to the SNPs and to the haplotypes described above, we considered the following covariates: gender, age, weight, clinical variables measured at baseline, duration of disease, presence of rheumatoid factor (RF) and concomitant treatment. First, univariate logistic regressions were performed to screen covariates. The models with and without a given covariate were compared using a log-likelihood ratio test, that is, by comparing twice the difference in log-likelihoods of the two model to the critical values of the χ2 statistic. We kept all the covariates for which the p-value of this test was less than 0.15. A multivariate regression model was then built including all candidate covariates selected in the previous step, and the final model was selected through backward selection, using log-likelihood ratio tests as previously. At this stage, covariates were kept in the model if the p-value of the test was less than 0.05.
RESULTS
Description of the cohort
The baseline characteristics of patients are presented in Table 2. The profile of clinical response of the 380 patients included in this pharmacogenetic study was the same as that of the entire ReAct population (6610 patients). At 12 weeks, 40% of the patients (n=151) showed an ACR50 response, 71% (n=263) an ACR20 response and 16% (n=59) an ACR70 response. MTX adjunction to ADA therapy was significantly associated with a better ACR50 response both in univariate (P=0.005) and multivariate analyses (odds ratio [OR], 1.68; 95% confidence interval [CI], 1.07–2.66; P=0.026). In the subgroup of patients not receiving methotrexate, there was a non significant trend for a better response when other DMARDs were administered in addition to ADA (38% of ACR50 responders versus 29% in patients given only ADA, NS).
Table 2.
Baseline characteristics of 381 genotyped patients and comparisons for the main RA characteristics between patients homozygous for the TNF GGC haplotype and the other haplotypes carriers.
| Characteristics | Baseline value All patients (N=380) | TNF haplotype * GGC homozygous (N=189) | Others TNF haplotypes (N=164) | P-value* (* GGC homozygous versus other) |
|---|---|---|---|---|
| Sex, % female/% male | 78.2/21.8 | 79.4/20.6 | 76.8/23.2 | 0.66 |
| Age, mean +/− SD years | 54.0 +/− 11.2 | 54.5 +/− 11.4 | 53.4 +/− 11.1 | 0.34 |
| Disease duration, mean weeks | 140.7 | 142.7 | 139.3 | 0.83 |
| MDAS, mean +/− SD (range) | 5.9 +/− 1.0 (3.2 – 8.9) | 5.9+/− 1.0 (3.2 – 8.9) | 5.9+/− 1.0 (3.2 – 8.3) | 0.87 |
| No. of tender joints (1–28) +/− SD | 13.2 +/− 6.5 | 13.2 +/− 6.5 | 13.3 +/− 6.4 | 0.93 |
| No. of swollen joints (1–28) +/− SD | 9.8 +/− 5.0 | 9.6+/− 5.2 | 10.3+/− 4.8 | 0.09 |
| RF positivity, % | 70.9 | 71.3 | 70.8 | 0.99 |
| CRP, mean mg/liter (range) | 26.5 (3.5 – 167.0) | 25.9 (3.5 – 146.0) | 28.3 (3.5 – 167.0) | 0.34 |
| ESR, mean mm/hour (range) | 31.6 (2.0 – 98.0) | 32.2 (2.0 – 95.0) | 30.6 (2.0 – 90.0) | 0.50 |
| ADA + MTX +,% | 47.9 | 43.4 | 51.2 | |
| ADA + other DMARDs | 25.3 | 27.0 | 25.0 | |
| ADA alone | 26.8 | 29.6 | 23.8 |
ADA= adalimumab
MDAS = Modified Disease Activity Score based on erythrocyte sedimentation rate and an evaluation of 28 joints
ESR = erythrocyte sedimentation rate
RF = rheumatoid factor
CRP = C-reactive protein
MTX = methotrexate treatment associated with
DMARD=disease-modifying antirheumatic drug
TNF haplotype (-238G, -308G and -857C
Genotype distributions were as follows: for TNF-238 G>A (N=364), 94% GG, 5% AG, and one patient had the rare AA genotype; for TNF-308 G>A (N=373), 71% GG, 26% AG, and 3% had the AA genotype; for TNF-857 C>T (N=361), 82.7% CC, 17% CT, and one patient had the rare TT genotype. These distributions were consistent with data from public databases for Caucasians (http://www.ncbi.nlm.nih.gov/projects/SNP).
SE could be determined in 326 patients. The distribution of the SE among patients was as follows: 25.4% had no copies, 47.6% 1 copy, and 27% 2 copies, for a total of 74.6% carriers.
