

Abstract
The identification and characterization of drugs for the treatment of cognitive disorders has been hampered by the absence of comprehensive hypotheses. Such hypotheses consist of (a) a precisely defined cognitive operation that fundamentally underlies a range of cognitive abilities and capacities and, if impaired, contributes to the manifestation of diverse cognitive symptoms; (b) defined neuronal mechanisms proposed to mediate the cognitive operation of interest; (c) evidence indicating that the putative cognition enhancer facilitates these neuronal mechanisms; (d) and evidence indicating that the cognition enhancer facilitates cognitive performance by modulating these underlying neuronal mechanisms. The evidence on the neuronal and attentional effects of nAChR agonists, specifically agonists selective for α4β2* nAChRs, has begun to support such a hypothesis. nAChR agonists facilitate the detection of signals by augmenting the transient increases in prefrontal cholinergic activity that are necessary for a signal to gain control over behavior in attentional contexts. The prefrontal microcircuitry mediating these effects include α4β2* nAChRs situated on the terminals of thalamic inputs and the glutamatergic stimulation of cholinergic terminals via ionotropic glutamate receptors. Collectively, this evidence forms the basis for hypothesis-guided development and characterization of cognition enhancers.
Keywords: cognition enhancers, cue detection, schizophrenia, cognitive disorders
1. Introduction
Drug-induced improvement of the cognitive capacities of patients suffering from schizophrenia, age-related cognitive decline, neurodegenerative disorders, or even of healthy subjects, has remained an elusive goal. For example, the renewed focus on the role of the cognitive impairments of schizophrenic patients for their ability to function in real-life settings stands in sobering contrast to the limited success of preclinical and clinical psychopharmacological research to develop cognition enhancers for co-treatment of this disorder [1–5].
During the last two decades a relatively large number of compounds have been suggested to enhance cognitive functions and capacities. However, clinical trials quite consistently failed to show robust efficacy of these compounds in healthy humans or patients. Our prior analysis of this situation blamed a research strategy that is characterized by a largely atheoretical collection of presumably beneficial cognitive effects. Moreover, preclinical research often employed a range of behavioral tests with only limited validity in terms of measuring the cognitive function(s) of interest. Likewise, the validity of animal models, such as the scopolamine model or the use of aged animals, in terms of reproducing clinically relevant cognitive impairments, was rarely clarified [6–8]. Tests that are well known for producing a high number of false-positives, such as testing anti-scopolamine effects on spontaneous behaviors, continue to be extensively used in this research (e.g., [6,7,9–12]). As an alternative to the collection of putatively beneficial effects, hypothesis-guided research would allow this field to break away from the rather non-productive reiteration of past research strategies. Such hypotheses are now emerging, specifically in the context of cognition enhancement produced by nicotinic acetylcholine receptor (nAChR) agonists (below).
Generally, hypothesis-guided approaches to research on drug-induced cognition enhancement may include the following components and goals:
- identification of the basic cognitive operation(s) that is (are) facilitated by a putative cognition enhancer
- determination of the behavioral and cognitive variables that reveal drug-induced enhancement of a defined cognitive operation
- determination of the neurobiological mechanisms that mediate the cognitive operation of interest
- demonstration that the putative cognition enhancer beneficially modulates these neurobiological mechanisms
- determination of the neuropharmacological mechanisms mediating the drug’s effects
- demonstration that in performing animals the administration of the putative cognition enhancer facilitates the processing of the target cognitive operation(s) by enhancing underlying neurobiological mechanisms.
The available evidence on the effects of nAChR agonists, particularly agonists acting at α4β2* nAChR, begins to support such a hypothesis. This hypothesis suggests that the beneficial cognitive effects of nAChR agonists can be attributed to the facilitation of the neuronal mechanisms that underlie cue detection (defined below), that cue detection is mediated by cue-evoked increases in cholinergic activity in the prefrontal cortex (PFC), that nAChR agonists augment such cholinergic transients, and that they do so via a local glutamatergic-cholinergic microcircuit. This hypothesis is consistent with the larger theory that pre-attentionally processed cues gain behavioral control via cholinergic modulation.
Importantly, this hypothesis may not explain the entire range of pro-cognitive effects that were demonstrated for nicotine and selective nAChR agonists. However, it explains how these compounds enhance fundamental attentional functions and attention-dependent cognitive processes including executive control capacities and learning and memory. In other words, the hypothesis described below is a reductionist approach that is capable of guiding future research efforts designed to develop even more efficacious treatments for cognitive impairments.
2. nAChR agonists enhance attention in healthy humans, patients and animals
Nicotine is likely one of the most extensively studied drugs in humans and its effects on cognitive abilities have been extensively reviewed (e.g., [13,14]). The following brief synopsis of this literature serves to reiterate the view that the beneficial cognitive effects of nicotine as well as the newer nAChR subtype-selective agonists are associated with, and may even be primarily attributed to, the enhancement of basic attentional operations. Furthermore, compared to selective nAChR agonists, the effects of nicotine generally are less robust and often more difficult to demonstrate - an issue that will require a neurobiological explanation (below).
In healthy, non-smoking or non-deprived smoking humans, acute or chronic administration of nicotine has been extensively demonstrated to facilitate attentional performance (e.g., [15–20]). Nicotine also benefits learning and memory performance, primarily by enhancing the attentional processing of items during encoding or rehearsal (e.g., [21–24]). However, the magnitude of the beneficial attentional effects of nicotine in healthy humans is limited and occasionally has failed to reach significance (e.g., [25,26]). The effects of nicotine were suggested to be larger when assessed by using laboratory tasks (such as the Stroop task) and less robust when tested using real-life tests involving multiple and competing demands on attentional performance [27]. These beneficial, albeit limited cognitive effects of nicotine in healthy humans (see also [28]) appear to generalize to effects in patients with impaired cognitive abilities, including patients with Alzheimer’s disease [29–35], schizophrenia [36,37] and ADHD [38–40].
Although the number of clinical studies on the efficacy of nAChR subtype-selective compounds has remained small, the beneficial cognitive effects of the α4β2* nAChR agonists ABT-418 [41], ispronicline [42] and ABT-089 [43], in patients with Alzheimer’s disease, age-associated memory impairment, or ADHD, respectively [44–46], appear more somewhat more robust than those of nicotine and, compared with the effects of nicotine, may manifest in interaction with lower demands on attentional effort (defined in [47]). As will be pointed out next, the greater efficacy of α4β2* nAChR agonists in humans is consistent with evidence from animal experiments.
Stolerman and colleagues extensively studied the effects of nicotine on the performance of rats in the 5-choice serial reaction time task. The demonstration of beneficial effects of nicotine depends on the setting of task parameters such as the duration of the intertrial interval [48] and the presence of performance-impairing distractors [49]. As is the case in healthy humans, the magnitude of the enhancing effects of nicotine in intact animals remains relatively limited [50] and varies across studies over multiple, repeated tests and following repeated administration ([51,52]; but see [53,54]).
We employed an operant sustained attention task (SAT; see Figure 1C) that differs from the 5-choice serial reaction time task primarily by the integration of explicit non-signal trials, thereby generating not just hits and misses but also correct rejections and false alarms (each response based on an active lever press). As will be pointed out further below, the random sequence of signal and non-signal trials was found to be key to understanding the functions of the cortical cholinergic input system and the mechanisms via which nAChR agonists affect attentional performance. (For details concerning the operant procedures, measures of performance, a version of the task developed for research in humans, and validity in terms of measuring sustained attention performance, see [5,55]). Although the acute administration of nicotine did not consistently produce significant enhancement of the performance by intact animals ([56]; Kozak, Howe, & Sarter, unpublished data), administration of the α4β2* nAChR agonist ABT-418 robustly and dose-dependently (bell-shaped dose-response curve) increased the animals’ hit rate [57]. Importantly this enhancement manifested in animals performing the standard SAT and did not require interactions with the performance-lowering effects of a distractor. Furthermore, and relevant for the hypothesized cholinergic mediation of the attentional effects of nAChR agonists (below), the enhancement of attentional performance produced by ABT-418 required the presence of an intact cortical cholinergic input system [57].
Figure 1.