In 346 patients in whom we determined the 3 SNPs of the TNFα promoter (-238A/G, -308A/G and -857C/T), we identified four main haplotypes (e.g., the haplotype GGC consisted of -238G, -308G and -857C). These most frequent haplotypes (GGC, GAC, GGT and AGC) accounted for more than 99% of the total number of haplotypes, with frequencies of occurrence of 74%, 14%, 8% and 3%, respectively. The rare AAC haplotype was found in only one patient.
Univariate and multivariate analysis of factors influencing ACR50 response to ADA
We used binary logistic regression to study the probability of achieving the ACR50 response to ADA therapy at 12 weeks. Univariate regression to screen for variables (with P < 0.15) to enter in the model in the second step revealed weight, body mass index (BMI), height, associated therapy with MTX, associated therapy with other DMARDs, presence of rheumatoid factor (RF), TNF polymorphisms −857 and −238, TNF haplotype (-238G, -308G and -857C), number of swollen joints and HAQ score at baseline. These covariates were retained as candidate covariates to build a multivariate model.
In the multivariate analysis, we found the following variables associated with ACR50 response: associated therapy with MTX (OR, 1.68; 95% CI, 1.07–2.66; P=0.026), presence of RF (OR, 1.91; 95% CI, 1.13–3.21; P=0.015), BMI (OR, 1.06; 95% CI, 1.01–1.11; P=0.01), baseline HAQ score (OR, 0.55; 95% CI, 0.37–0.82; P=0.003), and presence of TNF haplotypes other than the GGC homozygous haplotype (OR, 1.92; 95% CI, 1.21–3.02; P=0.005).
Influence of the SE and individual TNFα genotypes on ADA response
The SE was not identified in uni- or multivariate analysis as a factor influencing ACR50 response to ADA therapy at week 12. In fact, we found no association between ACR50 response to ADA and SE copy number or carrier status (Figure 2A and 2B) or any of the 3 TNFα gene polymorphisms -238A/G, -308A/G and -857C/T genotypes (Figure 2C, 2D and 2E) tested individually. However, we found a trend of an association between a lower response to ADA therapy at 12 weeks and -238GG, -308GG and -857CC genotypes (Figure 2C, 2D and 2E).
Figure 2.
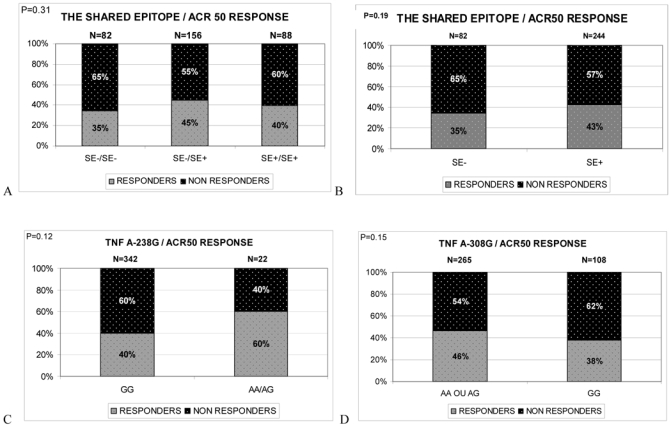
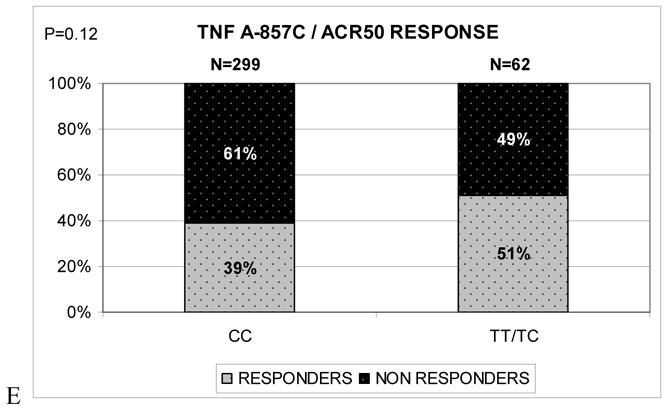
Genotype frequencies of TNFA gene polymorphisms and HLA DRB1 shared epitope (SE) among subjects with 50% response to ADA therapy according to the American College of Rheumatology (ACR) criteria (ACR50) (responders) and non responders at week 12 after treatment initiation. A. Shared epitope copy numbers (0, 1 or 2). B. Shared epitope carrier status. C. TNF -238 A/G genotypes; D. TNF -308 A/G genotypes; E. TNF -857 C/T genotypes
Influence of the TNFα haplotype on ADA response
In analyzing the influence of TNFα haplotype on response to ADA therapy, GGC haplotype carrier status showed significant results (Figure 3A). In the first analysis, we discarded rare haplotype combinations (N=27) represented fewer than 10 times: AGC/AAC (N=1), GAC/AGC (N=2), GAC/GAC (N=9), GGT/AGC (N=3), GGT/GAC (N=9), GGT/GGT (N=3). For subjects with the remaining four main haplotype combinations, those homozygous for the GGC haplotype (N=184) had a significantly lower ACR50 response rate (34%) than subjects with the three other