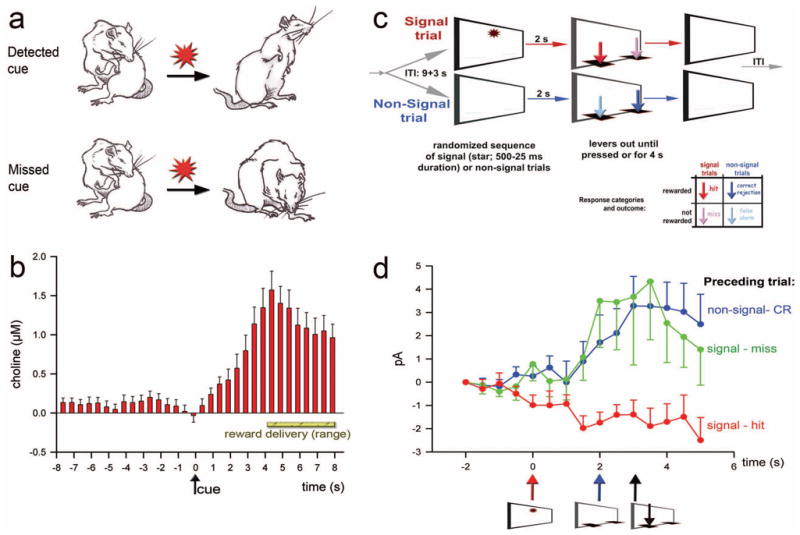
Prefrontal cholinergic transients mediating signal detection and attentional processing mode shifts. (a) Employing a cued appetitive response task, animals were trained to detect a rarely occurring cue that predicted reward delivery at one out of two reward ports (for details see [77]). Cue detection (as defined in the main text; [65]) was indicated by disengagement from ongoing behavior (typically grooming), orientation to and monitoring of the reward ports (see “detected cue”). As shown in (b) cues that were detected produced transient increases in cholinergic activity. No such transients were found in trials in which cues were missed. Importantly, reward was also delivered in such trials and thus animals eventually shifted from grooming behavior to port approach and reward retrieval. However, this was not associated with a cholinergic transient (for additional evidence and the effects of removal of prefrontal cholinergic inputs see [77]). (c) Main events of the sustained attention task (SAT). The task consists of a random sequence of signal and non-signal trials. Two seconds following an event the levers are extended, prompting the animal to report the presence or absence of a signal. Depending on the trial type, responses are classified as hits or misses and correct rejections or false alarms, respectively (see the color-coded response arrows in the outcome matrix). (d) Currents indicating ACh release in the prefrontal cortex during signal trials that yielded a hit (arrows indicate the time of signal presentation, lever extension, and correct lever press). Cholinergic transients were evoked by signals if the prior trial was a factual non-signal trial or a perceived non-signal trial (that is, a signal trial yielding a miss), but not if the prior trial also ended with a hit. Signals that resulted in misses never evoked transients, irrespective of the prior trial type (not shown; [78]) Collectively, these results indicate that prefrontal cholinergic transients mediate the shift from intrinsic or default-mode state-based activity to a state fostering signal detection. (a and b were reprinted from Neuron, Vol. 56, Parikh, V., Kozak, R., Martinez, V., Sarter, M., Prefrontal acetylcholine release controls cue detection on multiple time scales, p. 143 and 144. Copyright (2007) with permission from Elsevier.
Taken together, the evidence on the cognitive effects of nAChR agonists in humans and animals supports the view that these drugs act primarily by enhancing attentional processes and capacities, and that the effects of α4β2* nAChR agonists appear to be more robust than those of nicotine. The evidence concerning α7 nAChR agonists remains limited and conflicting and will not be addressed (e.g., [58–61]).
3. Transient increases in cholinergic activity mediate attentional performance
Following the validation of the immunotoxin 192-IgG-saporin as a tool for selectively removing the cortical cholinergic input system [62], several experiments demonstrated that such deafferentation selectively impairs the detection of signals in the SAT and similar tasks [63,64]. “Detection” is defined as a cognitive operation consisting of “…the entry of information concerning the presence of a signal into a system that allows the subject to report the existence of the signal by an arbitrary response indicated by the experimenter” [65]. Importantly, the animals’ ability to correctly reject non-signal events was unaffected (see Figure 1C for a schematic illustration of the task). Deafferentation-induced decreases in hits did not recover over several months of daily task practice. As will be discussed further below, the most recent evidence from electrochemical recordings of second-based changes of cholinergic activity in task-performing animals explains the selective effect of these cholinergic lesions on signal trial performance.
Studies using microdialysis to measure performance-associated ACh release in the cortex consistently demonstrated that attentional performance, as opposed to performing various operant control procedures that do not explicitly tax attentional processes, was associated with increases in frontal and parietal cortical ACh release. Furthermore, these studies suggested that performance-associated levels of ACh release do not indicate the level of attentional performance but rather the demands on attentional performance. Indeed, highest levels of performance-associated ACh release were observed as animals regained normal performance levels subsequent to behavioral or pharmacological performance challenges [66–71].
Because of their low temporal resolution (on the scale of minutes), measures of ACh release using microdialysis do not readily reveal the precise cognitive operations that are modulated or initiated by neurotransmitter release. However, the evidence from these studies confirmed the conclusions derived from the experiments on the attentional effects of cortical cholinergic denervation. Collectively, these studies demonstrated that cortical cholinergic activity, particularly in (right) prefrontal regions (see also [72–74]), mediates performance in tasks taxing attentional processes and resources. Furthermore, the contribution of the cholinergic system to attentional performance is associated specifically with the ability to detect signals.
Using enzyme-coated microelectrodes for the amperometric measurement of ACh release at a sub-second resolution [75,76] we demonstrated that the actual detection process is mediated via a transient increase in cholinergic activity in the medial PFC [77]. In our earlier studies we utilized a relatively simple cued appetitive response task in which the cue occurred rarely and predicted reward delivery at one out of two sites. As illustrated in Figure 1a, cue detection was defined based on cue-evoked behavior, while a miss was indicated by the failure of cues to evoke disengagement from ongoing behavior, typically grooming. Cues that elicited a transient increase in cholinergic activity were detected (Figure 1b). Conversely, cues that failed to evoke a cholinergic transient were missed. Importantly, in trials where cues were missed, reward was delivered, triggering port approach and reward delivery. However, reward-related behaviors were not associated with cholinergic transients. Based on these results and additional evidence described in Parikh et al., including the effects of prefrontal cholinergic deafferentation [77], we concluded that cue (or signal) detection requires a prefrontal cholinergic transient.
These findings also suggested that the striking increase in misses in SAT-performing animals that is caused by cholinergic deafferentation of the cortex is due to the lesion-induced absence of cholinergic transients. More recent evidence indicates an informative complication of this straight forward hypothesis [78]. As would be expected, in animals performing the SAT (Figure 1c), signals that fail to evoke cholinergic transients result in misses. However, unexpectedly, only about 70% of the signals that yielded hits evoked such cholinergic transients. The solution to this puzzle has been revealed by taking into account the preceding trial type. Signals resulting in hits evoke a cholinergic transient if the prior trial was a factual non-signal trial (a non-signal event resulting in a correct rejection) or a perceived non-signal trial (a signal trial that resulted in a miss). However, if the prior trial was a signal/hit then the subsequent signal does not evoke a cholinergic transient (Figure 1d). Collectively, these findings form the basis for a theoretical description of the cognitive operations that are mediated via cholinergic activity in the PFC.
4. Transient increases in cholinergic activity mediate cue detection: a target mechanism for cognition enhancers
The findings described above indicate that rather than mediating the actual process of detection, cholinergic transients support a processing mode shift. Non-signal trials do not require the detection of a signal and thus the execution of responses to non-signal events is dependent upon “intrinsic” or “associational” processing. In contrast, the process of detection requires that PFC circuitry elevates the processing of a pre-attentional representation of a signal to the level at which the signal is capable of influencing and even controlling ongoing cognitive and behavioral activity. The evidence suggests that a transient increase in cholinergic activity shifts the PFC network from mediating intrinsic processing, or from a processing state characterized by the absence of a specific task or demands on stimulus selection and stimulus processing (called the default-mode network; [79]), to a processing mode that fosters the integration of external signals into ongoing activity and thus detection (henceforth termed “up-shift”; see also [80]). To reiterate, if the network is already in the detection mode, as a result of a preceding hit, no additional cholinergic transient is required in order to generate a hit during the subsequent trial. Alternatively, if such a transient is required but does not occur, the response to the subsequent signal will be a miss. As deduced from this hypothesis, a post hoc analysis of the effects of cholinergic deafferentation on SAT performance indicated that the probability for a miss in (signal) trials that were preceded by non-signal/correct rejection trials is three times higher than the probability for a miss in trials preceded by a hit (Howe & Sarter, unpublished observations). In other words, the removal of cortical cholinergic inputs impairs attentional performance by reducing the probability for processing mode up-shifts.
The hypothesis that cholinergic transients permit upshifts and thus foster signal detection requires more comment. As the signal (or cue) evokes the cholinergic transient we need to propose an additional mechanism that, in the case of a miss, interferes with the cue-evoked orchestration of a cholinergic transient and thus fails to trigger an upshift. We found that misses were more likely if, prior to the signal, “background” cholinergic activity increased steadily [77]. Interestingly, human imaging data likewise indicated that pre-stimulus increases in the activity of the default network, perhaps reflecting a shift of attention toward intrinsic mentation, increased the likelihood for errors and attentional lapses [81–83].
Taken together, our evidence indicates the prefrontal cholinergic transients foster the shift from an intrinsic or default state of prefrontal neuronal activity to a mode that allows signals to be detected and to control behavior. Impairments in detection, reflecting failures to switch between default and detection networks or, in other words, to disengage from intrinsic processing, essentially contribute to, or even underlie, the cognitive impairments of schizophrenia, ADHD, aging, and other neuropsychiatric and neurodegenerative disorders (e.g., [84–89]). Thus, improving the probability for processing mode up-shifts is expected to directly enhance attentional performance and, benefit the overall cognitive status of patients. Mechanistically, drugs are hypothesized to enhance signal detection by augmenting the probability and/or magnitude of cue-evoked cholinergic transients. As will be described further below, the cognitive and cholinergic effects of nAChR agonists, particularly α4β2* nAChR agonists, are hypothesized to enhance cue detection by augmenting the amplitude of signal-evoked cholinergic transients.
5. Circuitry model for signal detection: prefrontal glutamatergic-cholinergic interactions
The following section describes major components of a parsimonious neuronal network model that explains how a preattentionally-processed cue evokes a cholinergic transient and thereby supports an up-shift. We will also address potential mechanisms responsible for misses and discuss the effects of nAChR agonists on this network further below.
The glutamatergic projections from the mediodorsal thalamus (MD) to the PFC [90] are not thought to “import” a primary sensory representation of the signal but rather a signal-evoked “attentional searchlight”. Attentional searchlight is a term that refers to a narrowing of the “place demanding attention” ([91]; see also [92]). As illustrated in Figure 2, the topographic projections from sensory regions to the thalamic reticular nucleus (TRN), which in turn contacts MD neurons, form essential components of the PFC afferent circuitry that “imports” pre-attentionally processed signals into the PFC [93–96]. The finding that attentional orienting is impaired following lesions of the TRN [97] is predicted by this scenario. The TRN receives cholinergic and non-cholinergic (not illustrated) inputs from the basal forebrain [98,99], thereby presumably enhancing the thalamic input in order to assist detection performance in the presence of distractors.
Figure 2.

Model describing the main components of the prefrontal cortex (PFC) circuitry mediating signal detection and processing mode up-shifts. The model combines well-documented evidence with parsimonious assumptions required to explain our electrochemical and performance data (see main text for details). The glutamatergic (GLU) inputs to the PFC, originating from the mediodorsal thalamic nucleus (MD) “import” a preattentionally processed representation of the signal into the PFC. MD neurons are part of a network that includes the thalamic reticular nucleus (TRN) and its topographic afferents from sensory cortical regions, and has been proposed to generate a signal-associated “attentional searchlight”, a term that refers to a pre-attentional narrowing of the “place demanding attention”. The signal-evoked glutamatergic transient in the cortex generates a cholinergic transient, via stimulation of ionotropic presynaptic glutamate receptors [100]. The cholinergic transient mediates the actual detection process or, depending on the task, a processing mode shift that fosters detection [77,78]. Prefrontal output neurons are presumed to be stimulated by ACh primarily via muscarinic (m)AChRs [129], thereby organizing the behavioral responses that indicate successful detection. The terminals of the MD inputs to the PFC are equipped with α4β2* nAChRs and nAChR agonists enhance detection performance and processing mode shifts primarily by positively modulating GLU release and thereby augmenting the amplitudes of the cholinergic transients that are essential for detection. While this glutamatergic-cholinergic interaction is sufficient to explain the presence of signal salience-dependent hit rates, it does not account for occasional misses of salient stimuli or the acute, detrimental effects of distractors and subsequent performance recovery. We speculate that as a result of, for example, task-unrelated increases in activity of this network prior to the presentation of signals, detection interference results from GABAergic inhibition of cholinergic terminals [105]. The cholinergic receptors situated on GABAergic interneurons are not clear but likely involve *β2* subunit containing nAChRs and also mAChRs [130,131]. These interneurons are also innervated by other cortical neurons (as symbolized by the connection from the neurons on top of the figure) and thus potentially allow increases in activity of the default-mode network to inhibit cholinergic terminals [132].
We know from our mechanistic studies that the generation of cholinergic transients in the medial PFC requires glutamate release and the stimulation of presynaptic ionotropic glutamate receptors that are presumably localized on cholinergic terminals [100]. Thus, consistent with the electrochemical recordings in performing animals (above), the neuronal representation of the pre-attentionally processed stimulus may evoke a cholinergic transient via such a glutamatergic-cholinergic interaction in the PFC, thereby allowing the stimulus to gain control over ongoing cognitive and behavioral activity and produce a hit response.
The signal duration-dependent hit rate in animals and humans performing the SAT task [55] may be explained by such a feed-forward mechanism, assuming that a more salient signal generates a greater glutamatergic transient and thus more likely a robust transient increase in cholinergic activity. However, explanations that would begin to indicate the mechanisms underlying misses of salient signals, the detection of signals in the absence of cholinergic transients as observed in signal trials preceded by hits (above), and the performance under distractor conditions, necessitate incorporation of additional pieces of evidence and assumptions.
As discussed above, our observation that increases in cholinergic activity prior to the presentation of a signal increase the likelihood for a miss may be related to the potentially interfering effects of increases in activity in the default-mode network (references above). It is not clear whether the activation of GABAergic interneurons [101] by multiple afferents, including inputs from other cortical regions [102], reflecting the departure of the network from the default-mode state, could interfere with signal-evoked recruitment of glutamatergic-cholinergic interactions (Figure 2). Indeed, the available evidence predicts that GABA release, likely from fast-spiking interneurons, silences cortical pyramidal as well as non-pyramidal cells (e.g., [103]) and inhibits ACh release from cholinergic terminals [104–106]. Although these effects would suffice to prevent the prefrontal circuit from up-shifting into the detection mode, activation of other neuromodulators, reflecting a departure from the default-mode state during the intertrial interval, also likely contributes to the suppression of cue-evoked glutamatergic-cholinergic activity [107]. Finally, we need to acknowledge that extremely little is known about the contribution of non-cholinergic afferents from the basal forebrain; these GABAergic and presumably glutamatergic projections may be recruited separately from the cholinergic system and may strongly influence the state of the detection-mediating circuitry in the PFC (e.g., [108,109]).
While this model explains the signal-duration-dependency of misses and inspires hypotheses concerning the mechanisms underlying failures to detect even rather salient stimuli, we can only speculate about the mechanisms that are responsible for the observation that during hit-hit sequences, the second signal does not require a cholinergic transient to be detected. Perhaps other afferent neuromodulators, particularly the noradrenergic input system, maintain the network in the detection mode as a result of a prior hit (e.g., [110–112]).
Finally, we know that performance challenges such as distractors increase levels of ACh release above those seen during standard SAT performance, and that such cholinergic activity broadly increases the activity of PFC neurons [66,113]. Again, the model illustrated in Figure 2 suggests that activation of GABAergic mechanisms may be involved in the effects of distractors on performance. Furthermore, based on our evidence on the mesolimbic regulation of the cholinergic inputs to the PFC [114–116], the model suggests that in response to erroneous performance, the prefrontal projections to mesolimbic regions, particularly the nucleus accumbens and the ventral tegmentum, both of which project to the basal forebrain, are capable of mobilizing the cholinergic system in response to the detection of errors and non-reward, in order to support residual performance and performance recovery [47]. However, we do not presently know how such mechanisms might affect the characteristics of cholinergic transients and/or the more tonic levels of cholinergic activity.
6. nAChR agonists evoked and modulate cholinergic transients
Administration of brief pulses of nAChR agonists into the medial PFC generates cholinergic transients that mirror, in terms of amplitude and decay rate, the transients observed in animals performing tasks involving detection processes [100]. Moreover, administration of α4β2* nAChR agonists evokes substantially sharper signals when compared with those evoked by nicotine (Figure 3). The slower decay rate of cholinergic transients evoked by nicotine appears to be due in part to the stimulation of α7 nAChR [100] and local dopamine release (Ward, Parikh, Sarter, unpublished data; see also [117]).
Figure 3.
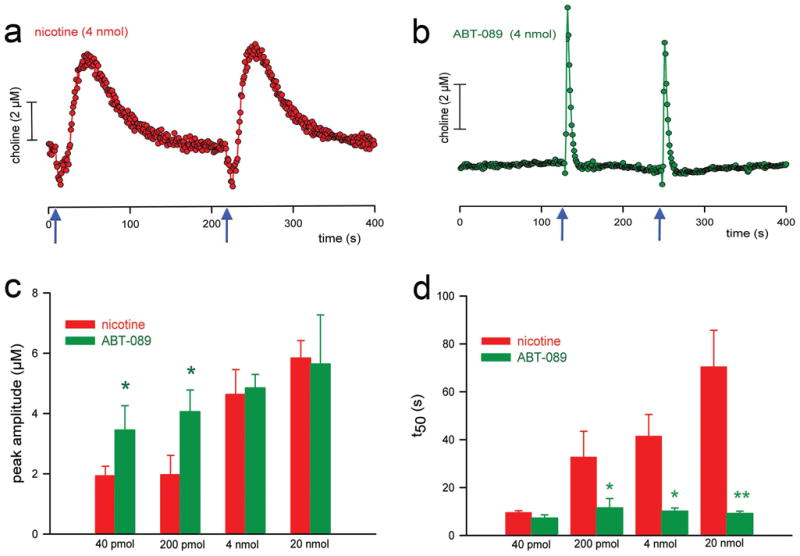
Cholinergic transients evoked by local, prefrontal application (pressure ejections) of nicotine and the α4β2* nAChR agonist ABT-089. (a, b) Self-referenced traces depicting cholinergic transients produced by two pressure ejections (see arrows on the abscissa for timepoints) of nicotine or ABT-089. Compared with the transients evoked by nicotine, ABT-089 was more potent with respect to the amplitudes of the cholinergic transients (c). Furthermore, decay rates of cholinergic transients produced by nicotine were strikingly higher and dose-dependent when compared with ABT-089 (for additional, mechanistic data see [100]). The “sharper” and larger signals of α4β2* nAChR agonists are hypothesized to underlie the more robust beneficial attentional effects of these compounds. (Reprinted from the Journal of Neuroscience, Parikh et al. 2008, with permission of the Journal of Neuroscience/Society for Neuroscience).