main combined haplotypes: GGC/GAC (47%; N=77), GGC/GGT (53%; N=45), and GGC/AGC (71%; N=14) (P=0.0041, Pc=0.02) (Figure 3A). This observation highly suggested a recessive effect of the GGC haplotype on response to treatment. In fact, the response rate for homozygous GGC haplotype carriers (34%; N=184) was significantly lower than the response rate observed when pooling all other haplotype combinations, including the rare ones (50%; N=162) (P=0.003, after Bonferroni correction Pc=0.015) (Figure 3B).
Figure 3.
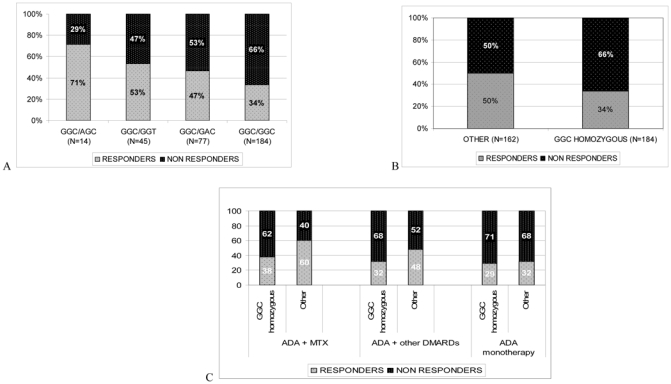
Haplotype combinations frequencies among ADA ACR50 responders and non responders at week 12. A. Comparison of the four main haplotypes of the TNF locus. B. Comparison of the GGC homozygous haplotype with all the other haplotypes. C. Comparison of the GGC homozygous haplotype with all the other haplotypes by treatment with ADA plus methotrexate (MTX), ADA plus any other disease-modifying antirheumatic drug (DMARD) or ADA alone (monotherapy).
A similar but non significant trend was observed for ACR20 and ACR70 responses to ADA therapy at week 12: The GGC/GGC group showed a 69% ACR20 response versus 76% for the other haplotype carriers (P=0.14) and a 15% ACR70 response versus 19% for the other haplotype carriers (P=0.3). The lack of significant difference regarding ACR20 and ACR70 responses can be explained by a lack of statistical power since responders and non responders are unbalanced with these criteria. Conversely, ACR50 response at week 12 provides the best statistical power to demonstrate an effect with a distribution of responders and nonresponders approaching 50% of the whole population.
As well as our finding of the homozygous effect of the GGC haplotype, we noted a “trans” effect of certain haplotypes present on the opposite chromosome. Alone, the “trans” association of the AGC haplotype with the GGC haplotype appeared to be associated with a better response to ADA therapy: 71% for the GGC/AGC combination (Figure 3A) versus 40% for all the other haplotype combinations, including the rare ones. The difference between those groups was not significant after Bonferroni correction (P=0.019; Pc=0.095). Nevertheless, few patients were AGT haplotype carriers (10 ACR50 responders and 4 ACR50 nonresponders), so this analysis was therefore underpowered.
Baseline characteristics did not significantly differ between GGC homozygous patients and other haplotype carriers, especially in DAS28 RA disease-activity criteria (Table 2).
Since about half of our sample received MTX therapy with ADA, we further analyzed the GGC homozygosity effect according to treatment: ADA plus MTX, ADA plus any other DMARD, and ADA alone. Surprisingly, the lower ACR50 response rate was present mainly in the group of patients receiving ADA plus MTX (n=182): 38% ACR50 response versus 60% for the other haplotype carriers (P=0.006; Pc=0.03) (Figure 3C). For the ADA plus other DMARD group (n=96), we observed a similar, although not significant, trend: 32% ACR50 response versus 48% for the other haplotype carriers (Figure 3C). For the ADA-alone group (n=102), the GGC homozygous haplotype had no significant effect (29% versus 32% for the other haplotype carriers) (Figure 3C).
In analyzing the time-course evolution of ACR50 response among treatment groups, for patients receiving ADA plus MTX, the response differed between GGC haplotype homozygous and other haplotype carriers as soon as week 2 after treatment initiation and increased until week 12 (Figure 4A). For patients receiving ADA plus any other DMARD, the response showed a trend in favor of a difference between haplotype groups over time, which might have become significant with longer follow-up, which was not available in the ReAct protocol (Figure 4B). For patients receiving ADA alone, the response did not significantly differ between haplotype groups over time (Figure 4C).
Figure 4.
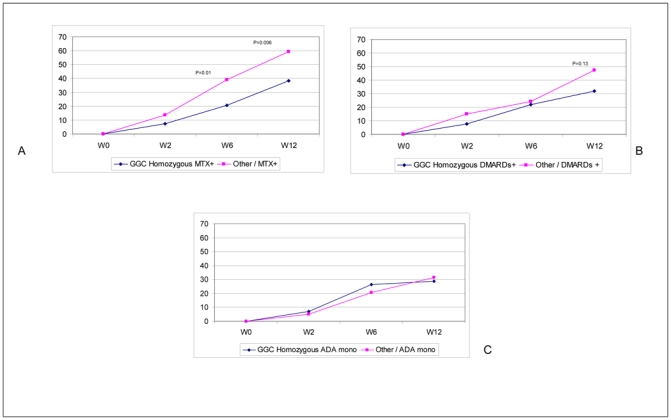
Time course of ACR50 response according to treatment and GGC homozygous haplotype carrier status. A. Patients treated with ADA plus methotrexate (MTX); B. Patients treated with ADA plus any other disease-modifying antirheumatic drug (DMARD); C. Patients treated with ADA alone; other=other than GGC homozygous haplotype; W=week.
Influence of the SE on the negative effect of GGC homozygosity on ADA response
The TNFα locus is located near HLA DRB1, so we analyzed the extent to which the GGC haplotype was in LD with the SE. Although some alleles belonging to the SE were in LD with the some TNF gene alleles, the overall LD between the GGC haplotype and the SE alleles was weak, with a Somers D′ equal to 0.24.
Our extended haplotype reconstructions resulted in 82 SE-TNF haplotype combinations. Careful examination of these haplotypes showed the GGC haplotype mainly associated with HLA DRB1*0101 and *0401 alleles of the SE, which is not surprising because GGC is the most frequent TNFα haplotype and DRB1*0101 and *0401 are the most frequent HLA DRB1 alleles among Caucasians. Most of these extended haplotypes were associated with an approximate 40% ACR50 response (range 36–42%), except for the 0701-GGC haplotype (n=29; 24% ACR50 response) and the 0405-GGC haplotype (n=23; 30% ACR50 response).
The rate of ACR50 responders among the GGC haplotype carriers was independent of the number of SE copies (40% with 2 SE copies; 41% with 0 or 1 SE copies). Thus, SE does not play a role in the lower response rate to ADA at week 12 observed in patients carrying the TNFα homozygous GGC haplotype.
DISCUSSION
This large pharmacogenetic study has investigated the effect of TNFα gene polymorphisms on response to ADA in RA patients. Although the presence of individual TNFα SNPs was not associated with a specific pattern of response to ADA, haplotype analysis provided convincing data suggesting that the ancestral haplotype of the TNFα promoter, GGC, the most frequent among Caucasians [15], was associated with a lower response to ADA when present in the homozygous form.
Pharmacogenetic studies of the influence of TNF genes on response to TNF blocker treatment in RA have given discordant results (Table 1). Two studies reported an association of the TNFA-308GG genotype with better response to infliximab [3] or etanercept [8] but concerned only a limited number of patients: 59 and 22, respectively. A recent study suggested that the TNFA -308GG genotype was associated with a better response to TNF blockers (ADA, infliximab or etanercept) at week 24, independent of rheumatic disease (i.e., RA, psoriatic arthritis or ankylosing spondylitis) [12]. Again, this study involved a limited number of patients (N=88), of whom only 54 had RA. Studies involving larger sample sizes [N=151 with etanercept (2) and N=198 with infliximab (11)] did not confirm these results, nor did ours, with 381 patients receiving ADA therapy. A study of 70 Asian patients did not find any association of TNF-308A/G with response to etanercept [9].