The mechanisms via which nAChR agonists, particularly α4β2* nAChR agonists, evoke cholinergic transients map well onto the circuitry model shown in Figure 2. α4β2* nAChRs are situated on the terminals of thalamic afferents [101,118]. α4β2* nAChR agonists evoke glutamatergic transients which, via presynaptic NMDA and AMPA receptor stimulation, generate cholinergic transients [100]. Blockade of α4β2* nAChRs attenuates nicotine-evoked glutamatergic and cholinergic transients [100]. Furthermore, local blockade of these ionotropic glutamate receptors prevents nAChR agonists from evoking cholinergic transients [100].
As discussed in the Introduction, when compared with nicotine, α4β2* nAChR agonists more robustly improve attentional performance of animals and humans. Given the key role of α4β2* nAChRs in the generation of cholinergic transients (Figure 2), we hypothesize that the greater potency, in terms of amplitude, of selective α4β2* nAChR agonist-evoked glutamatergic and cholinergic transients [100] underlies their greater cognitive efficacy (see also below). We also speculate that the slower decay rate of nicotine-evoked cholinergic transients may not support the detection process as optimally as the high-amplitude, “sharp” cholinergic transients evoked by selective α4β2* nAChR agonists [100]. However, this latter point remains to be demonstrated.
The effects of systemically administered nicotine on detection performance and the amplitude of simultaneously recorded cholinergic transients constitute the final piece of evidence in support of the present hypothesis. The evidence illustrated in Figure 4 indicates that nicotine-induced increases in detection rate and decreases in response times are associated with positively modulated cholinergic transients (Howe, Sarter, & Parikh, in preparation). Thus, in the presence of nicotine, the amplitudes of cholinergic transients are greater and therefore the detection process is more efficacious and faster (see also [119–122]). Forthcoming research will clarify whether the additional slowing of the decay rate of cholinergic transients produced by nicotine (Figure 4) contributes to the limitations of the beneficial effects of nicotine and, conversely, whether “sharper” transients, as produced by α4β2* nAChR agonists, more robustly enhance attentional performance.
Figure 4.

Effects of systemic administration of nicotine on the detection of cues in a cued appetitive response task and on cholinergic transients mediating cue detection [77,133]. The figure shows self-referenced traces indicating acetylcholine release in animals performing a cued appetitive response task (see Figure 1a), during trials involving cue detection (upper pair) and in which the cue was missed (lower pair). Administration of 0.4 mg/kg nicotine augmented the amplitude of detected cue-evoked cholinergic transients and also slowed the decay rate of these transients. Nicotine did not affect the flat traces recored during trials with missed cues. Nicotine increased the number of trials in which the cue was detected (see Figure 1) by 33%. Furthermore, the latency from cue detection to reward retrieval was reduced from 3.4±0.75 s (saline) to 2.4±0.8 s (nicotine).
7. Conclusions, gaps and generalizations
The ability to select and discriminate stimuli and to advance their processing so that they gain control over ongoing cognitive and behavioral activity represent fundamental components of attentional performance. Failures to detect and/or to shift out of the default-processing mode contribute to the decline of attentional performance and attention-dependent cognitive processes, including learning. Accumulating evidence indicates that prefrontal glutamatergic-cholinergic interactions represent an essential mechanism for detection and processing mode shifts. If cholinergic transients are not allowed to manifest, signals will be missed. Agonists at nAChR enhance the probability and amplitude of signal-evoked cholinergic transients and thereby facilitate attentional performance. Selective α4β2* nAChR agonists appear more efficacious, owing to the presence of α4β2* nAChRs on the terminals of thalamic inputs. Importantly, repeated stimulation of these receptors may lead to their up-regulation [123,124], perhaps explaining why several experiments found more robust beneficial cognitive effects of nicotine following chronic treatment [54].
Collectively, this evidence begins to form the basis for a hypothesis describing how nAChR agonists enhance cognition. This hypothesis consists of the following components. First, it describes a defined cognitive operation that is the target of enhancement (signal detection and associated processing mode up-shift). Second, the hypothesis postulates a neurobiological mechanism that mediates this operation (a transient increase in cholinergic activity). Third, it suggests that a group of compounds facilitate these operations (selective α4β2* nAChR agonists). Fourth, the hypothesis is supported by evidence indicating that these compounds enhance signal detection and attentional performance by augmenting cholinergic transients that mediate this cognitive operation (Figure 4).
Important aspects of this hypothesis remain unsettled, including the question of whether the slow decay rates of cholinergic signals evoked by nicotine are detrimental, limiting the beneficial effects of non-selective nAChR agonists. Furthermore, the potential presence and role of more tonic increases (based on minutes) in cholinergic activity remain unclear. Additional issues requiring research concern the mechanisms underlying misses of salient signals, the effects of distractors, and the maintenance of the detection mode during signal trials preceded by hits. Although these are important gaps, the available evidence appears sufficient to guide the development of nAChR agonists as cognition enhancers.
Given current research efforts concerning cortical α7 nAChRs, we need to reiterate that conclusive knowledge about the cognitive and neurobiological effects of these compounds presently remains too limited to generate hypotheses or integrate the effects of these compounds into the current model (Figure 2; but see [125]). At this point our evidence suggests primarily that the slower decay rate of cholinergic transients evoked by nicotine is largely due to stimulation of α7 nAChRs [100] but, again, the functional implications of this effect remain speculative.
The hypothesis that cholinergic transients mediate detection processes and processing mode shifts, and that nAChR agonists enhance these cognitive operations, is likely to generalize to other types of shifts between cognitive operations. For example, cortical cholinergic deafferentation also impairs the performance in a cross-modal divided attention task. The detrimental effects of the deafferentation were restricted to a component of the task in which the modality of the next stimulus was unpredictable while animals’ performance was unaffected during blocks of trials consisting of stimuli from just one modality [126]. These findings indicate that modality shifts in attentional contexts likewise require cholinergic activity and thus are potentially subject to nAChR-induced enhancement. Moreover, it will be of interest to determine whether task switching performance, using paradigms that have become productive instruments in research on cognitive aging (e.g., [127]), likewise benefit from stimulation of nAChRs [128].
Although beneficial attentional effects of nAChR agonists have been extensively documented, the specific cognitive operation and underlying neuronal mechanisms that are enhanced by these drugs are only now becoming clear. The hypothesis described above predicts that drugs that stimulate α4β2* nAChR in the prefrontal cortex activate a glutamatergic-cholinergic mechanism that enhances the detection of signals in attentional contexts. This hypothesis indicates neurobiological and behavioral measures that therefore can be employed to search for and characterize new nAChR ligands for the treatment of a wide range of cognitive disorders.