Many explanations may account for such discrepancies, but the small number (less than 80 patients) of subjects in the positive studies is the best probable explanation, since the 3 negative studies each included more than 150 patients. Moreover, parameters such as the response criteria chosen as primary endpoint (DAS28 decrease, ACR20 or ACR50 response) and the delay of efficacy for the primary endpoint (range from 6 to 24 months from baseline) differed among studies [2, 4, 7, 8, 12].
Interestingly, few of these studies focused on TNF haplotypes as a factor that could influence response to TNF blockers. One study analyzed extended haplotypes comprising HLA DRB1 alleles, TNFα 488/-238/-308 polymorphisms and lymphotoxin α 720/365/249 polymorphisms. This study involved a US Caucasian population and reported an association of 2 extended haplotypes with better response to etanercept in RA [2]. Unfortunately, the authors did not analyze the effect of TNFα polymorphism haplotypes alone on response to etanercept, which precluded any comparison with our results. Another study analyzing the effect of TNFα haplotypes on response to TNF blockers involved an Asian population [9]. The different ethnic origin of the patients in this last report limits a comparison with our results. Nevertheless, extended haplotypes were inferred from five TNFα promoter polymorphisms (−1031, −863, −857, −308, −238). The frequency of haplotypes carrying -238G, -308G, and -857C alleles was 71% in the Asian population, very close to the 73% observed in our Caucasian population, which suggests a similar frequency of the TNF (-238G, -308G, and -857C) haplotype in both Caucasian and Asian populations. A TNF haplotypic effect on etanercept response was not evidenced in this study, but no study has investigated a homozygous effect of the main haplotypes with this treatment. Interestingly, patients homozygous for the -857C-allele were poor responders to etanercept. In fact, when ACR20 nonresponders were compared with ACR70 responders, the proportion of responders was higher in the TT-CT group than in the CC group (OR, 12; 95% CI, 1.2–120; P=0.033). The result was not significant after Bonferroni correction, but the sample size studied was low (N=70 patients). The result mirrored in part the effect of the (-238G, -308G, and -857C) homozygous haplotype associated with the lower response to ADA we observed.
The functional effect of TNFα promoter haplotypes has not been reported to date. In fact, most of the functional studies of the TNFα promoter focused on separate analysis of SNPs and mainly on the -308A/G polymorphism. Most published studies reported high TNFα production, among various cell types and stimulation conditions, in the A allele carriers [21–24]. These results, even if not confirmed by others [25–27], are mainly accepted by the scientific community. We found that the TNF (-238G, -308G, and -857C) homozygous haplotype was associated with a lower response to ADA therapy than were other haplotypes. In agreement with a high TNFα production with presence of -308A alleles, poorer responders might be those who produce low levels of TNFα. In fact, recent studies suggest that subjects with high response to TNF blockers could also have high TNFα bioactivity [28] or high TNFα synovial levels at baseline [29].
The effect of the TNF (-238G, -308G, and -857C) homozygous haplotype on ADA response was restricted to patients also receiving MTX, with a trend to association with ADA plus any other DMARD treatment, and was absent in patients receiving ADA alone. The reason for such observation remains unclear. A specific effect of TNF promoter polymorphisms on response to MTX itself cannot be excluded. This point has never been addressed before in the literature. MTX plus ADA provided significantly higher response than ADA alone. Such results are markedly enhanced in GGC haplotype homozygous carriers and could help physicians in treatment management. In fact, the necessity for MTX treatment with ADA therapy appears critical in GGC haplotype homozygous carriers, to reach a good ACR50 response rate at week 12.
Discrepancies among published results involve the implication of the SE in response to TNF blockers in RA (Table 1). One study reported increased response to etancercept in patients with 2 copies of the SE (OR, 4.3; 95% CI, 1.8–10.3) (N=151) [2] and a second did not replicate such results on a larger studied population (N=198) [11]. Regardless, our results among 322 patients do not show any effect of SE copy number on response to ADA.
In conclusion, this pharmacogenetic study is remarkable because of the size of the population studied as well as the quality of the clinical data recorded within the ReAct study. It provides robust data indicating that a single TNFα locus haplotype (-238G/-308G/-857C), when carried on both chromosomes, is associated with a lower response to ADA in RA patients treated with a combination of ADA and MTX than patients with other haplotypes. The mechanism of action of this genetic predisposition (a specific effect on response to MTX itself or an effect on TNFα bioactivity and/or TNFα serum levels) needs further investigations.
Acknowledgments
This work was promoted by the Club Rhumatismes et Inflammation with a grant from Abbott France.
We thank Axelle Belaube and Laura Contreras, Abbot France, for help in conduct of this pharmacogenomic study. We thank the following collaborators for their participation in DNA collection: Pr Pierre Bourgeois (Hôpital La Pitié-Salpétrière – Paris), Pr Alain Cantagrel (Hôpital Rangueil – Toulouse), Pr Bernard Combe (Hôpital Lapeyronie – Montpellier), Pr Thierry Schaeverbeke (Hôpital Pellegrin – Bordeaux), Pr Maxime Dougados (Hôpital Cochin – Paris), Dr Jean-Paul Eschard (Hôpital Sébastopol - Reims), Pr Liana Euller-Ziegler (Hôpital de L’Archet 1 – Nice), Pr Patrice Fardellone (Hôpital Nord - Amiens), Pr René-Marc Flipo (Hôpital B Roger Salengro - Lille), Pr Pascal Hilliquin (Centre Hospitalier Sud Francilien - Corbeil Essonnes), Pr Robert Juvin (Hôpital Albert Michallon – La Tronche), Pr André Kahan (Hôpital Cochin - Paris), Pr Pierre Lafforgue (Hôpital de la Conception – Marseille), Pr Christian Marcelli (Hôpital de la Côte de Nacre – Caen), Pr Isabelle Chary-Valckenaere (Hôpital du Brabois - Vandoeuvre les Nancy), Dr Xavier Puechal (Centre hospitalier – Le Mans), Pr Jean-Michel Ristori (Hôpital Gabriel Montpied – Clermont-Ferrand), Pr Jean Sibilia (Hôpital de Hautepierre – Strasbourg), Pr Philippe Goupille (Hôpital Trousseau – Tours), Pr Aleth Perdriger (Hôpital Sud Fontenoy – Rennes), Pr Eric Houvenagel (Centre hospitalier Saint-Philibert – Lomme), Pr Christian Alexandre (Hopital de Bellevue, Saint-Etienne), and Dr Alain Heraud (Centre hospitalier Robert Boulin – Libourne).
References
- 1.Padyukov L, Lampa J, Heimburger M, Ernestam S, Cederholm T, Lundkvist I, et al. Genetic markers for the efficacy of tumour necrosis factor blocking therapy in rheumatoid arthritis. Ann Rheum Dis. 2003 Jun;62(6):526–9. doi: 10.1136/ard.62.6.526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Criswell LA, Lum RF, Turner KN, Woehl B, Zhu Y, Wang J, et al. The influence of genetic variation in the HLA-DRB1 and LTA-TNF regions on the response to treatment of early rheumatoid arthritis with methotrexate or etanercept. Arthritis Rheum. 2004 Sep;50(9):2750–6. doi: 10.1002/art.20469. [DOI] [PubMed] [Google Scholar]
- 3.Mascheretti S, Hampe J, Kuhbacher T, Herfarth H, Krawczak M, Folsch UR, et al. Pharmacogenetic investigation of the TNF/TNF-receptor system in patients with chronic active Crohn’s disease treated with infliximab. Pharmacogenomics J. 2002;2(2):127–36. doi: 10.1038/sj.tpj.6500091. [DOI] [PubMed] [Google Scholar]
- 4.Mugnier B, Balandraud N, Darque A, Roudier C, Roudier J, Reviron D. Polymorphism at position -308 of the tumor necrosis factor alpha gene influences outcome of infliximab therapy in rheumatoid arthritis. Arthritis Rheum. 2003 Jul;48(7):1849–52. doi: 10.1002/art.11168. [DOI] [PubMed] [Google Scholar]
- 5.Cuchacovich M, Ferreira L, Aliste M, Soto L, Cuenca J, Cruzat A, et al. Tumour necrosis factor-alpha (TNF-alpha) levels and influence of -308 TNF-alpha promoter polymorphism on the responsiveness to infliximab in patients with rheumatoid arthritis. Scand J Rheumatol. 2004;33(4):228–32. doi: 10.1080/03009740410005863. [DOI] [PubMed] [Google Scholar]
- 6.Martinez A, Salido M, Bonilla G, Pascual-Salcedo D, Fernandez-Arquero M, de Miguel S, et al. Association of the major histocompatibility complex with response to infliximab therapy in rheumatoid arthritis patients. Arthritis Rheum. 2004 Apr;50(4):1077–82. doi: 10.1002/art.20154. [DOI] [PubMed] [Google Scholar]