Acknowledgments
We thank Drs. Sarah Baran (University of Michigan) and Michael W. Decker (Abbott Laboratories) for comments on a draft of this manuscript. Our research was supported by PHS Grants MH080332 and MH080426. W.M.H. was supported by the PHS Training Grant T32 DA007267.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Barch DM, Carter CS, Arnsten A, Buchanan RW, Cohen JD, Geyer M, et al. Selecting Paradigms From Cognitive Neuroscience for Translation into Use in Clinical Trials: Proceedings of the Third CNTRICS Meeting. Schizophr Bull. 2009;35:109–14. doi: 10.1093/schbul/sbn163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Carter CS, Barch DM. Cognitive neuroscience-based approaches to measuring and improving treatment effects on cognition in schizophrenia: the CNTRICS initiative. Schizophr Bull. 2007;33:1131–37. doi: 10.1093/schbul/sbm081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hagan JJ, Jones DN. Predicting drug efficacy for cognitive deficits in schizophrenia. Schizophr Bull. 2005;31:830–53. doi: 10.1093/schbul/sbi058. [DOI] [PubMed] [Google Scholar]
- 4.Marder SR, Fenton W. Measurement and Treatment Research to Improve Cognition in Schizophrenia: NIMH MATRICS initiative to support the development of agents for improving cognition in schizophrenia. Schizophr Res. 2004;72:5–9. doi: 10.1016/j.schres.2004.09.010. [DOI] [PubMed] [Google Scholar]
- 5.Nuechterlein KH, Luck SJ, Lustig C, Sarter M. CNTRICS final task selection: control of attention. Schizophr Bull. 2009;35:182–96. doi: 10.1093/schbul/sbn158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sarter M, Hagan J, Dudchenko P. Behavioral screening for cognition enhancers: from indiscriminate to valid testing: Part I. Psychopharmacology (Berl) 1992;107:144–59. doi: 10.1007/BF02245132. [DOI] [PubMed] [Google Scholar]
- 7.Sarter M, Hagan J, Dudchenko P. Behavioral screening for cognition enhancers: from indiscriminate to valid testing: Part II. Psychopharmacology (Berl) 1992;107:461–73. doi: 10.1007/BF02245257. [DOI] [PubMed] [Google Scholar]
- 8.Sarter M, Bruno JP, Parikh V. Abnormal neurotransmitter release underlying behavioral and cognitive disorders: toward concepts of dynamic and function-specific dysregulation. Neuropsychopharmacology. 2007;32:1452–61. doi: 10.1038/sj.npp.1301285. [DOI] [PubMed] [Google Scholar]
- 9.Sarter M. Taking stock of cognition enhancers. Trends Pharmacol Sci. 1991;12:456–61. doi: 10.1016/0165-6147(91)90636-7. [DOI] [PubMed] [Google Scholar]
- 10.Sarter M. Preclinical research into cognition enhancers. Trends Pharmacol Sci. 2006;27:602–08. doi: 10.1016/j.tips.2006.09.004. [DOI] [PubMed] [Google Scholar]
- 11.Sarter M, Podell M. Preclinical psychopharmacology of AIDS-associated dementia: lessons to be learned from the cognitive psychopharmacology of other dementias. J Psychopharmacol. 2000;14:197–204. doi: 10.1177/026988110001400302. [DOI] [PubMed] [Google Scholar]
- 12.Woolley ML, Waters KA, Gartlon JE, Lacroix LP, Jennings C, Shaughnessy F, et al. Evaluation of the pro-cognitive effects of the AMPA receptor positive modulator, 5-(1-piperidinylcarbonyl)-2,1,3-benzoxadiazole (CX691), in the rat. Psychopharmacology (Berl) 2009;202:343–54. doi: 10.1007/s00213-008-1325-2. [DOI] [PubMed] [Google Scholar]
- 13.Levin ED. Nicotinic receptor subtypes and cognitive function. J Neurobiol. 2002;53:633–40. doi: 10.1002/neu.10151. [DOI] [PubMed] [Google Scholar]
- 14.Kassel JD. Smoking and attention: a review and reformulation of the stimulus-filter hypothesis. Clin Psychol Rev. 1997;17:451–78. doi: 10.1016/s0272-7358(97)00032-9. [DOI] [PubMed] [Google Scholar]
- 15.Warburton DM. Commentary on: “Effects of scopolamine and nicotine on human rapid information processing performance.” Psychopharmacology (1984) 82:147–150. Nicotine improves information processing: saying the unthinkable. Psychopharmacology (Berl) 2002;162:345–48. doi: 10.1007/s00213-002-1087-1. [DOI] [PubMed] [Google Scholar]
- 16.Mancuso G, Warburton DM, Melen M, Sherwood N, Tirelli E. Selective effects of nicotine on attentional processes. Psychopharmacology (Berl) 1999;146:199–204. doi: 10.1007/s002130051107. [DOI] [PubMed] [Google Scholar]
- 17.Le Houezec J, Halliday R, Benowitz NL, Callaway E, Naylor H, Herzig K. A low dose of subcutaneous nicotine improves information processing in non-smokers. Psychopharmacology (Berl) 1994;114:628–34. doi: 10.1007/BF02244994. [DOI] [PubMed] [Google Scholar]
- 18.Warburton DM, Mancuso G. Evaluation of the information processing and mood effects of a transdermal nicotine patch. Psychopharmacology (Berl) 1998;135:305–10. doi: 10.1007/s002130050514. [DOI] [PubMed] [Google Scholar]
- 19.Levin ED, Conners CK, Silva D, Hinton SC, Meck WH, March J, et al. Transdermal nicotine effects on attention. Psychopharmacology (Berl) 1998;140:135–41. doi: 10.1007/s002130050750. [DOI] [PubMed] [Google Scholar]
- 20.Newhouse PA, Potter A, Singh A. Effects of nicotinic stimulation on cognitive performance. Curr Opin Pharmacol. 2004;4:36–46. doi: 10.1016/j.coph.2003.11.001. [DOI] [PubMed] [Google Scholar]
- 21.Rusted JM, Sawyer R, Jones C, Trawley SL, Marchant NL. Positive effects of nicotine on cognition: the deployment of attention for prospective memory. Psychopharmacology (Berl) 2009;202:93–102. doi: 10.1007/s00213-008-1320-7. [DOI] [PubMed] [Google Scholar]
- 22.Marchant NL, Trawley S, Rusted JM. Prospective memory or prospective attention: physiological and pharmacological support for an attentional model. Int J Neuropsychopharmacol. 2008;11:401–11. doi: 10.1017/S146114570700819X. [DOI] [PubMed] [Google Scholar]
- 23.Rusted JM, Warburton DM. Facilitation of memory by post-trial administration of nicotine: evidence for an attentional explanation. Psychopharmacology (Berl) 1992;108:452–55. doi: 10.1007/BF02247420. [DOI] [PubMed] [Google Scholar]
- 24.Warburton DM, Skinner A, Martin CD. Improved incidental memory with nicotine after semantic processing, but not after phonological processing. Psychopharmacology (Berl) 2001;153:258–63. doi: 10.1007/s002130000565. [DOI] [PubMed] [Google Scholar]
- 25.Giessing C, Fink GR, Rosler F, Thiel CM. fMRI data predict individual differences of behavioral effects of nicotine: a partial least square analysis. J Cogn Neurosci. 2007;19:658–70. doi: 10.1162/jocn.2007.19.4.658. [DOI] [PubMed] [Google Scholar]
- 26.Giessing C, Thiel CM, Rosler F, Fink GR. The modulatory effects of nicotine on parietal cortex activity in a cued target detection task depend on cue reliability. Neuroscience. 2006;137:853–64. doi: 10.1016/j.neuroscience.2005.10.005. [DOI] [PubMed] [Google Scholar]
- 27.Rusted JM, Caulfield D, King L, Goode A. Moving out of the laboratory: does nicotine improve everyday attention? Behav Pharmacol. 2000;11:621–29. doi: 10.1097/00008877-200011000-00009. [DOI] [PubMed] [Google Scholar]
- 28.Lindgren M, Stenberg G, Rosen I. Effects of nicotine in a bimodal attention task. Neuropsychobiology. 1998;38:42–49. doi: 10.1159/000026515. [DOI] [PubMed] [Google Scholar]
- 29.Sunderland T, Tariot PN, Newhouse PA. Differential responsivity of mood, behavior, and cognition to cholinergic agents in elderly neuropsychiatric populations. Brain Res. 1988;472:371–89. doi: 10.1016/0006-8993(88)91227-9. [DOI] [PubMed] [Google Scholar]
- 30.Wilson AL, Langley LK, Monley J, Bauer T, Rottunda S, McFalls E, et al. Nicotine patches in Alzheimer’s disease: pilot study on learning, memory, and safety. Pharmacol Biochem Behav. 1995;51:509–14. doi: 10.1016/0091-3057(95)00043-v. [DOI] [PubMed] [Google Scholar]
- 31.White HK, Levin ED. Four-week nicotine skin patch treatment effects on cognitive performance in Alzheimer’s disease. Psychopharmacology (Berl) 1999;143:158–65. doi: 10.1007/s002130050931. [DOI] [PubMed] [Google Scholar]
- 32.Newhouse PA, Sunderland T, Tariot PN, Blumhardt CL, Weingartner H, Mellow A, et al. Intravenous nicotine in Alzheimer’s disease: a pilot study. Psychopharmacology (Berl) 1988;95:171–75. doi: 10.1007/BF00174504. [DOI] [PubMed] [Google Scholar]
- 33.Jones GM, Sahakian BJ, Levy R, Warburton DM, Gray JA. Effects of acute subcutaneous nicotine on attention, information processing and short-term memory in Alzheimer’s disease. Psychopharmacology (Berl) 1992;108:485–94. doi: 10.1007/BF02247426. [DOI] [PubMed] [Google Scholar]
- 34.Sahakian B, Jones G, Levy R, Gray J, Warburton D. The effects of nicotine on attention, information processing, and short-term memory in patients with dementia of the Alzheimer type. Br J Psychiatry. 1989;154:797–800. doi: 10.1192/bjp.154.6.797. [DOI] [PubMed] [Google Scholar]
- 35.Newhouse PA, Potter A, Kelton M, Corwin J. Nicotinic treatment of Alzheimer’s disease. Biol Psychiatry. 2001;49:268–78. doi: 10.1016/s0006-3223(00)01069-6. [DOI] [PubMed] [Google Scholar]
- 36.Barr RS, Culhane MA, Jubelt LE, Mufti RS, Dyer MA, Weiss AP, et al. The effects of transdermal nicotine on cognition in nonsmokers with schizophrenia and nonpsychiatric controls. Neuropsychopharmacology. 2008;33:480–90. doi: 10.1038/sj.npp.1301423. [DOI] [PubMed] [Google Scholar]
- 37.Smith RC, Warner-Cohen J, Matute M, Butler E, Kelly E, Vaidhyanathaswamy S, et al. Effects of nicotine nasal spray on cognitive function in schizophrenia. Neuropsychopharmacology. 2006;31:637–43. doi: 10.1038/sj.npp.1300881. [DOI] [PubMed] [Google Scholar]
- 38.Potter AS, Newhouse PA. Acute nicotine improves cognitive deficits in young adults with attention-deficit/hyperactivity disorder. Pharmacol Biochem Behav. 2008;88:407–17. doi: 10.1016/j.pbb.2007.09.014. [DOI] [PubMed] [Google Scholar]
- 39.Potter AS, Newhouse PA, Bucci DJ. Central nicotinic cholinergic systems: a role in the cognitive dysfunction in attention-deficit/hyperactivity disorder? Behav Brain Res. 2006;175:201–11. doi: 10.1016/j.bbr.2006.09.015. [DOI] [PubMed] [Google Scholar]
- 40.Wilens TE, Decker MW. Neuronal nicotinic receptor agonists for the treatment of attention-deficit/hyperactivity disorder: focus on cognition. Biochem Pharmacol. 2007;74:1212–23. doi: 10.1016/j.bcp.2007.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Arneric SP, Sullivan JP, Briggs CA, Donnelly-Roberts D, Anderson DJ, Raszkiewicz JL, et al. (S)-3-methyl-5-(1-methyl-2-pyrrolidinyl) isoxazole (ABT 418): a novel cholinergic ligand with cognition-enhancing and anxiolytic activities: I. In vitro characterization. J Pharmacol Exp Ther. 1994;270:310–18. [PubMed] [Google Scholar]
- 42.Lippiello P, Letchworth SR, Gatto GJ, Traina VM, Bencherif M. Ispronicline: a novel alpha4beta2 nicotinic acetylcholine receptor-selective agonist with cognition-enhancing and neuroprotective properties. J Mol Neurosci. 2006;30:19–20. doi: 10.1385/JMN:30:1:19. [DOI] [PubMed] [Google Scholar]
- 43.Sullivan JP, Donnelly-Roberts D, Briggs CA, Anderson DJ, Gopalakrishnan M, Xue IC, et al. ABT-089 [2-methyl-3-(2-(S)-pyrrolidinylmethoxy)pyridine]: I. A potent and selective cholinergic channel modulator with neuroprotective properties. J Pharmacol Exp Ther. 1997;283:235–46. [PubMed] [Google Scholar]
- 44.Potter A, Corwin J, Lang J, Piasecki M, Lenox R, Newhouse PA. Acute effects of the selective cholinergic channel activator (nicotinic agonist) ABT-418 in Alzheimer’s disease. Psychopharmacology (Berl) 1999;142:334–42. doi: 10.1007/s002130050897. [DOI] [PubMed] [Google Scholar]
- 45.Dunbar GC, Inglis F, Kuchibhatla R, Sharma T, Tomlinson M, Wamsley J. Effect of ispronicline, a neuronal nicotinic acetylcholine receptor partial agonist, in subjects with age associated memory impairment (AAMI) J Psychopharmacol. 2007;21:171–78. doi: 10.1177/0269881107066855. [DOI] [PubMed] [Google Scholar]
- 46.Wilens TE, Verlinden MH, Adler LA, Wozniak PJ, West SA. ABT-089, a neuronal nicotinic receptor partial agonist, for the treatment of attention-deficit/hyperactivity disorder in adults: results of a pilot study. Biol Psychiatry. 2006;59:1065–70. doi: 10.1016/j.biopsych.2005.10.029. [DOI] [PubMed] [Google Scholar]
- 47.Sarter M, Gehring WJ, Kozak R. More attention must be paid: the neurobiology of attentional effort. Brain Res Rev. 2006;51:145–60. doi: 10.1016/j.brainresrev.2005.11.002. [DOI] [PubMed] [Google Scholar]
- 48.Mirza NR, Stolerman IP. Nicotine enhances sustained attention in the rat under specific task conditions. Psychopharmacology (Berl) 1998;138:266–74. doi: 10.1007/s002130050671. [DOI] [PubMed] [Google Scholar]
- 49.Hahn B, Shoaib M, Stolerman IP. Nicotine-induced enhancement of attention in the five-choice serial reaction time task: the influence of task demands. Psychopharmacology (Berl) 2002;162:129–37. doi: 10.1007/s00213-002-1005-6. [DOI] [PubMed] [Google Scholar]
- 50.Stolerman IP, Mirza NR, Hahn B, Shoaib M. Nicotine in an animal model of attention. Eur J Pharmacol. 2000;393:147–54. doi: 10.1016/s0014-2999(99)00886-9. [DOI] [PubMed] [Google Scholar]
- 51.Blondel A, Simon H, Sanger DJ, Moser P. The effect of repeated nicotine administration on the performance of drug-naive rats in a five-choice serial reaction time task. Behav Pharmacol. 1999;10:665–73. doi: 10.1097/00008877-199911000-00013. [DOI] [PubMed] [Google Scholar]
- 52.Blondel A, Sanger DJ, Moser PC. Characterisation of the effects of nicotine in the five-choice serial reaction time task in rats: antagonist studies. Psychopharmacology (Berl) 2000;149:293–305. doi: 10.1007/s002130000378. [DOI] [PubMed] [Google Scholar]
- 53.Hahn B, Stolerman IP. Nicotine-induced attentional enhancement in rats: effects of chronic exposure to nicotine. Neuropsychopharmacology. 2002;27:712–22. doi: 10.1016/S0893-133X(02)00348-2. [DOI] [PubMed] [Google Scholar]
- 54.Amitai N, Markou A. Chronic nicotine improves cognitive performance in a test of attention but does not attenuate cognitive disruption induced by repeated phencyclidine administration. Psychopharmacology (Berl) 2009;202:275–86. doi: 10.1007/s00213-008-1246-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Demeter E, Sarter M, Lustig C. Rats and humans paying attention: cross-species task development for translational research. Neuropsychology. 2008;22:787–99. doi: 10.1037/a0013712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Turchi J, Holley LA, Sarter M. Effects of nicotinic acetylcholine receptor ligands on behavioral vigilance in rats. Psychopharmacology (Berl) 1995;118:195–205. doi: 10.1007/BF02245840. [DOI] [PubMed] [Google Scholar]
- 57.McGaughy J, Decker MW, Sarter M. Enhancement of sustained attention performance by the nicotinic acetylcholine receptor agonist ABT-418 in intact but not basal forebrain-lesioned rats. Psychopharmacology (Berl) 1999;144:175–82. doi: 10.1007/s002130050991. [DOI] [PubMed] [Google Scholar]
- 58.Grottick AJ, Haman M, Wyler R, Higgins GA. Reversal of a vigilance decrement in the aged rat by subtype-selective nicotinic ligands. Neuropsychopharmacology. 2003;28:880–87. doi: 10.1038/sj.npp.1300102. [DOI] [PubMed] [Google Scholar]
- 59.Grottick AJ, Higgins GA. Effect of subtype selective nicotinic compounds on attention as assessed by the five-choice serial reaction time task. Behav Brain Res. 2000;117:197–208. doi: 10.1016/s0166-4328(00)00305-3. [DOI] [PubMed] [Google Scholar]
- 60.Hahn B, Sharples CG, Wonnacott S, Shoaib M, Stolerman IP. Attentional effects of nicotinic agonists in rats. Neuropharmacology. 2003;44:1054–67. doi: 10.1016/s0028-3908(03)00099-6. [DOI] [PubMed] [Google Scholar]
- 61.Young JW, Finlayson K, Spratt C, Marston HM, Crawford N, Kelly JS, et al. Nicotine improves sustained attention in mice: evidence for involvement of the alpha7 nicotinic acetylcholine receptor. Neuropsychopharmacology. 2004;29:891–900. doi: 10.1038/sj.npp.1300393. [DOI] [PubMed] [Google Scholar]
- 62.Holley LA, Wiley RG, Lappi DA, Sarter M. Cortical cholinergic deafferentation following the intracortical infusion of 192 IgG-saporin: a quantitative histochemical study. Brain Res. 1994;663:277–86. doi: 10.1016/0006-8993(94)91274-2. [DOI] [PubMed] [Google Scholar]
- 63.McGaughy J, Kaiser T, Sarter M. Behavioral vigilance following infusions of 192 IgG-saporin into the basal forebrain: selectivity of the behavioral impairment and relation to cortical AChE-positive fiber density. Behav Neurosci. 1996;110:247–65. doi: 10.1037//0735-7044.110.2.247. [DOI] [PubMed] [Google Scholar]
- 64.McGaughy J, Everitt BJ, Robbins TW, Sarter M. The role of cortical cholinergic afferent projections in cognition: impact of new selective immunotoxins. Behav Brain Res. 2000;115:251–63. doi: 10.1016/s0166-4328(00)00262-x. [DOI] [PubMed] [Google Scholar]
- 65.Posner MI, Snyder CRR, Davidson BJ. Attention and the detection of signals. Journal of Experimental Psychology: General. 1980;109:160–74. [PubMed] [Google Scholar]
- 66.Kozak R, Bruno JP, Sarter M. Augmented prefrontal acetylcholine release during challenged attentional performance. Cereb Cortex. 2006;16:9–17. doi: 10.1093/cercor/bhi079. [DOI] [PubMed] [Google Scholar]
- 67.Kozak R, Martinez V, Young D, Brown H, Bruno JP, Sarter M. Toward a neuro-cognitive animal model of the cognitive symptoms of schizophrenia: disruption of cortical cholinergic neurotransmission following repeated amphetamine exposure in attentional task-performing, but not non-performing, rats. Neuropsychopharmacology. 2007;32:2074–86. doi: 10.1038/sj.npp.1301352. [DOI] [PubMed] [Google Scholar]