- 7.Mugnier B, Roudier J. Tumor necrosis factor alpha haplotypes versus tumor necrosis factor alpha -308 G/A polymorphism in the prediction of infliximab treatment efficacy in rheumatoid arthritis. Arthritis Rheum. 2004 Dec;50(12):4075–6. doi: 10.1002/art.20801. author reply 6–7. [DOI] [PubMed] [Google Scholar]
- 8.Fonseca JE, Carvalho T, Cruz M, Nero P, Sobral M, Mourao AF, et al. Polymorphism at position -308 of the tumour necrosis factor alpha gene and rheumatoid arthritis pharmacogenetics. Ann Rheum Dis. 2005 May;64(5):793–4. doi: 10.1136/ard.2004.028167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kang CP, Lee KW, Yoo DH, Kang C, Bae SC. The influence of a polymorphism at position -857 of the tumour necrosis factor alpha gene on clinical response to etanercept therapy in rheumatoid arthritis. Rheumatology (Oxford) 2005 Apr;44(4):547–52. doi: 10.1093/rheumatology/keh550. [DOI] [PubMed] [Google Scholar]
- 10.Lee YH, Rho YH, Choi SJ, Ji JD, Song GG. Association of TNF-alpha -308 G/A polymorphism with responsiveness to TNF-alpha-blockers in rheumatoid arthritis: a meta-analysis. Rheumatol Int. 2006 Aug 15; doi: 10.1007/s00296-006-0175-7. [DOI] [PubMed] [Google Scholar]
- 11.Marotte H, Pallot-Prades B, Grange L, Tebib J, Gaudin P, Alexandre C, et al. The shared epitope is a marker of severity associated with selection for, but not with response to, infliximab in a large rheumatoid arthritis population. Ann Rheum Dis. 2006 Mar;65(3):342–7. doi: 10.1136/ard.2005.037150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Seitz M, Wirthmuller U, Moller B, Villiger PM. The -308 tumour necrosis factor-{alpha} gene polymorphism predicts therapeutic response to TNF{alpha}-blockers in rheumatoid arthritis and spondyloarthritis patients. Rheumatology (Oxford) 2007 Jan;46(1):93–6. doi: 10.1093/rheumatology/kel175. [DOI] [PubMed] [Google Scholar]
- 13.Arnett FC, Edworthy SM, Bloch DA, McShane DJ, Fries JF, Cooper NS, et al. The American Rheumatism Association 1987 revised criteria for the classification of rheumatoid arthritis. Arthritis Rheum. 1988 Mar;31(3):315–24. doi: 10.1002/art.1780310302. [DOI] [PubMed] [Google Scholar]
- 14.Prevoo ML, van’t Hof MA, Kuper HH, van Leeuwen MA, van de Putte LB, van Riel PL. Modified disease activity scores that include twenty-eight-joint counts. Development and validation in a prospective longitudinal study of patients with rheumatoid arthritis. Arthritis Rheum. 1995 Jan;38(1):44–8. doi: 10.1002/art.1780380107. [DOI] [PubMed] [Google Scholar]
- 15.Mullighan CG, Fanning GC, Chapel HM, Welsh KI. TNF and lymphotoxin-alpha polymorphisms associated with common variable immunodeficiency: role in the pathogenesis of granulomatous disease. J Immunol. 1997 Dec 15;159(12):6236–41. [PubMed] [Google Scholar]
- 16.Simmonds MJ, Heward JM, Howson JM, Foxall H, Nithiyananthan R, Franklyn JA, et al. A systematic approach to the assessment of known TNF-alpha polymorphisms in Graves’ disease. Genes Immun. 2004 Jun;5(4):267–73. doi: 10.1038/sj.gene.6364066. [DOI] [PubMed] [Google Scholar]
- 17.Gregersen PK, Silver J, Winchester RJ. The shared epitope hypothesis. An approach to understanding the molecular genetics of susceptibility to rheumatoid arthritis. Arthritis Rheum. 1987 Nov;30(11):1205–13. doi: 10.1002/art.1780301102. [DOI] [PubMed] [Google Scholar]
- 18.Gorman JD, Lum RF, Chen JJ, Suarez-Almazor ME, Thomson G, Criswell LA. Impact of shared epitope genotype and ethnicity on erosive disease: a meta-analysis of 3,240 rheumatoid arthritis patients. Arthritis Rheum. 2004 Feb;50(2):400–12. doi: 10.1002/art.20006. [DOI] [PubMed] [Google Scholar]