- 68.Arnold HM, Burk JA, Hodgson EM, Sarter M, Bruno JP. Differential cortical acetylcholine release in rats performing a sustained attention task versus behavioral control tasks that do not explicitly tax attention. Neuroscience. 2002;114:451–60. doi: 10.1016/s0306-4522(02)00292-0. [DOI] [PubMed] [Google Scholar]
- 69.Himmelheber AM, Sarter M, Bruno JP. Operant performance and cortical acetylcholine release: role of response rate, reward density, and non-contingent stimuli. Brain Res Cogn Brain Res. 1997;6:23–36. doi: 10.1016/s0926-6410(97)00014-1. [DOI] [PubMed] [Google Scholar]
- 70.Dalley JW, McGaughy J, O’Connell MT, Cardinal RN, Levita L, Robbins TW. Distinct changes in cortical acetylcholine and noradrenaline efflux during contingent and noncontingent performance of a visual attentional task. J Neurosci. 2001;21:4908–14. doi: 10.1523/JNEUROSCI.21-13-04908.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Passetti F, Dalley JW, O’Connell MT, Everitt BJ, Robbins TW. Increased acetylcholine release in the rat medial prefrontal cortex during performance of a visual attentional task. Eur J Neurosci. 2000;12:3051–58. doi: 10.1046/j.1460-9568.2000.00183.x. [DOI] [PubMed] [Google Scholar]
- 72.Apparsundaram S, Martinez V, Parikh V, Kozak R, Sarter M. Increased capacity and density of choline transporters situated in synaptic membranes of the right medial prefrontal cortex of attentional task-performing rats. J Neurosci. 2005;25:3851–56. doi: 10.1523/JNEUROSCI.0205-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Martinez V, Sarter M. Lateralized attentional functions of cortical cholinergic inputs. Behav Neurosci. 2004;118:984–91. doi: 10.1037/0735-7044.118.5.984. [DOI] [PubMed] [Google Scholar]
- 74.Dalley JW, Theobald DE, Bouger P, Chudasama Y, Cardinal RN, Robbins TW. Cortical cholinergic function and deficits in visual attentional performance in rats following 192 IgG-saporin-induced lesions of the medial prefrontal cortex. Cereb Cortex. 2004;14:922–32. doi: 10.1093/cercor/bhh052. [DOI] [PubMed] [Google Scholar]
- 75.Parikh V, Pomerleau F, Huettl P, Gerhardt GA, Sarter M, Bruno JP. Rapid assessment of in vivo cholinergic transmission by amperometric detection of changes in extracellular choline levels. Eur J Neurosci. 2004;20:1545–54. doi: 10.1111/j.1460-9568.2004.03614.x. [DOI] [PubMed] [Google Scholar]
- 76.Parikh V, Sarter M. Cortical choline transporter function measured in vivo using choline-sensitive microelectrodes: clearance of endogenous and exogenous choline and effects of removal of cholinergic terminals. J Neurochem. 2006;97:488–503. doi: 10.1111/j.1471-4159.2006.03766.x. [DOI] [PubMed] [Google Scholar]
- 77.Parikh V, Kozak R, Martinez V, Sarter M. Prefrontal acetylcholine release controls cue detection on multiple timescales. Neuron. 2007;56:141–54. doi: 10.1016/j.neuron.2007.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Howe WM, Parikh V, Martinez V, Sarter M. Prefrontal cholinergic switching from associational processing to cue detection: evidence from sub-second measures of prefrontal cholinergic neurotransmission, using choline-sensitive microelectrodes, in animals performing an operant sustained attention task. Society for Neuroscience Annual Meeting. 2007 [Google Scholar]
- 79.Buckner RL, Andrews-Hanna JR, Schacter DL. The brain’s default network: anatomy, function, and relevance to disease. Ann N Y Acad Sci. 2008;1124:1–38. doi: 10.1196/annals.1440.011. [DOI] [PubMed] [Google Scholar]
- 80.Sridharan D, Levitin DJ, Menon V. A critical role for the right fronto-insular cortex in switching between central-executive and default-mode networks. Proc Natl Acad Sci U S A. 2008;105:12569–74. doi: 10.1073/pnas.0800005105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Eichele T, Debener S, Calhoun VD, Specht K, Engel AK, Hugdahl K, et al. Prediction of human errors by maladaptive changes in event-related brain networks. Proc Natl Acad Sci U S A. 2008;105:6173–78. doi: 10.1073/pnas.0708965105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Weissman DH, Roberts KC, Visscher KM, Woldorff MG. The neural bases of momentary lapses in attention. Nat Neurosci. 2006;9:971–78. doi: 10.1038/nn1727. [DOI] [PubMed] [Google Scholar]
- 83.Singh KD, Fawcett IP. Transient and linearly graded deactivation of the human default-mode network by a visual detection task. Neuroimage. 2008;41:100–12. doi: 10.1016/j.neuroimage.2008.01.051. [DOI] [PubMed] [Google Scholar]
- 84.Sarter M, Nelson CL, Bruno JP. Cortical cholinergic transmission and cortical information processing in schizophrenia. Schizophr Bull. 2005;31:117–38. doi: 10.1093/schbul/sbi006. [DOI] [PubMed] [Google Scholar]
- 85.MSCL . Attentional functions in learning and memory. In: LR S, editor. Encyclopedia of Neuroscience. Oxford: Academic Press; 2009. pp. 639–45. [Google Scholar]
- 86.Broyd SJ, Demanuele C, Debener S, Helps SK, James CJ, Sonuga-Barke EJ. Default-mode brain dysfunction in mental disorders: A systematic review. Neurosci Biobehav Rev. 2008 doi: 10.1016/j.neubiorev.2008.09.002. [DOI] [PubMed] [Google Scholar]
- 87.Persson J, Lustig C, Nelson JK, Reuter-Lorenz PA. Age differences in deactivation: a link to cognitive control? J Cogn Neurosci. 2007;19:1021–32. doi: 10.1162/jocn.2007.19.6.1021. [DOI] [PubMed] [Google Scholar]
- 88.Bishop SJ. Trait anxiety and impoverished prefrontal control of attention. Nat Neurosci. 2009;12:92–98. doi: 10.1038/nn.2242. [DOI] [PubMed] [Google Scholar]
- 89.Lustig C, Snyder AZ, Bhakta M, O’Brien KC, McAvoy M, Raichle ME, et al. Functional deactivations: change with age and dementia of the Alzheimer type. Proc Natl Acad Sci U S A. 2003;100:14504–09. doi: 10.1073/pnas.2235925100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Sarter M, Markowitsch HJ. Collateral innervation of the medial and lateral prefrontal cortex by amygdaloid, thalamic, and brain-stem neurons. J Comp Neurol. 1984;224:445–60. doi: 10.1002/cne.902240312. [DOI] [PubMed] [Google Scholar]
- 91.Crick F. Function of the thalamic reticular complex: the searchlight hypothesis. Proc Natl Acad Sci U S A. 1984;81:4586–90. doi: 10.1073/pnas.81.14.4586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Pinault D. The thalamic reticular nucleus: structure, function and concept. Brain Res Brain Res Rev. 2004;46:1–31. doi: 10.1016/j.brainresrev.2004.04.008. [DOI] [PubMed] [Google Scholar]
- 93.McAlonan K, Cavanaugh J, Wurtz RH. Attentional modulation of thalamic reticular neurons. J Neurosci. 2006;26:4444–50. doi: 10.1523/JNEUROSCI.5602-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Zikopoulos B, Barbas H. Prefrontal projections to the thalamic reticular nucleus form a unique circuit for attentional mechanisms. J Neurosci. 2006;26:7348–61. doi: 10.1523/JNEUROSCI.5511-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Zikopoulos B, Barbas H. Circuits formultisensory integration and attentional modulation through the prefrontal cortex and the thalamic reticular nucleus in primates. Rev Neurosci. 2007;18:417–38. doi: 10.1515/revneuro.2007.18.6.417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Zikopoulos B, Barbas H. Parallel driving and modulatory pathways link the prefrontal cortex and thalamus. PLoS ONE. 2007;2:e848. doi: 10.1371/journal.pone.0000848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Weese GD, Phillips JM, Brown VJ. Attentional orienting is impaired by unilateral lesions of the thalamic reticular nucleus in the rat. J Neurosci. 1999;19:10135–39. doi: 10.1523/JNEUROSCI.19-22-10135.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Jourdain A, Semba K, Fibiger HC. Basal forebrain and mesopontine tegmental projections to the reticular thalamic nucleus: an axonal collateralization and immunohistochemical study in the rat. Brain Res. 1989;505:55–65. doi: 10.1016/0006-8993(89)90115-7. [DOI] [PubMed] [Google Scholar]
- 99.Bickford ME, Gunluk AE, Van Horn SC, Sherman SM. GABAergic projection from the basal forebrain to the visual sector of the thalamic reticular nucleus in the cat. J Comp Neurol. 1994;348:481–510. doi: 10.1002/cne.903480402. [DOI] [PubMed] [Google Scholar]
- 100.Parikh V, Man K, Decker MW, Sarter M. Glutamatergic contributions to nicotinic acetylcholine receptor agonist-evoked cholinergic transients in the prefrontal cortex. J Neurosci. 2008;28:3769–80. doi: 10.1523/JNEUROSCI.5251-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Lucas-Meunier E, Monier C, Amar M, Baux G, Fregnac Y, Fossier P. Involvement of Nicotinic and Muscarinic Receptors in the Endogenous Cholinergic Modulation of the Balance between Excitation and Inhibition in the Young Rat Visual Cortex. Cereb Cortex. 2009 doi: 10.1093/cercor/bhn258. [DOI] [PubMed] [Google Scholar]
- 102.Jiang L, Role LW. Facilitation of cortico-amygdala synapses by nicotine: activity-dependent modulation of glutamatergic transmission. J Neurophysiol. 2008;99:1988–99. doi: 10.1152/jn.00933.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Galarreta M, Hestrin S. Spike transmission and synchrony detection in networks of GABAergic interneurons. Science. 2001;292:2295–99. doi: 10.1126/science.1061395. [DOI] [PubMed] [Google Scholar]
- 104.McClure-Begley TD, King NM, Collins AC, Stitzel JA, Wehner JM, Butt CM. Acetylcholine-Stimulated [3H]GABA Release from Mouse Brain Synaptosomes is Modulated by {alpha}4{beta}2 and {alpha}4{alpha}5{beta}2 Nicotinic Receptor Subtypes. Mol Pharmacol. 2009 doi: 10.1124/mol.108.052274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Rasmusson DD, Smith SA, Semba K. Inactivation of prefrontal cortex abolishes cortical acetylcholine release evoked by sensory or sensory pathway stimulation in the rat. Neuroscience. 2007;149:232–41. doi: 10.1016/j.neuroscience.2007.06.057. [DOI] [PubMed] [Google Scholar]
- 106.Kimura F, Baughman RW. Distinct muscarinic receptor subtypes suppress excitatory and inhibitory synaptic responses in cortical neurons. J Neurophysiol. 1997;77:709–16. doi: 10.1152/jn.1997.77.2.709. [DOI] [PubMed] [Google Scholar]
- 107.Atzori M, Kanold PO, Pineda JC, Flores-Hernandez J, Paz RD. Dopamine prevents muscarinic-induced decrease of glutamate release in the auditory cortex. Neuroscience. 2005;134:1153–65. doi: 10.1016/j.neuroscience.2005.05.005. [DOI] [PubMed] [Google Scholar]
- 108.Lin CS, Nicolelis MA, Schneider JS, Chapin JK. A major direct GABAergic pathway from zona incerta to neocortex. Science. 1990;248:1553–56. doi: 10.1126/science.2360049. [DOI] [PubMed] [Google Scholar]
- 109.Lin SC, Nicolelis MA. Neuronal ensemble bursting in the basal forebrain encodes salience irrespective of valence. Neuron. 2008;59:138–49. doi: 10.1016/j.neuron.2008.04.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Seu E, Lang A, Rivera RJ, Jentsch JD. Inhibition of the norepinephrine transporter improves behavioral flexibility in rats and monkeys. Psychopharmacology (Berl) 2009;202:505–19. doi: 10.1007/s00213-008-1250-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.McGaughy J, Ross RS, Eichenbaum H. Noradrenergic, but not cholinergic, deafferentation of prefrontal cortex impairs attentional set-shifting. Neuroscience. 2008;153:63–71. doi: 10.1016/j.neuroscience.2008.01.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Aston-Jones G, Cohen JD. An integrative theory of locus coeruleus-norepinephrine function: adaptive gain and optimal performance. Annu Rev Neurosci. 2005;28:403–50. doi: 10.1146/annurev.neuro.28.061604.135709. [DOI] [PubMed] [Google Scholar]
- 113.Gill TM, Sarter M, Givens B. Sustained visual attention performance-associated prefrontal neuronal activity: evidence for cholinergic modulation. J Neurosci. 2000;20:4745–57. doi: 10.1523/JNEUROSCI.20-12-04745.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Neigh GN, Arnold HM, Rabenstein RL, Sarter M, Bruno JP. Neuronal activity in the nucleus accumbens is necessary for performance-related increases in cortical acetylcholine release. Neuroscience. 2004;123:635–45. doi: 10.1016/j.neuroscience.2003.10.006. [DOI] [PubMed] [Google Scholar]
- 115.Zmarowski A, Sarter M, Bruno JP. NMDA and dopamine interactions in the nucleus accumbens modulate cortical acetylcholine release. Eur J Neurosci. 2005;22:1731–40. doi: 10.1111/j.1460-9568.2005.04333.x. [DOI] [PubMed] [Google Scholar]
- 116.Zmarowski A, Sarter M, Bruno JP. Glutamate receptors in nucleus accumbens mediate regionally selective increases in cortical acetylcholine release. Synapse. 2007;61:115–23. doi: 10.1002/syn.20354. [DOI] [PubMed] [Google Scholar]
- 117.Livingstone PD, Srinivasan J, Kew JN, Dawson LA, Gotti C, Moretti M, et al. alpha7 and non-alpha7 nicotinic acetylcholine receptors modulate dopamine release in vitro and in vivo in the rat prefrontal cortex. Eur J Neurosci. 2009 doi: 10.1111/j.1460-9568.2009.06613.x. [DOI] [PubMed] [Google Scholar]
- 118.Lambe EK, Picciotto MR, Aghajanian GK. Nicotine induces glutamate release from thalamocortical terminals in prefrontal cortex. Neuropsychopharmacology. 2003;28:216–25. doi: 10.1038/sj.npp.1300032. [DOI] [PubMed] [Google Scholar]
- 119.Lambe EK, Olausson P, Horst NK, Taylor JR, Aghajanian GK. Hypocretin and nicotine excite the same thalamocortical synapses in prefrontal cortex: correlation with improved attention in rat. J Neurosci. 2005;25:5225–29. doi: 10.1523/JNEUROSCI.0719-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Disney AA, Aoki C, Hawken MJ. Gain modulation by nicotine in macaque v1. Neuron. 2007;56:701–13. doi: 10.1016/j.neuron.2007.09.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Kawai H, Lazar R, Metherate R. Nicotinic control of axon excitability regulates thalamocortical transmission. Nat Neurosci. 2007;10:1168–75. doi: 10.1038/nn1956. [DOI] [PubMed] [Google Scholar]
- 122.Quarta D, Naylor CG, Morris HV, Patel S, Genn RF, Stolerman IP. Different effects of ionotropic and metabotropic glutamate receptor antagonists on attention and the attentional properties of nicotine. Neuropharmacology. 2007;53:421–30. doi: 10.1016/j.neuropharm.2007.05.023. [DOI] [PubMed] [Google Scholar]
- 123.Moroni M, Zwart R, Sher E, Cassels BK, Bermudez I. alpha4beta2 nicotinic receptors with high and low acetylcholine sensitivity: pharmacology, stoichiometry, and sensitivity to long-term exposure to nicotine. Mol Pharmacol. 2006;70:755–68. doi: 10.1124/mol.106.023044. [DOI] [PubMed] [Google Scholar]
- 124.Nashmi R, Xiao C, Deshpande P, McKinney S, Grady SR, Whiteaker P, et al. Chronic nicotine cell specifically upregulates functional alpha 4* nicotinic receptors: basis for both tolerance in midbrain and enhanced long-term potentiation in perforant path. J Neurosci. 2007;27:8202–18. doi: 10.1523/JNEUROSCI.2199-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Bitner RS, Bunnelle WH, Anderson DJ, Briggs CA, Buccafusco J, Curzon P, et al. Broad-spectrum efficacy across cognitive domains by alpha7 nicotinic acetylcholine receptor agonism correlates with activation of ERK1/2 and CREB phosphorylation pathways. J Neurosci. 2007;27:10578–87. doi: 10.1523/JNEUROSCI.2444-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Turchi J, Sarter M. Cortical acetylcholine and processing capacity: effects of cortical cholinergic deafferentation on crossmodal divided attention in rats. Cogn Brain Res. 1997;6:147–58. doi: 10.1016/s0926-6410(97)00027-x. [DOI] [PubMed] [Google Scholar]
- 127.Kennedy KM, Raz N. Aging white matter and cognition: Differential effects of regional variations in diffusion properties on memory, executive functions, and speed. Neuropsychologia. 2009 doi: 10.1016/j.neuropsychologia.2009.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Thiel CM, Fink GR. Effects of the cholinergic agonist nicotine on reorienting of visual spatial attention and top-down attentional control. Neuroscience. 2008;152:381–90. doi: 10.1016/j.neuroscience.2007.10.061. [DOI] [PubMed] [Google Scholar]
- 129.Nelson CL, Sarter M, Bruno JP. Prefrontal cortical modulation of acetylcholine release in posterior parietal cortex. Neuroscience. 2005;132:347–59. doi: 10.1016/j.neuroscience.2004.12.007. [DOI] [PubMed] [Google Scholar]
- 130.Bandyopadhyay S, Sutor B, Hablitz JJ. Endogenous acetylcholine enhances synchronized interneuron activity in rat neocortex. J Neurophysiol. 2006;95:1908–16. doi: 10.1152/jn.00881.2005. [DOI] [PubMed] [Google Scholar]
- 131.Azam L, Winzer-Serhan U, Leslie FM. Co-expression of alpha7 and beta2 nicotinic acetylcholine receptor subunit mRNAs within rat brain cholinergic neurons. Neuroscience. 2003;119:965–77. doi: 10.1016/s0306-4522(03)00220-3. [DOI] [PubMed] [Google Scholar]
- 132.Materi LM, Semba K. Inhibition of synaptically evoked cortical acetylcholine release by intracortical glutamate: involvement of GABAergic neurons. Eur J Neurosci. 2001;14:38–46. doi: 10.1046/j.0953-816x.2001.01619.x. [DOI] [PubMed] [Google Scholar]
- 133.Howe WM, Parikh V, Gritton H, Giuliano C, Ward J, Sarter M. Prefrontal cholinergic transients indicating cue detection as a target for cognition enhancers. Society for Neuroscience Annual Meeting. 2008;388:26. [Google Scholar]