- 19.Stephens JC, Schneider JA, Tanguay DA, Choi J, Acharya T, Stanley SE, et al. Haplotype variation and linkage disequilibrium in 313 human genes. Science. 2001 Jul 20;293(5529):489–93. doi: 10.1126/science.1059431. [DOI] [PubMed] [Google Scholar]
- 20.Stephens M, Donnelly P. A comparison of bayesian methods for haplotype reconstruction from population genotype data. Am J Hum Genet. 2003 Nov;73(5):1162–9. doi: 10.1086/379378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bouma G, Crusius JB, Oudkerk Pool M, Kolkman JJ, von Blomberg BM, Kostense PJ, et al. Secretion of tumour necrosis factor alpha and lymphotoxin alpha in relation to polymorphisms in the TNF genes and HLA-DR alleles. Relevance for inflammatory bowel disease. Scand J Immunol. 1996 Apr;43(4):456–63. doi: 10.1046/j.1365-3083.1996.d01-65.x. [DOI] [PubMed] [Google Scholar]
- 22.Louis E, Franchimont D, Piron A, Gevaert Y, Schaaf-Lafontaine N, Roland S, et al. Tumour necrosis factor (TNF) gene polymorphism influences TNF-alpha production in lipopolysaccharide (LPS)-stimulated whole blood cell culture in healthy humans. Clin Exp Immunol. 1998 Sep;113(3):401–6. doi: 10.1046/j.1365-2249.1998.00662.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tang GJ, Huang SL, Yien HW, Chen WS, Chi CW, Wu CW, et al. Tumor necrosis factor gene polymorphism and septic shock in surgical infection. Crit Care Med. 2000 Aug;28(8):2733–6. doi: 10.1097/00003246-200008000-00008. [DOI] [PubMed] [Google Scholar]
- 24.Fernandes H, Koneru B, Fernandes N, Hameed M, Cohen MC, Raveche E, et al. Investigation of promoter polymorphisms in the tumor necrosis factor-alpha and interleukin-10 genes in liver transplant patients. Transplantation. 2002 Jun 27;73(12):1886–91. doi: 10.1097/00007890-200206270-00006. [DOI] [PubMed] [Google Scholar]
- 25.Huizinga TW, Westendorp RG, Bollen EL, Keijsers V, Brinkman BM, Langermans JA, et al. TNF-alpha promoter polymorphisms, production and susceptibility to multiple sclerosis in different groups of patients. J Neuroimmunol. 1997 Feb;72(2):149–53. doi: 10.1016/s0165-5728(96)00182-8. [DOI] [PubMed] [Google Scholar]
- 26.Kubota T, McNamara DM, Wang JJ, Trost M, McTiernan CF, Mann DL, et al. Effects of tumor necrosis factor gene polymorphisms on patients with congestive heart failure. VEST Investigators for TNF Genotype Analysis. Vesnarinone Survival Trial. Circulation. 1998 Jun 30;97(25):2499–501. doi: 10.1161/01.cir.97.25.2499. [DOI] [PubMed] [Google Scholar]
- 27.de Jong BA, Westendorp RG, Bakker AM, Huizinga TW. Polymorphisms in or near tumour necrosis factor (TNF)-gene do not determine levels of endotoxin-induced TNF production. Genes Immun. 2002 Feb;3(1):25–9. doi: 10.1038/sj.gene.6363824. [DOI] [PubMed] [Google Scholar]
- 28.Marotte H, Maslinski W, Miossec P. Circulating tumour necrosis factor-alpha bioactivity in rheumatoid arthritis patients treated with infliximab: link to clinical response. Arthritis Res Ther. 2005;7(1):R149–55. doi: 10.1186/ar1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wijbrandts CA, Dijkgraaf MG, Kraan MC, Dinant H, Vos K, Lems WF, Wolbink GJ, Sijpkens D, Dijkmans AC, Tak PP. The Response To TNF-alpha Blockade In Rheumatoid Arthritis Is In Part Dependent On TNF-alpha Expression In The Synovium. Arthritis Rheum. 2006;54(9) doi: 10.1136/ard.2007.080440. suppl. L2. [DOI] [PMC free article] [PubMed] [Google Scholar]